
Abstract
Over the past 5 years, improvements in the design of antibody–drug conjugates (ADCs) have enabled major advances that have reshaped the treatment of several advanced-stage solid tumours. Considering the intended rationale behind the design of ADCs, which is to achieve targeted delivery of cytotoxic molecules by linking them to antibodies targeting tumour-specific antigens, ADCs would be expected to be less toxic than conventional chemotherapy. However, most ADCs are still burdened by off-target toxicities that resemble those of the cytotoxic payload as well as on-target toxicities and other poorly understood and potentially life-threatening adverse effects. Given the rapid expansion in the clinical indications of ADCs, including use in curative settings and various combinations, extensive efforts are ongoing to improve their safety. Approaches currently being pursued include clinical trials optimizing the dose and treatment schedule, modifications of each ADC component, identification of predictive biomarkers for toxicities, and the development of innovative diagnostic tools. In this Review, we describe the determinants of the toxicities of ADCs in patients with solid tumours, highlighting key strategies that are expected to improve tolerability and enable improvements in the treatment outcomes of patients with advanced-stage and those with early stage cancers in the years to come.
Key points
-
Indications for the use of antibody–drug conjugates (ADCs) are rapidly expanding, with development progressively moving from the advanced-stage to the early stage setting, and from monotherapy to combination strategies.
-
Despite being designed with the rationale of expanding the therapeutic indices of conventional chemotherapies, most ADCs have a toxicity profile similar to that of their cytotoxic payload.
-
Unconventional and potentially life-threatening toxicities can also be observed with certain ADCs, requiring an increased understanding of these events and the optimization of diagnostic and management practices.
-
Multiple pharmacological modification strategies are being pursued in an attempt to improve the tolerability of ADCs, including molecular alterations of the antibody moiety, linker and/or cytotoxic payload.
-
Exploration of different doses within randomized trials and investigation of response-adapted dosing strategies could enable optimized use of ADCs, which could maximize their therapeutic value for each indication.
-
Extensive efforts to identify biomarkers of toxicities in patients receiving ADCs and to develop diagnostic tools enabling the anticipation and/or early detection of toxicities are currently ongoing.
Similar content being viewed by others

Introduction
The targeted delivery of cytotoxic payloads through antibody–drug conjugates (ADCs) is rapidly emerging as a highly effective strategy to treat multiple types of cancer1. A total of 12 ADCs are currently approved by the FDA and/or EMA for patients with various cancers2,3, with 6 of these approved for solid tumour indications. Importantly, most of these approvals have occurred in the past 4 years, reflecting an expanding pipeline of promising agents that is expected to lead to further approvals in the near future4. Together with improving outcomes for patients with a range of tumour types, ADCs are challenging the current approach to the categorization of cancer: HER2-targeted ADCs have shown activity in breast and gastric cancers deemed to be HER2-negative by traditional criteria5,6,7,8,9,10, leading to the definition of a novel ‘HER2-low’ subset of targetable tumours11. Although only preliminary data are currently available, this novel subset might expand further across tumour types based on positive results from the DESTINY-Pantumor02 phase II trial12. The cross-histological activity of ADCs and the expansion of their reach beyond canonical biomarker-defined subtypes have a crucial repercussion: a large and rapidly increasing proportion of patients with cancer are becoming eligible for treatment with ADCs. This is particularly true for patients with advanced-stage cancers but also includes some patients with early stage solid tumours, a setting in which multiple trials testing ADCs are currently ongoing in an attempt to prevent disease recurrence and thus improve cure rates.
ADCs were originally designed with the aim of improving the therapeutic index of conventional chemotherapies by enabling the selective delivery of cytotoxic payloads to cancer cells expressing a tumour-associated antigen targeted by the antibody component of the conjugate13. However, this objective has not been clearly achieved by the current generation of ADCs: indeed, after normalizing for cytotoxin content, ADCs have been found to have a similar maximum-tolerated dose to that of their unconjugated cytotoxic payload, and have not yet been proven to lower the minimum effective dose required for antitumour activity. Rather, data suggest that, when dosed at or near their maximum-tolerated doses, certain ADCs have enhanced antitumour activity compared to that of the closely related unconjugated cytotoxic compound14. In numerical terms, a meta-analysis of data from 169 clinical trials testing ADCs found that these agents are associated with treatment-related adverse events in >90% of patients (including grade ≥3 events in 40%), with extensive variability in the types of toxicities and their incidence depending on the specific composition of the conjugate15. Furthermore, safety concerns have led to discontinuation of the clinical development of numerous initially promising ADCs. For example, the delta-like protein 3 (DLL3)-targeted ADC rovalpituzumab tesirine, which reached phase III testing in patients with small-cell lung cancer, was found to be less tolerable and to provide inferior overall survival compared to topotecan16, leading to discontinuation of further development. Similarly, the EGFR-targeted construct depatuxizumab mafodotin17 was discontinued after the emergence of relevant toxicities plus no improvement in overall survival when added to standard-of-care therapy in a phase III trial involving patients with glioblastoma.
Given the large proportion of patients with cancer currently receiving ADCs as well as the expected expansion in the use of these compounds to patients with early stage disease, improving our understanding of the adverse effects of ADCs is becoming increasingly important to enable safe implementation in clinical practice and to optimize the design of novel conjugates. In this Review, we discuss current knowledge of the toxicities of ADCs in patients with solid tumours, highlighting ongoing efforts to optimize the safety of these agents in clinical practice.
ADCs as targeted chemotherapy
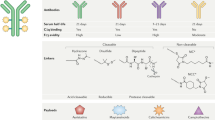
Despite extensive differences in structure, all approved ADCs share three key elements: an engineered monoclonal antibody targeting a tumour-associated antigen, a cytotoxic payload and a molecular linker connecting the payload with the antibody13. Such a structure has the rationale of merging the selectivity of an antibody with the antitumour activity of cytotoxic molecules with the aim of delivering the highest possible dose of the cytotoxic drug to the tumour while also sparing non-malignant tissues. In addition to delivering a cytotoxic payload, ADCs have been shown to retain the antitumour effects of the antibodies, including inhibition of oncogenic signalling pathways and enhancement of antitumour immunity through the induction of antibody-dependent cellular cytotoxicity18. In this framework, ADCs can neither be solely intended as chemotherapies nor as targeted therapies; they rather represent a complex combination of these two treatment modalities, often considered to be ‘targeted chemotherapies’. This combination is reflected in both the antitumour activity and toxicities of this class of agents (Fig. 1 and Table 1).

Thus far, six antibody–drug conjugates (ADCs) have been approved by the FDA and/or EMA for patients with solid tumours. This figure depicts the composition of each of these ADCs (in terms of targeted antigen, type of monoclonal antibody, payload and linker), the currently approved indications and the most common toxicities observed with each agent. The adverse effect profile of each ADC is often a mixture of on-target and off-target effects, with the latter generally determining the maximum-tolerated dose. Common adverse effects seen to varying degrees across many ADCs include fatigue, alopecia, cytopenias and gastrointestinal disturbances, although individual ADCs can also be associated with more unique toxicities related to the stability of the compound and the complex interactions of various different components of the conjugate. CINV, chemotherapy-induced nausea and vomiting; GGFG, Gly-Gly-Phe-Gly; DAR, drug-to-antibody ratio; DXd, deruxtecan; ILD, interstitial lung disease; MCC, maleimidomethyl cyclohexane-1-carboxylate; MMAE, monomethyl auristatin E; TOPO1, topoisomerase 1; Trop2, trophoblast cell-surface antigen 2; VCit, valine-citrulline.
Given their large molecular size, ADCs are generally administered via intravenous injection, either as a bolus dose or an infusion; subcutaneous administration of ADCs has not yet been proven to be safe, and data from in vivo experiments suggest that this route of administration could be associated with reduced activity and severe skin toxicities for some ADCs19. After injection, ADCs are ideally meant to remain intact while in circulation and to only release their cytotoxic payload within or in close proximity to the targeted tumour cells. Each ADC characteristic has a crucial role in achieving this objective, with small modifications in structure often leading to dramatic changes in the pharmacokinetic and/or pharmacodynamic profile of the agent (Table 2). After injection, ADCs typically circulate in the bloodstream as a dynamic mixture of the intact ADC (>90% of the composition)20, the unconjugated drug (or drug–linker) and the dissociated antibody13. From the circulation, ADCs progressively diffuse into the interstitial spaces of body tissues, ultimately reaching the targeted tumour cells at a fraction estimated to be approximately 0.1% for solid tumours21,22. The limited fraction of ADCs reaching tumour cells underscores the need to utilize potent cytotoxic molecules, which, with few exceptions, are generally unsuitable for unconjugated administration. Once an ADC reaches the tumour microenvironment (TME), it is generally thought to bind the target antigen expressed on the cancer cell surface, undergo internalization within endosomes, and release the payload via chemical or enzymatic cleavage in the lysosomes, ultimately leading to either necrosis or apoptosis depending on the mechanism of action of the payload and the concentration reached in the target site23. More hydrophobic payloads (such as monomethyl auristatin E (MMAE) and exatecan derivatives) are able to diffuse outside of the target cells following intracellular deconjugation from the antibody, thus exerting a ‘bystander killing’ effect on antigen-negative cells that is thought to enhance the antitumour activity of certain ADCs24,25,26. This effect can be associated with increased efficacy and killing of cancer cells in tumours with limited or heterogeneous expression of the target antigen27, although it also constitutes a determinant of toxicity28: the released payload can also enter adjacent non-malignant cells via passive diffusion or transporter-mediated uptake, potentially leading to off-target cytotoxicity28. Beyond the traditionally accepted mechanism of targeted payload release within tumour cells, certain ADCs do not require antigen engagement and internalization in order to release the cytotoxic payload29. For example, sacituzumab govitecan was shown to release its SN-38 payload extracellularly within the TME, and this mechanism possibly explains both the activity and toxicities seen with this and other ADCs30.
As mentioned above, most parts of the ADC do not reach tumour cells and are progressively degraded and then eliminated via a combination of specific and non-specific mechanisms, including target-mediated clearance, Fcγ receptor (FcγR)-mediated uptake and/or pinocytosis by macrophages located in a variety of tissues31,32. Importantly, before undergoing degradation and excretion, payload–linker complexes released from ADCs utilizing thiol-maleimide chemistries (accounting for most of the approved ADCs) can react with the free cysteine residues of serum albumin, thus producing long-lived albumin–linker–payload adducts14,33. Although still incompletely characterized, this phenomenon might have implications for both the activity and toxicity profile of certain ADCs via albumin-conjugate tumour uptake and non-specific disposition in non-malignant tissues that ultimately prolongs the half-life of the cytotoxic payload after each infusion14,34.
Dissecting the toxicities of ADCs
ADCs are modular agents and small modifications of any of their key components can lead to major changes in clinical profile35,36,37,38 (Fig. 2). In this section, we dissect the relative contributions of each ADC component and the role of patient characteristics in determining the type and severity of the toxicities observed in patients receiving these agents.

The adverse effect profile of antibody–drug conjugates (ADCs) is often dominated by off-target, off-tumour toxicities that usually resemble those of the cytotoxic payload carried by the conjugate. Therefore, payload selection can have major implications for the expected toxicity profile of ADCs. However, the type, incidence and severity of these toxicities are also a function of the stability of the linker used to bind the payload to the antibody, with a less-stable linker expected to lead to increased chemotherapy-related toxicities owing to higher peak concentrations of released payload, and a more-stable linker sometimes associated with an increased incidence of unexpected adverse effects such as ocular toxicities. The antibody component of ADCs can contribute to the toxicity profile through the induction of on-target, off-tumour toxicities as well as via engagement of Fcγ receptors on immune cells. Lastly, extensive interpatient variability in the toxicity of the same ADC can be observed depending on multiple patient-related factors, including baseline organ function, the presence of comorbidities, pharmacogenomic polymorphisms, body composition and ethnicity. Cit, citrulline; GSH, glutathione; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; Trop2, trophoblast cell-surface antigen 2; Val, valine. aHighly stable linkers have been associated with an increase in the incidence of certain adverse effects, including ocular toxicities, neurotoxicities or hepatotoxicities, depending on the specific agent.
Payload
On the basis of the rationale behind the development of ADCs, the identity of the targeted antigen would be expected to determine the toxicity profile of the drug. However, clinical experience indicates that most of the adverse events associated with ADCs remain similar in spectrum, incidence and severity to those of the payload backbone14,15, and different ADCs that share the same payload often have similar toxicity profiles regardless of differences in the target antigen39. These toxicities can be broadly divided into off-target, off-tumour effects that are unrelated to the antigen targeted by the antibody vehicle, and on-target, off-tumour effects resulting from engagement of the antibody with cognate antigen located in non-malignant tissues.
Off-target, off-tumour toxicities dominate the toxicity profile of most ADCs15, often resulting in an adverse effect profile resembling that of the backbone payload14,15. For example, ADCs carrying auristatins (synthetic analogues of dolastatin with antimitotic activity relating to F-actin stabilization) commonly lead to peripheral neuropathies. For example, any-grade peripheral neuropathies have been reported in 46.3% of patients with advanced-stage urothelial carcinoma receiving enfortumab vedotin40, which is similar to the incidence of such events in patients receiving unconjugated dolastatin (any-grade peripheral neuropathies have been reported in 36% of patients with solid tumours receiving dolastatin 10)41. ADCs carrying an antimitotic microtubule-targeting maytansine derivative (DM1) payload can induce hepatotoxicity and thrombocytopenia (any-grade forms of both events occur in approximately 30% of patients with breast cancer receiving trastuzumab ematansine (T-DM1))42 similar to unconjugated maytansine (any-grade hepatotoxicity in up to 43% of patients with solid tumours and thrombocytopenia in up to 10%)43,44. ADCs with camptothecin-class topoisomerase I inhibitor payloads (such as SN-38, belotecan and deruxtecan (DXd)) commonly cause alopecia, diarrhoea and neutropenia (any-grade forms of these events have been observed in >50% of patients with triple-negative breast cancer (TNBC))45,46, similar to irinotecan (any-grade forms of these events reported in >50% of patients with colorectal cancer)47. The key mechanism of off-target, off-tumour toxicities with the currently approved ADCs is thought to, at least in part, relate to the premature deconjugation of the payload within the systemic circulation, which leads to the diffusion of free cytotoxic payload into extratumoural compartments14. Such payloads are generally lipophilic molecules, which are able to permeate plasma membranes and enter non-target, non-malignant cells48. As previously mentioned, part of the payload can also become bound to serum albumin and other circulating thiol-containing plasma proteins, which can increase the half-life of the payload–linker complex and potentially lead to deposition of the payload in non-malignant tissues14,34. Beyond payload detachment from the ADC, other mechanisms have been suggested to mediate the exposure of non-malignant cells to the cytotoxic payload, including non-specific endocytosis of the intact ADC within non-malignant cells24 as well as off-target, receptor-mediated uptake resulting from the interaction of the Fc domain of the antibody backbone with Fc receptors expressed by immune cells24. These latter mechanisms might be more relevant to highly stable ADCs, which are associated with limited premature deconjugation of the payload and release into the circulation and are thus more likely to encounter non-malignant tissues as intact ADCs49. Regardless of the mechanism, the extent of off-target exposure of non-malignant cells to the payload ultimately determines the tolerability of the agent, making the choice of the payload a crucial decision in the design of any ADC.
Linker
As mentioned above, the main mechanism leading to off-target toxicities in patients receiving ADCs probably relates to the timing and localization of the release of the payload from the conjugate. These features are largely determined by the stability and pharmacological structure of the linker, which can therefore have major implications for the toxicity profile of the ADC49,50.
An ideal linker is sufficiently stable to deliver the payload to the intended site, yet also labile enough to release an effective amount of payload either within or in close proximity to the tumour. Non-cleavable linkers, such as maleimidomethyl cyclohexane-1-carboxylate (as utilized in T-DM1), require intracellular degradation of the ADC to release the payload, resulting in stability in the circulation (relative to ADCs with cleavable linkers). Following antigen binding and intracellular release of the payload, the presence of charged lysines or cysteines limits the cell membrane permeability of the unconjugated payload and renders it unlikely to re-enter the circulation or diffuse into bystander cells35. Conversely, cleavable linkers have chemically or enzymatically labile chemical structures, which can lead to payload release through several mechanisms, including acidic degradation (hydrazones and carbonates), cleavage by plasma and lysosomal proteases (peptides and glucuronides), or thiol-disulfide exchange reactions (disulfides)51. In general, less-stable linkers lead to earlier release of free payload into the circulation, with a higher peak cytotoxin concentration and an increase in typical chemotherapy-related toxicities (such as cytopenias, alopecia and/or gastrointestinal toxicities)52. However, more-stable linkers can lead to prolonged circulation of the intact ADC, with the payload released later and in variable body locations beyond the circulation14,52. This aspect might account for the peculiar toxicity profiles of certain highly stable ADCs, several of which have been found to have limited chemotherapy-related toxicities but unexpectedly high incidences of ocular toxicities49,53,54,55. These findings suggest that a balance should be pursued when attempting to determine ADC stability, and that excessive payload release and payload retention by the ADC can both lead to unintended toxicities.
Beyond linker stability, both the specific chemistry used to link payloads with antibodies and the drug-to-antibody ratio (DAR) can have implications for the toxicity profile of ADCs. Stochastic conjugation, namely the non-selective linking of payloads to lysine or cysteine residues located in the antibody, produces a mixture of ADCs with heterogenous DARs56, which can lead to variations in the toxicity of the conjugate50. The manufacture of T-DM1 involves stochastic lysine bioconjugation, which generates a heterogenous admixture of conjugates with an average DAR of 3.5, with the payload located at variable conjugation sites around the antibody35. The manufacturing processes for trastuzumab deruxtecan (T-DXd) and sacituzumab govitecan instead involve stochastic cysteine conjugation, which enables drug conjugation at eight different regions of the antibody, enabling a DAR of up to 8, with reduced heterogeneity compared with lysine conjugation50. All other approved ADCs, including those for haematological indications, are produced using stochastic conjugation methods involving either lysine or cysteine50. Strategies designed to enable more consistent payload conjugation are an area of active development and will be further discussed later in the Review.
Antibody
Much of the toxicity of ADCs is determined by the linker–payload complex14, although even ADCs with the same payload and linker can differ substantially in their adverse effect profile owing to on-target, off-tumour toxicities. These events are related to the engagement of a specific target or the accumulation of payload in non-malignant tissues expressing the ADC target and thus vary broadly depending on the target antigen. This phenomenon was initially observed with the LeY-targeted conjugate BR96–doxorubicin, which was associated with gastrointestinal tract toxicities (where LeY is highly expressed) and less frequently with the haematological or cardiac toxicities typically seen with unconjugated doxorubicin57. Cardiotoxicities can be observed with T-DM1 or T-DXd and are largely related to the on-target, off-tumour effects of trastuzumab58 as reflected by the observation that such toxicities are less frequent with other DXd-based ADCs. Instead, the trophoblast cell-surface antigen 2 (Trop2)-targeted ADC datopotamab deruxtecan (Dato-DXd) is associated with high incidences of rash and stomatitis59, similar to other Trop2-targeted ADCs (such as PF-06664178)60, possibly owing to high levels of Trop2 expression in skin and mucosal tissues61. The HER3-targeted ADC patritumab deruxtecan (HER3-DXd) is associated with a high risk of thrombocytopenia62, an adverse effect that has also been observed with unconjugated patritumab63. Similarly, despite many MMAE (also known as vedotin)-based ADCs having similar toxicity profiles (typically characterized by anaemia, neutropenia and peripheral neuropathy)40,64,65, which are mostly related to their payload, more unique adverse effects can be observed depending on the targeted antigen. For example, the anti-nectin 4 ADC enfortumab vedotin is associated with severe skin toxicities and dysgeusia40, probably owing to high levels of nectin 4 expression in the skin and salivary glands66, whereas treatment with tisotumab vedotin is associated with a substantial risk of bleeding64, which might be related to targeting tissue factor (also known as coagulation factor III)67. In certain scenarios, on-target toxicities might dominate the safety profile of the ADC: this is the case for the EphA2-targeted MMAF-based ADC MEDI-547, which is associated with life-threatening bleeding and coagulation events, even at low doses, that are potentially related to the on-target engagement of EphA2, a receptor implicated in neoangiogenesis68,69. To minimize the risk of such on-target toxicities, a careful selection of ADC targets is warranted with preference recommended for antigens with differential levels of expression between tumour cells (ideally high expression) and non-malignant cells (ideally low or absent expression)70.
Beyond inducing on-target toxicities through their antigen-binding domains, the antibody component of the ADC can also lead to toxicities through interactions of their Fc domain with FcγRs expressed on immune cells or other non-malignant cells24. This mechanism has been suggested to be implicated in the induction of interstitial lung disease (ILD) by deruxtecan-based ADCs owing to the uptake of these conjugates by alveolar macrophages71, which express high levels of FcγR72,73. Induction of ILD has indeed been observed, with variable incidences, with several ADCs seemingly regardless of the targeted antigen (including HER2, HER3 and Trop2)74 and has not been observed in monkeys exposed to high doses of unconjugated DXd71, suggesting that this effect might not be related to the targeting of any specific epitope nor to the payload alone. Similarly, engagement of the antibody Fc domain with FcγRs expressed on megakaryocytes might have a role in T-DM1-induced thrombocytopenia75, although controversies remain regarding this mechanism76.
Overall, although antibody-related toxicities do not generally dominate the adverse effect profile of ADCs, several instances of severe on-target, off-tumour toxicities or toxicities mediated by the Fc portion of the antibody have been observed. These observations highlight the complexity of the mechanism of action of these compounds and the relevance of each ADC component to the tolerability profile.
Patient-related factors
Beyond the differences in toxicity profiles observed among various ADCs, a relevant degree of heterogeneity also exists in the spectrum and grade of adverse effects occurring in different patients who receive the same ADC. Multiple patient-related factors can affect the pharmacokinetics and pharmacodynamics of these agents, including baseline organ function, the presence of comorbidities, polymorphisms in enzymes involved in the metabolism of ADCs or their catabolites, and the body composition of each patient.
Impaired baseline organ function and/or specific comorbidities can affect both the metabolism of the ADC and the level of susceptibility to further organ damage. Impaired baseline renal function and lower baseline oxygen saturation have both been associated with a higher risk of pulmonary toxicities from T-DXd77, whereas impaired liver function has been found to increase the level of exposure to the cytotoxin released by enfortumab vedotin78. As we will discuss later, polymorphisms in genes encoding enzymes involved in the metabolism of ADCs and/or their payloads can result in increased toxicities as observed in patients with deleterious polymorphisms in UGT1A1 who received sacituzumab govitecan79,80. Body weight and composition can both alter the metabolism of ADCs, potentially producing variability in toxicity profiles. Changes in body weight and changes in albumin concentration have both been consistently shown to affect the pharmacokinetics of T-DM1, T-DXd, tisotumab vedotin, enfortumab vedotin, mirvetuximab soravtansine and several other ADCs78,81,82,83,84. Lastly, ethnicity has been found to affect ADC metabolism: for example, patients of Japanese ethnicity have a 20% increase in mean serum T-DXd concentration compared to patients from other countries81, a finding that might explain the higher incidences of ILD observed in this patient population77.
Adverse effects associated with combination strategies
On the basis of their mechanism of action, ADCs offer multiple opportunities for combination strategies, with the aim of achieving either additive or synergistic antitumour activity. Together with the potential to improve clinical activity, however, these combinations also harbour the risk of increasing the toxicity of the regimen, either owing to overlapping adverse effects or unexpected synergies. In this section, we recapitulate the available toxicity data on ADC combinations administered alongside different classes of anticancer agents (Tables 3 and 4).
ADCs combined with chemotherapy
Combining different types of chemotherapies is an established method of overcoming resistance and achieving improved treatment outcomes85. Moreover, certain chemotherapies have been suggested to synergize with ADCs: gemcitabine, for example, has been shown to upregulate HER2 expression on cancer cells in vitro via activation of NF-κB signalling, thus enhancing the activity of T-DM1 (ref. 86). Combining ADCs with chemotherapy, however, comes with certain challenges related to overlapping toxicities.
Some of the data in this context originate from a trial testing T-DM1 in combination with docetaxel (with or without pertuzumab) in patients with HER2-positive breast cancer. Among patients with metastatic breast cancer, this combination led to several dose-limiting toxicities (DLTs) and grade ≥3 adverse events in around 80% of patients, with more than half of all patients having neutropenia, fatigue, epistaxis, stomatitis, nausea and diarrhoea87. T-DM1 has also been combined with capecitabine in a randomized phase II trial enrolling patients with HER2-positive metastatic breast cancer, demonstrating no improvement in response rate and more than doubling in the percentage of patients discontinuing treatment when adding capecitabine to T-DM1 monotherapy (from 16.3% to 35.8%)88. T-DXd was combined with 5-fluorouracil (5-FU) or capecitabine in the DESTINY-Gastric03 trial that enrolled 25 patients with metastatic HER2-positive gastric cancer. Dose-limiting stomatitis was observed in two patients in addition to a high incidence of grade ≥3 adverse events in the 5-FU combination arm (in 93.3% of patients), with most patients requiring reductions in the 5-FU dose; no DLTs were observed in the capecitabine arm, although the incidence of grade ≥3 adverse effects was high (90%), and 80% of patients required capecitabine dose reductions89. Dato-DXd was tested in combination with platinum-based chemotherapy and pembrolizumab, resulting in grade ≥3 toxicities in 60% of patients, with nausea, anaemia, fatigue and stomatitis all commonly observed90. Finally, mirvetuximab soravtansine was combined with carboplatin in a phase Ib trial, resulting in a high incidence of any-grade nausea (in 67% of patients) as well as any-grade vomiting, diarrhoea, ocular toxicities, fatigue and cytopenias, all in ≥50% of patients91.
In summary, data from most trials seem to suggest a non-negligible increase in toxicities when combining ADCs with conventional chemotherapy, likely owing to overlapping toxicities related to the off-target, off-tumour effects of ADC payloads.
ADCs combined with endocrine therapy
Endocrine therapy is a common treatment strategy with the general objective of inhibiting the growth of hormone-dependent cancers by either blocking hormone production or the ability of hormones to promote tumour cell growth. Such agents are generally well tolerated and are most frequently (but not exclusively) administered to patients with either breast or prostate cancer92. The non-overlapping toxicity profiles of most endocrine therapies has enabled them to be combined with chemotherapy, and more recently with ADCs. In the phase III KATHERINE trial, which compared adjuvant T-DM1 to trastuzumab in patients with HER2-positive breast cancer with residual disease after neoadjuvant HER2-directed therapy, concomitant adjuvant endocrine therapy was permitted in both arms. The incidence of any-grade toxicities was similar among patients receiving T-DM1 with versus without endocrine therapy, as was the incidence of grade ≥3 adverse events (26.0% versus 24.9%), serious adverse events (12.9% versus 12.2%) and adverse events leading to T-DM1 dose reduction (11.0% versus 15.0%)93. Endocrine therapy has also been tested in combination with T-DXd, both in patients with early stage and advanced-stage HER2-low breast cancers. The randomized phase II TALENT trial, in which patients with early stage HER2-low breast cancer received neoadjuvant T-DXd either with or without anastrozole, demonstrated a comparable toxicity profile in both arms94. Similarly, in the phase Ib DESTINY-Breast08 trial, the addition of anastrozole or fulvestrant to T-DXd did not lead to DLTs and was associated with a toxicity profile similar to that of T-DXd alone in patients with metastatic HER2-low breast cancer95. Overall, combining ADCs with endocrine therapy does not seem to be associated with an increase in toxicities, consistent with the different adverse effect profiles of each agent alone as well as the favourable toxicity profile of most endocrine therapies relative to other systemic anticancer therapies.
ADCs combined with immunotherapy
ADCs share with chemotherapy the potential to elicit immunogenic cell death96,97,98 while also harbouring a potentially immune-stimulating function through the Fc domain of the antibody99, thus providing a rationale for combination with immune-checkpoint inhibitors (ICIs)100. This rationale is supported by the observation that the adverse effect profiles of ADCs and ICIs are generally non-overlapping, suggesting that combination therapy is feasible101. Several early phase and late-phase trials have tested various combinations of ICIs and ADCs, including at least one large randomized study. The phase III KATE2 trial tested the addition of atezolizumab to T-DM1 in patients with pretreated HER2-positive metastatic breast cancer, demonstrating a moderate increase in toxicities in the combination arm: one treatment-related death occurred in a patient receiving T-DM1 plus atezolizumab, and the incidence of clinically serious adverse events (33% versus 19%) as well as that of most adverse effects, with particular mention of pyrexia (35% versus 16%, including several cases that led to hospitalization), were all increased with the addition of atezolizumab102. Randomized data are also available for enfortumab vedotin with versus without pembrolizumab in a study involving 149 patients with advanced-stage urothelial carcinoma: the addition of pembrolizumab led to an increase in the incidence of clinically serious treatment-related adverse events (23.7% versus 15.1%), fatal treatment-related adverse events (3.9% versus 2.7%) and a general increase in the incidence of all treatment-related adverse events, with particular mention to severe skin reactions103. Nonetheless, this combination received accelerated FDA approval in April 2023 for patients with locally advanced or metastatic urothelial carcinoma who are ineligible for cisplatin-containing chemotherapy based on the compelling efficacy of this combination. The non-randomized design of most other studies testing ADCs plus ICIs precludes definitive answers to many of the outstanding research questions. Nonetheless, no concerning signals of synergistic toxicity have been observed thus far with the combination of ICIs and T-DXd8,104,105, Dato-DXd106 or sacituzumab govitecan107, all of which have a mostly additive toxicity profile. These safety profiles include no notable differences in both the rate and severity of ILD observed with DXd-based ADCs, which do not seem to increase following the addition of ICIs104,106,108,109. Further data in this space are expected from ongoing randomized phase III trials (NCT05629585, NCT05382286 and NCT05633654), which are expected to provide clarification on the toxicity profiles of regimens combining ICIs with ADCs beyond T-DM1.
ADCs combined with targeted therapies
Among currently approved ADCs, T-DM1 is the agent with the largest amount of evidence available on activity and safety when administered in combination with targeted agents. The combination of T-DM1 with the HER2 tyrosine kinase inhibitor tucatinib was tested in a phase Ib trial and was found to be tolerable, albeit with frequent gastrointestinal and hepatic toxicities, with 37% of the patients having at least one clinically serious adverse event and 56% of patients requiring tucatinib dose interruptions110. Data from the ongoing phase III HER2CLIMB-02 trial testing T-DM1 with versus without tucatinib in patients with locally advanced or metastatic HER2-positive breast cancer are expected to be reported soon, and these might clarify the added toxicity of the combination over that of T-DM1 alone. T-DM1 has also been tested in combination with intermittent CDK4 and CDK6 inhibition using ribociclib in a phase Ib trial, with no DLTs observed, although 58% of patients required ribociclib dose reductions owing to thrombocytopenia or neutropenia and one patient had grade 2 QTcF prolongation111. Sacituzumab govitecan was combined with the PARP inhibitor talazoparib in a phase Ib trial enrolling patients with metastatic TNBC, leading to multiple DLTs owing to severe myelosuppression described in the first study report (most of the enrolled patients had febrile neutropenia)112. Lastly, the addition of the anti-VEGFA antibody bevacizumab to mirvetuximab soravtansine was evaluated in a phase Ib trial involving patients with platinum-resistant ovarian cancer113, resulting in a toxicity profile comparable to that of the ADC alone114, although grade 1–2 pneumonitis was observed with the addition of bevacizumab in six (9%) patients versus none with mirvetuximab soravtansine alone.
Emerging strategies to optimize the safety of ADCs
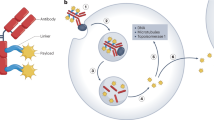
In the short time frame of 4 years, the number of ADCs approved as treatments for patients with cancer has doubled, resulting in a rapidly expanding population of patients exposed to these agents. This rapid pace of development warrants extensive efforts in the development of practices to minimize the risks associated with receiving ADCs. Several strategies have been pursued to either prevent or optimally manage the toxicities associated with ADCs in clinical practice (Fig. 3).

Multiple strategies designed to improve the safety profile of antibody–drug conjugates (ADCs) and thus increase their therapeutic index are currently being developed. a, Dose-optimization strategies such as treatment duration modifications and fractionated dosing, treatment-response dose adjustments, dose capping, and designing randomized dose-finding studies. b, Drug-engineering approaches, including but not limited to the generation of probody–drug conjugates, silencing the Fc portion of the ADC to avoid Fc receptor-mediated toxicities and novel site-specific conjugation technologies. c, Using pharmacogenomic profiling to inform on the risks of developing adverse events in each individual patient and tailoring ADC administration accordingly. d, Using wearable biosensors to detect and monitor specific toxicities associated with ADCs.
Dose-optimization strategies
Given the dose dependency of many of the toxicities associated with ADCs, considerable attention has been dedicated to the optimization of both dose and administration schedules, in an attempt to improve the therapeutic index115. Five classic dose-optimization strategies have been adopted for this purpose, including body weight dose capping, treatment duration capping, optimization of dose frequency, response-guided dose adaptation and randomized dose-finding studies.
Dose capping
The rationale for dose capping derives from the influence of body weight on the pharmacokinetic profile of ADCs, which can lead to risks of overdosing for patients with greater body weight. A dose cap of 125 mg was adopted for the treatment of patients with urothelial carcinoma with enfortumab vedotin after three fatal adverse events reported to be related to treatment were observed with this ADC in patients with baseline body weights ≥100 kg (ref. 116). Analogously, dosing based on adjusted ideal body weight was adopted for mirvetuximab soravtansine in order to reduce the incidence of ocular adverse events117. In general, dose capping should always be considered when, during the development of an ADC, concerning adverse effects are observed among patients with body weight above the ideal range and/or pharmacokinetic modelling suggests a relevant effect of patient body weight on drug exposure.
Capping of treatment duration
Capping of treatment duration aims to minimize the incidence of chronic and potentially permanent adverse events such as peripheral neuropathy. This approach is well established for unconjugated chemotherapies, such as taxanes118 or platinum salts119, and has also proven beneficial with ADCs. For example, data from parametric time-to-event analyses indicate that the predicted risk of developing grade ≥2 peripheral neuropathy with polatuzumab vedotin increases by ≥50% when eight versus six treatment cycles are administered120. These data supported the approval of polatuzumab vedotin for patients with relapsed and/or refractory diffuse large B cell lymphoma at a dose of 1.8 mg/kg every 3 weeks for a maximum of six cycles121. This approach has not yet been extensively tested in patients with solid tumours but has the potential to offer improved tolerability and safety across several different ADCs and cancer types.
Fractionated administration
The frequency of administration is another crucial variable that, when modulated, might improve the safety of ADCs. Administration of ADCs using fractionated dosing schedules can result in exposure to the same cumulative dose of the drug albeit with a lower peak plasma concentration (Cmax) compared to that achieved with a single but higher dose, potentially ameliorating adverse events driven by the Cmax. The first successful example of this strategy was achieved with the first-ever approved ADC, the CD33-targeted agent gemtuzumab ozogamicin. This compound was first granted accelerated approval at the dose of 9 mg/m2 every 2 weeks for patients with relapsed CD33-positive acute myeloid leukaemia in 2000 (ref. 122). However, a high incidence of liver toxicity and veno-occlusive disease was reported in real-world studies, leading to its withdrawal from the market in 2010 (ref. 122). On the basis of subsequent evidence of improved safety, gemtuzumab ozogamicin was reapproved in 2017 with a fractionated, reduced-dose dosing schedule (days 1, 4, and 7 of each induction cycle)2,122. Another example of a successful dose frequency adjustment that improved ADC safety is provided by the CD22-targeted ADC inotuzumab ozogamicin, which was found to be more tolerable and equally effective when administered at fractionated weekly rather than three-weekly or four-weekly doses, leading to the current approval for patients with acute lymphoblastic leukaemia at an induction dose of 1.8 mg/m2 administered as three-weekly doses, with recommended dosing for subsequent cycles dependent on the initial response123,124,125. Similar approaches with ADCs currently used to treat patients with solid tumours might be warranted, particularly given the uncertain implications of Cmax for the activity of most ADCs115.
Treatment response-guided dose adjustments
Response-guided dose adjustments provide a method of modulating the extent of exposure to an ADC based on the initial response of each patient. This adaptive dosing strategy is currently used for patients receiving inotuzumab ozogamicin, for which the initial dose of 1.8 mg/m2 is reduced to 1.6 mg/m2 in subsequent cycles in patients who enter complete remission in order to reduce the risk of toxicities123. Testing this strategy in patients with solid tumours seems most suitable for indications in which responses to the ADC are frequent and long remissions can be expected such as patients with HER2-positive metastatic breast cancer receiving T-DXd. In the phase III DESTINY-Breast03 trial involving patients with trastuzumab-refractory HER2-positive metastatic breast cancer, treatment with T-DXd resulted in complete remissions in 21% of patients, with more than half of all patients in the T-DXd arm being free of progression at 2 years126. Given the known association between a complete response and long-lasting remission observed in this disease127, the evaluation of response-guided T-DXd dosing might be reasonable, and similar attempts could be pursued with other ADCs.
Randomized dose-finding studies
Randomized dose-finding studies are prospective trials designed to evaluate more than one dosage of a compound, with the purpose of identifying an optimal dose that maximizes the therapeutic index. Randomized dose-finding studies enabled the optimal dose of T-DXd to be identified for patients with HER2-positive breast cancer and those with HER2-mutant non-small-cell lung cancer (NSCLC): for both indications, randomized trials were conducted to compare the benefit-to-risk profile of 5.4 mg/kg and 6.4 mg/kg administered as three-weekly doses. In the setting of HER2-positive breast cancer, 115 patients were randomized to receive T-DXd at either 5.4 mg/kg or 6.4 mg/kg, and each group had a similar objective response rate (ORR; 56.5% and 61.5%, respectively), albeit with 12% and 21% of patients developing ILD, and a substantial increase in the incidence of grade ≥3 treatment-emergent adverse events at the higher dose (39% and 58%, respectively)128. With regard to HER2-mutant metastatic NSCLC, 80 patients were randomized 2:1 to receive T-DXd at doses of either 5.4 mg/kg or 6.4 mg/kg, and both groups had similar ORRs (53.8% and 57.7%, respectively), albeit with a near doubling of the incidence of grade ≥3 adverse events (from 31.7% to 58.0%), including an increased incidence of ILD with the higher dose (5.9% at 5.4 mg/kg versus 14% at 6.4 mg/kg)129. T-DXd was approved for patients with HER2-mutant metastatic NSCLC at the dose of 5.4 mg/kg based on these data. Receiving a higher dose of T-DXd was confirmed to be associated with a higher risk of ILD in a pooled analysis of data from 1,150 patients receiving the drug, of whom 44% had breast cancer and the remaining 56% had solid tumours of other histologies77. Similar attempts at randomized dose-finding studies might be reasonable for other ADCs and indications, particularly for those in which fatal adverse events and/or considerable grade ≥3 toxicities are observed either in trials or clinical practice.
Optimizing ADC design
ADC engineering and other optimization strategies can provide important methods of maximizing both the efficacy and safety of these agents. Indeed, innovations in the design of each ADC component can enable fine-tuning of the pharmacological properties, with potential implications for tolerability.
Innovations in the antibody moiety
Most of the currently approved and investigational ADCs are directed against targets that are, to varying degrees, heterogeneously expressed in non-malignant tissues. The generation of probody–drug conjugates (PDCs), consisting of conditionally activated ADCs in which the antigen-binding regions of the antibody are masked by protease-cleavable peptides that enable selective unmasking and activation within the TME, provides one example of an engineering strategy that might reduce the incidence of on-target, off-tumour toxicities130. PDCs are indeed expected to remain largely intact in non-malignant tissues while being effectively cleaved by tumour-associated proteases in malignant tissues in which the masking peptide is released, enabling the antibody to bind the target antigen and the payload to reach tumour cells130. This approach has the additional advantage of potentially overcoming the binding site barrier seen in most solid tumours owing to delayed antigen binding and improved tumour penetration, which could lead to improved efficacy and an improved therapeutic index131,132. Of note, early clinical data with PDCs have yet to reveal clinically relevant improvements in the safety profile as observed in a phase I/II study in which patients with advanced-stage solid tumours received the CD166-targeted PDC praluzatamab ravtansine: ocular toxicities occurred in 43% of patients and 37% of patients had ≥1 treatment-emergent grade ≥3 adverse event14,133. Nonetheless, multiple other PDCs are in development, with additional data required to draw conclusions on the clinical utility of this technology. Beyond masking of the binding regions of the antibody, attempts to silence the antibody Fc domain with the aim of reducing off-target, off-tumour toxicities related to the Fc-mediated uptake of the ADC by immune cells are currently ongoing134,135.
A different strategy that might enable improved ADC delivery to the tumour site and potentially reduce the incidence of on-target toxicity involves conjugation of the payload to bispecific rather than conventional antibodies. By targeting two different antigens, bispecific antibodies might have increased selectivity and improved internalization in tumour cells compared with conventional antibodies136,137,138,139. The first clinical data on the safety and activity of a bispecific ADC were presented in September 2022, with the HER2-targeting ADC ZW49 (directed against two non-overlapping HER2 epitopes) demonstrating an ORR of 31% in patients with advanced-stage HER2-expressing solid tumours, albeit with a non-negligible toxicity profile (mostly owing to ocular toxicities, including keratitis in 42%)140. Several other bispecific ADCs are currently being developed, including, among others, those targeting different HER2 epitopes, HER2 and Trop2, HER2 and PRLR, HER2 and CD63, EGFR and MUC1, and EGFR and HER3 (refs. 141,142,143,144,145).
Innovations in linker technology
Multiple strategies designed to improve the stability of ADCs in systemic circulation are currently being developed in an attempt to optimize safety. Most of these efforts focus on the site-specific rather than stochastic conjugation of payloads to antibodies, via either engineered cysteines146,147,148, the introduction of unnatural amino acids149,150, and/or the addition of selenocysteine, glutamine or aldehyde tags151. These methods enable the preparation of homogenous populations of ADCs with predictable DARs and sites of payload attachment, which might be associated with an improved tolerability profile and more predictable pharmacokinetics. Preclinical evidence suggests that these conjugation strategies confer improved safety profiles, and clinical experimentation is currently ongoing56. Nonetheless, as previously mentioned, clinical data suggest that even excess linker stability can be associated with severe toxicities as observed in the clinical trials of SYD985, ARX788 and A166, among other ADCs45,54,55. A different engineering strategy involves adding short polyethylene glycol (PEG) moieties to the linker, which might reduce the incidence of non-specific ADC uptake via an increase in ADC hydrophilicity, ultimately reducing the incidence of off-target toxicities152. Preclinical data suggest retained activity and improved pharmacokinetic properties for ADCs developed using this strategy153,154,155, and clinical testing is currently ongoing.
Innovations in the payload
In an attempt to improve the therapeutic index of single-payload ADCs, novel constructs featuring two different types of payload linked to the same monoclonal antibody have been developed. Together with potentially expanding the spectrum of activity of the ADC, the combination of payloads with multiple and non-overlapping mechanisms of action within the same conjugate might also lead to improved tolerability. Preclinical data have confirmed the feasibility and high antitumour activity of this approach with constructs such as a HER2-targeted antibody conjugated to both MMAE and MMAF and an FGF2-targeted antibody conjugated to MMAE and α-amanitin156,157,158,159, although no clinical data are currently available. Furthermore, novel payloads, beyond anti-microtubule agents, topoisomerase I inhibitors and DNA-intercalating agents, are being explored. These payloads include other cytotoxic molecules (such as topoisomerase II inhibitors or agents that inhibit transcription or translation)48 as well as ADCs with unconventional payloads such as immunostimulatory molecules, heterobifunctional protein degraders and tyrosine kinase inhibitors48. A detailed description of each of these strategies is beyond the scope of this Review; however, it is worth highlighting that non-conventional payloads might be subject to toxicity issues similar to those observed with canonical cytotoxin-conjugated ADCs and might warrant careful experimentation and fine-tuning. As an illustration, preliminary data from a phase I study testing a HER2-targeted, immune-stimulating ADC carrying a Toll-like receptor 7 agonist were reported in December 2022 and revealed high incidences of cytokine-release syndrome, pyrexia, nausea, vomiting and headache (>30% each), consistent with the off-target effects that might be expected from release of the immunostimulatory payload160. Moreover, the development of the immune-stimulating STING-agonist ADC XMT-2056 was halted in March 2023 after a fatal adverse effect deemed to be related to the conjugate was observed in the second patient treated at the initial dose level in the dose-escalation portion of the trial161. In these settings, leveraging the knowledge accumulated through the development of conventional ADCs is warranted in order to develop safer innovative ADCs that are able to effectively expand the range of therapeutic options.
The co-administration of ADCs with payload-neutralizing antibody fragments is another innovation designed to improve the tolerability of ADCs. By binding to unconjugated circulating payload molecules, these payload-neutralizing fragments are designed to reduce the amount of cytotoxin reaching non-malignant tissues and thus reduce the extent of off-target toxicities in patients receiving ADCs. Preclinical data indicate a reduction in chemotherapy-related toxicities, amelioration of body weight loss in mouse models and retained activity when a HER2-targeted MMAE-based ADC is co-administered with the humanized anti-MMAE Fab fragment ABC3315 in xenograft models162. Clinical investigations will be required to clarify whether this strategy can effectively improve tolerability without affecting the antitumour activity of ADCs.
Pharmacogenomics
The identification of patients with the greatest risk of developing adverse events upon treatment with ADCs is an important step in improving the clinical management of such patients. In a post hoc analysis of data from the ASCENT trial, in which patients with metastatic TNBC received sacituzumab govitecan, deleterious UGT1A1 polymorphisms were noted to correlate with the occurrence of adverse events reported to be related to treatment80. The payload of sacituzumab govitecan (SN-38) is a lipophilic molecule that is largely cleared via glucuronidation by UGT1A1. Data from previous studies indicate that patients with colorectal cancer receiving irinotecan who have loss-of-function UGT1A1 polymorphisms have increased exposure to SN-38, resulting in higher incidences of both neutropenia and diarrhoea163,164. This effect was also observed among patients homozygous for the UGT1A1*28 polymorphism receiving the combination of capecitabine and irinotecan, who were found to be 14.2 times more likely to develop febrile neutropenia than those with wild-type UGT1A1 (ref. 165). In ASCENT, the incidences of grade ≥3 neutropenia, febrile neutropenia and diarrhoea were all higher among patients who were homozygous for UGT1A1*28 compared with patients who were either heterozygous for this allele or were homozygous for the wild-type allele80. Similarly, patients who were homozygous for UGT1A1*28 had an increased incidence of toxicities on sacituzumab govitecan in the phase I/II IMMU-132-01 basket trial79. Given the relatively low frequency of the homozygous UGT1A1*28 genotype (<10%)163, these data have not yet led to recommendation for routine testing of UGT1A1 polymorphisms, although monitoring patients who are known to be homozygous for UGT1A1*28 more closely for the development of toxicities is currently recommended. In addition to polymorphisms in genes encoding enzymes involved in payload metabolism, polymorphisms affecting genes that encode enzymes involved in payload release or in those encoding Fc receptors might also have a role in determining the safety profile and tolerability of a given ADC in specific patient subgroups. Overall, these data suggest that pharmacogenomic parameters can have a relevant effect on the safety profile of certain ADCs. As novel ADCs are being developed, considering the inclusion of pharmacogenomic profiling when designing early phase trials could be a reasonable approach that ensures that the safety profile is not overly affected in specific populations and/or by inter-individual genetic variations.
Diagnostic tools
Another potential approach for the early detection and better management of ADC-induced adverse events involves the use of wearable biosensors (WBSs). These devices have the potential to provide real-time information through the continuous, non-invasive measurement of biological factors, including vital parameters and biofluids (such as sweat, saliva and interstitial fluid). In the context of ADC-related toxicities, the use of WBSs might support the early identification of treatment-induced ILD by detecting changes in oxygen saturation levels and/or heart and breathing rates. Moreover, forced vital capacity assessments using home spirometry (such as a Bluetooth-enabled hand-held spirometer), pulse oximetry or cough monitors (such as the Leicester Cough Monitor or VitaloJAK) have emerged as potential methods of monitoring for ILD166,167,168. The rapid development of these technologies might enable early diagnosis of ILD in patients at risk, help identify acute exacerbation of ILD and support real-time clinical decision-making, although several barriers to adoption of these measures also exist, including costs and the need for international, evidence-based guidelines regarding the use of home-based measurements.
ADCs in the early stage setting
ADCs have reshaped the treatment algorithms for the management of several forms of advanced-stage solid tumours over the past decade13. However, the greatest clinical benefit from this class of agents might be achieved in patients with early stage disease who often receive chemotherapy in an attempt to prevent disease recurrence and who might benefit from the improved delivery of cytotoxic agents provided by ADCs, ideally including cure for a higher percentage of patients. Several trials are currently ongoing in this space (Supplementary Table), with one ADC (T-DM1) already approved for patients with early stage HER2-positive breast cancer169. Importantly, this setting raises unique challenges in terms of toxicities: although increased incidences of short-term toxicities are commonly considered acceptable to achieve the goal of cure, permanent toxicities that might have long-term effects on quality of life are less acceptable and caution is needed to ensure that fatal toxicities can be avoided170,171. In this context, early stopping rules in trials testing ADCs in the early stage setting may be warranted.
The post-neoadjuvant setting, where ADCs can provide a rescue strategy for patients with residual disease at surgery (those without a pathological complete response) following preoperative systemic treatment is an appealing setting in which to test novel ADCs. The only ADC approved currently for this scenario is T-DM1, which was shown to improve disease-free survival in the phase III KATHERINE trial169. In this study, toxicities were more frequent with adjuvant T-DM1 compared with trastuzumab (grade ≥3 toxicities occurred in 25.7% versus 15.4% of patients) although no fatal toxicities were observed; peripheral neuropathy was also more frequent with T-DM1 (18.6% versus 6.9%), which resolved in most patients during follow-up. Given the favourable risk-to-benefit profile, T-DM1 has rapidly become the standard-of-care adjuvant therapy for patients with residual invasive disease following neoadjuvant therapy and is recommended for this indication in most international guidelines172,173. Concomitantly, this success has raised the hope that novel ADCs could further improve outcomes in this high-risk setting. The ongoing phase III DESTINY-Breast05 trial (NCT04622319) is comparing post-neoadjuvant T-DXd to T-DM1 in a population similar to that of KATHERINE. Of note, T-DXd resulted in fatal ILD in 1–2% of patients in several studies conducted in the advanced-stage setting77, a finding that indicates a need for extensive caution in the use of this agent in earlier settings. Sacituzumab govitecan is also being tested in the post-neoadjuvant setting within the phase III SASCIA trial (NCT04595565), in which patients with HER2-negative breast cancer and residual invasive disease after neoadjuvant chemotherapy are being randomized to either sacituzumab govitecan or treatment of physician choice. At the first interim safety analysis, patients receiving sacituzumab govitecan had an increased incidence of grade 3–4 adverse effects (66% versus 20% with treatment of physician choice) as well as more dose delays and treatment discontinuations, although no fatal toxicities were observed174. Analogously, the phase III OptimICE-RD (NCT05633654) and Tropion-Breast03 (NCT05629585) trials are currently testing sacituzumab govitecan and Dato-DXd in this setting, respectively.
ADCs are also under investigation in the preoperative setting. In the phase III KRISTINE trial, neoadjuvant T-DM1 plus pertuzumab failed to improve the outcomes of patients with HER2-positive breast cancer compared with chemotherapy and dual HER2 blockade with trastuzumab plus pertuzumab175. Neoadjuvant T-DXd (6–8 cycles) was evaluated in patients with HER2-low breast cancer in the phase II TALENT trial: most patients completed treatment and underwent surgery, although 12% came off study prior to treatment completion, either owing to death (one patient), withdrawal of consent (two patients) or meeting the discontinuation criteria94. The phase III DESTINY-Breast11 study (NCT05113251) is currently testing T-DXd as a substitute for either all or part of the traditional neoadjuvant chemotherapy regimen for HER2-positive breast cancer, whereas the phase II TRUDI trial (NCT05795101) is testing the combination of neoadjuvant T-DXd plus durvalumab for patients with non-metastatic, HER2-expressing inflammatory breast cancer. Neoadjuvant sacituzumab govitecan is being tested in patients with TNBC, with a first report coming from the randomized phase II NeoSTAR trial demonstrating a limited need for dose reductions (6%), no discontinuations owing to adverse events and no fatal toxicities46. Neoadjuvant enfortumab vedotin (three cycles) was evaluated for patients with urothelial carcinoma ineligible for cisplatin: 86% (19 of 22) of patients were able to complete treatment, with 18% having one or more grade 3–4 treatment-related adverse event and three fatal adverse events, although none of these were reported to be related to treatment176.
Window-of-opportunity trials, a study design in which newly diagnosed patients receive a short course of treatment with a novel therapy before surgery or before initiating standard neoadjuvant treatment, usually with biological surrogates as end points, provide a novel method of testing potential neoadjuvant therapies, including ADCs177. Such trials might expedite drug development by providing evidence of the biological effects of the novel ADC in patients with treatment-naive tumours within a short time frame. For example, HER3-DXd was evaluated in the window-of-opportunity TOT-HER3 trial (NCT04610528), in which a single dose of HER3-DXd was administered before surgery to 78 patients with hormone receptor-positive breast cancer. This study demonstrated an increase in CelTIL score, which indicates an increase in tumour-infiltrating lymphocytes and/or a decrease in tumour cellularity, after treatment among responders as well as a lack of an association between baseline HER3 expression and treatment response178. These data informed the design of the ongoing randomized phase II SOLTI-VALENTINE trial (NCT05569811), which is testing the activity of six neoadjuvant cycles of HER3-DXd (with or without letrozole) or chemotherapy in patients with hormone receptor-positive breast cancer.
A final novel strategy involves the administration of ADCs to previously untreated patients in the adjuvant setting. This strategy was tested in the randomized phase II ATEMPT trial, in which 1 year of adjuvant T-DM1 led to a 5-year invasive disease-free survival of 97% among patients with resected stage I HER2-positive breast cancer179. One year of adjuvant T-DM1 was found to be safe, with an incidence of clinically relevant toxicities not significantly different to that observed with paclitaxel and trastuzumab (46% versus 47%; P = 0.83)180. Cardiac toxicities with adjuvant T-DM1 were also extremely rare in ATEMPT (in <1% of patients)181. A second study (ATEMPT 2.0, NCT04893109) is currently ongoing and is designed to determine whether a shorter course of T-DM1 (six cycles) can decrease the incidence of toxicities while retaining clinical activity.
Additional trials testing ADCs in the early stage setting are currently ongoing in patients with various tumour types (Supplementary Table). Nonetheless, the incidences of high-grade toxicities and treatment discontinuations observed with novel ADCs in this setting are non-negligible, indicating that a careful balance of potential risks versus benefits is warranted. This balance might be best optimized in the post-neoadjuvant setting, with the selective treatment of patients with residual disease after neoadjuvant therapy, a feature that has been associated with inferior outcomes across several cancer types182,183,184,185,186,187.
Conclusions
ADCs have improved upon the activity of traditional chemotherapy regimens in a range of solid tumours. Despite their ideally targeted mechanism of action, most ADCs still confer frequent and sometimes life-threatening toxicities. Given the rapid expansion in their indications, an awareness of these adverse effects and their management is crucial as are efforts to prevent and mitigate ADC-related toxicities. These include careful attention to the risks of synergistic or overlapping toxicities when combining ADCs with other anticancer agents as well as caution in the development and testing of novel ADCs in early stage disease, in which a dedicated balance of risks and benefits is warranted for each indication. Innovations in ADC design, pharmacogenomic testing and WBSs as well as increased attention to the optimization of ADC dosing through dedicated prospective trials might help to leverage the potential of this highly promising class of anticancer drugs, which is still far from being fully explored.
References
Tarantino, P. et al. Antibody-drug conjugates: smart chemotherapy delivery across tumor histologies. CA Cancer J. Clin. https://doi.org/10.3322/caac.21705 (2021).
Fu, Z., Li, S., Han, S., Shi, C. & Zhang, Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal. Transduct. Target. Ther. 7, 93 (2022).
de la Torre, B. G. & Albericio, F. The pharmaceutical industry in 2022: an analysis of FDA drug approvals from the perspective of molecules. Molecules 28, 1038 (2023).
do Pazo, C., Nawaz, K. & Webster, R. M. The oncology market for antibody-drug conjugates. Nat. Rev. Drug Discov. 20, 583–584 (2021).
Modi, S. et al. Antitumor activity and safety of trastuzumab deruxtecan in patients with HER2-low-expressing advanced breast cancer: results from a phase Ib study. J. Clin. Oncol. 38, 1887–1896 (2020).
Banerji, U. et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2-expressing breast cancer: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 20, 1124–1135 (2019).
Wang, J. et al. RC48-ADC, a HER2-targeting antibody-drug conjugate, in patients with HER2-positive and HER2-low expressing advanced or metastatic breast cancer: a pooled analysis of two studies. J. Clin. Oncol. 39, 1022–1022 (2021).
Schmid, P. et al. BEGONIA: phase 1b/2 study of durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC) — initial results from arm 1, d+paclitaxel (P), and arm 6, d+trastuzumab deruxtecan (T-DXd). J. Clin. Oncol. 39, 1023–1023 (2021).
Jiang, Z. et al. A multiple center, open-label, single-arm, phase II clinical trial of MRG002, an HER2-targeted antibody-drug conjugate, in patients with HER2-low expressing advanced or metastatic breast cancer. J. Clin. Oncol. 40, 1102–1102 (2022).
Yamaguchi, K. et al. Trastuzumab deruxtecan in anti–-human epidermal growth factor receptor 2 treatment-naive patients with human epidermal growth factor receptor 2-low gastric or gastroesophageal junction adenocarcinoma: exploratory cohort results in a phase II trial. J. Clin. Oncol. 41, 816–825 (2023).
Tarantino, P. et al. HER2-low breast cancer: pathological and clinical landscape. J. Clin. Oncol. 38, 1951–1962 (2020).
AstrazZeneca. Enhertu Showed Clinically Meaningful and Durable Responses Across Multiple HER2-Expressing Tumour Types in DESTINY-PanTumor02 Phase II Trial https://www.astrazeneca.com/media-centre/press-releases/2023/enhertu-destiny-pantumor02-shows-positive-results.html (2023).
Drago, J. Z., Modi, S. & Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 18, 327–344 (2021).
Colombo, R. & Rich, J. R. The therapeutic window of antibody drug conjugates: a dogma in need of revision. Cancer Cell 40, 1255–1263 (2022).
Zhu, Y., Liu, K., Wang, K. & Zhu, H. Treatment-related adverse events of antibody-drug conjugates in clinical trials: a systematic review and meta-analysis. Cancer 129, 283–295 (2023).
Blackhall, F. et al. Efficacy and safety of rovalpituzumab tesirine compared with topotecan as second-line therapy in DLL3-high SCLC: results from the phase 3 TAHOE study. J. Thorac. Oncol. 16, 1547–1558 (2021).
Lassman, A. B. et al. Depatuxizumab mafodotin in EGFR-amplified newly diagnosed glioblastoma: a phase III randomized clinical trial. Neuro-Oncology 25, 339–350 (2022).
Junttila, T. T., Li, G., Parsons, K., Phillips, G. L. & Sliwkowski, M. X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 128, 347–356 (2011).
Chang, H.-P., Le, H. K. & Shah, D. K. Pharmacokinetics and pharmacodynamics of antibody-drug conjugates administered via subcutaneous and intratumoral routes. Pharmaceutics 15, 1132 (2023).
Jin, Y., Schladetsch, M. A., Huang, X., Balunas, M. J. & Wiemer, A. J. Stepping forward in antibody-drug conjugate development. Pharmacol. Ther. 229, 107917 (2022).
Epenetos, A. A., Snook, D., Durbin, H., Johnson, P. M. & Taylor-Papadimitriou, J. Limitations of radiolabeled monoclonal antibodies for localization of human neoplasms. Cancer Res. 46, 3183–3191 (1986).
Mach, J.-P. et al. Tumor localization of radio-labeled antibodies against carcinoembryonic antigen in patients with carcinoma. N. Engl. J. Med. 303, 5–10 (1980).
Marei, H. E., Cenciarelli, C. & Hasan, A. Potential of antibody–drug conjugates (ADCs) for cancer therapy. Cancer Cell Int. 22, 255 (2022).
Mahalingaiah, P. K. et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 200, 110–125 (2019).
Giugliano, F., Corti, C., Tarantino, P., Michelini, F. & Curigliano, G. Bystander effect of antibody-drug conjugates: fact or fiction? Curr. Oncol. Rep. 24, 809–817 (2022).
Khera, E. et al. Cellular-resolution imaging of bystander payload tissue penetration from antibody-drug conjugates. Mol. Cancer Ther. 21, 310–321 (2022).
Suzuki, M. et al. Visualization of intratumor pharmacokinetics of [fam-] trastuzumab deruxtecan (DS-8201a) in HER2 heterogeneous model using phosphor-integrated dots imaging analysis. Clin. Cancer Res. 27, 3970–3979 (2021).
Okeley, N. M. et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 16, 888–897 (2010).
Staudacher, A. H. & Brown, M. P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br. J. Cancer 117, 1736–1742 (2017).
Kopp, A. et al. Antibody–drug conjugate sacituzumab govitecan drives efficient tissue penetration and rapid intracellular drug release. Mol. Cancer Ther. 22, 102–111 (2023).
Lucas, A. T. et al. Factors affecting the pharmacology of antibody-drug conjugates. Antibodies https://doi.org/10.3390/antib7010010 (2018).
Han, T. H. & Zhao, B. Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab. Dispos. 42, 1914–1920 (2014).
Wei, C. et al. Where did the linker-payload go? A quantitative investigation on the destination of the released linker-payload from an antibody-drug conjugate with a maleimide linker in plasma. Anal. Chem. 88, 4979–4986 (2016).
Nilsen, J. et al. Human and mouse albumin bind their respective neonatal Fc receptors differently. Sci. Rep. 8, 14648 (2018).
Lewis Phillips, G. D. et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 68, 9280–9290 (2008).
Polson, A. G. et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: target and linker-drug selection. Cancer Res. 69, 2358–2364 (2009).
Hamblett, K. J. et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 10, 7063–7070 (2004).
Sun, X. et al. Effects of drug-antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjug. Chem. 28, 1371–1381 (2017).
Nguyen, T. D., Bordeau, B. M. & Balthasar, J. P. Mechanisms of ADC toxicity and strategies to increase ADC tolerability. Cancers 15, 713 (2023).
Powles, T. et al. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N. Engl. J. Med. 384, 1125–1135 (2021).
Pitot, H. C. et al. Phase I trial of dolastatin-10 (NSC 376128) in patients with advanced solid tumors. Clin. Cancer Res. 5, 525–531 (1999).
Verma, S. et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 367, 1783–1791 (2012).
Franklin, R. et al. A phase I-II study of maytansine utilizing a weekly schedule. Cancer 46, 1104–1108 (1980).
Eagan, R. T., Ingle, J. N., Rubin, J., Frytak, S. & Moertel, C. G. Early clinical study of an intermittent schedule for maytansine (NSC-153858): brief communication. J. Natl Cancer Inst. 60, 93–96 (1978).
Bardia, A. et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N. Engl. J. Med. 384, 1529–1541 (2021).
Spring, L. et al. Phase 2 study of response-guided neoadjuvant sacituzumab govitecan (IMMU-132) in patients with localized triple-negative breast cancer: results from the NeoSTAR trial. J. Clin. Oncol. 40, 512–512 (2022).
Rougier, P. et al. Phase II study of irinotecan in the treatment of advanced colorectal cancer in chemotherapy-naive patients and patients pretreated with fluorouracil-based chemotherapy. J. Clin. Oncol. 15, 251–260 (1997).
Conilh, L., Sadilkova, L., Viricel, W. & Dumontet, C. Payload diversification: a key step in the development of antibody–drug conjugates. J. Hematol. Oncol. 16, 3 (2023).
Colombo, R., Barnscher, S. D. & Rich, J. R. Abstract 1538: revisiting the dogma of antibody drug conjugates (ADCs): emerging data challenge the benefit of linker stability and the primacy of payload delivery. Cancer Res. 83, 1538–1538 (2023).
Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs 8, 659–671 (2016).
Bargh, J. D., Isidro-Llobet, A., Parker, J. S. & Spring, D. R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 48, 4361–4374 (2019).
Tarcsa, E., Guffroy, M. R., Falahatpisheh, H., Phipps, C. & Kalvass, J. C. Antibody-drug conjugates as targeted therapies: are we there yet? A critical review of the current clinical landscape. Drug Discov. Today Technol. 37, 13–22 (2020).
Zhang, J. et al. Phase I study of A166, an antibody–drug conjugate in advanced HER2-expressing solid tumours. NPJ Breast Cancer 9, 28 (2023).
Zhang, J. et al. Phase I trial of a novel anti-HER2 antibody–drug conjugate, ARX788, for the treatment of HER2-positive metastatic breast cancer. Clin. Cancer Res. 28, 4212–4221 (2022).
Saura Manich, C. et al. LBA15 Primary outcome of the phase III SYD985.002/TULIP trial comparing [vic-]trastuzumab duocarmazine to physician’s choice treatment in patients with pre-treated HER2-positive locally advanced or metastatic breast cancer. Ann. Oncol. 32, S1288 (2021).
Matsuda, Y. & Mendelsohn, B. A. An overview of process development for antibody-drug conjugates produced by chemical conjugation technology. Expert Opin. Biol. Ther. 21, 963–975 (2021).
Saleh, M. N. et al. Phase I trial of the anti-Lewis Y drug immunoconjugate BR96-doxorubicin in patients with Lewis Y-expressing epithelial tumors. J. Clin. Oncol. 18, 2282–2292 (2000).
Fu, Z., Liu, J., Li, S., Shi, C. & Zhang, Y. Treatment-related adverse events associated with HER2-targeted antibody-drug conjugates in clinical trials: a systematic review and meta-analysis. eClinicalMedicine https://doi.org/10.1016/j.eclinm.2022.101795 (2023).
Krop, I. et al. Abstract GS1-05: Datopotamab deruxtecan in advanced/metastatic HER2- breast cancer: results from the phase 1 TROPION-PanTumor01 study. Cancer Res. https://doi.org/10.1158/1538-7445.Sabcs21-gs1-05 (2022).
King, G. T. et al. A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Invest. N. Drugs 36, 836–847 (2018).
Stepan, L. P. et al. Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: potential implications as a cancer therapeutic target. J. Histochem. Cytochem. 59, 701–710 (2011).
Krop, I. E. et al. Results from the phase 1/2 study of patritumab deruxtecan, a HER3-directed antibody-drug conjugate (ADC), in patients with HER3-expressing metastatic breast cancer (MBC). J. Clin. Oncol. 40, 1002–1002 (2022).
Wakui, H. et al. Phase 1 and dose-finding study of patritumab (U3-1287), a human monoclonal antibody targeting HER3, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 73, 511–516 (2014).
Coleman, R. L. et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 22, 609–619 (2021).
Shi, F. et al. Disitamab vedotin: a novel antibody-drug conjugates for cancer therapy. Drug Deliv. 29, 1335–1344 (2022).
Challita-Eid, P. M. et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 76, 3003–3013 (2016).
Unruh, D. & Horbinski, C. Beyond thrombosis: the impact of tissue factor signaling in cancer. J. Hematol. Oncol. 13, 93 (2020).
Annunziata, C. M. et al. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Invest. N. Drugs 31, 77–84 (2013).
Boyd, A. W., Bartlett, P. F. & Lackmann, M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 13, 39–62 (2014).
Schettini, F. et al. Identification of cell surface targets for CAR-T cell therapies and antibody-drug conjugates in breast cancer. ESMO Open 6, 100102 (2021).
Kumagai, K. et al. Interstitial pneumonitis related to trastuzumab deruxtecan, a human epidermal growth factor receptor 2-targeting Ab-drug conjugate, in monkeys. Cancer Sci. 111, 4636–4645 (2020).
Berger, M. et al. Tissue-specific Fc gamma and complement receptor expression by alveolar macrophages determines relative importance of IgG and complement in promoting phagocytosis of Pseudomonas aeruginosa. Pediatr. Res. 35, 68–77 (1994).
Bruggeman, C. W. et al. Tissue-specific expression of IgG receptors by human macrophages ex vivo. PLoS One 14, e0223264 (2019).
Tarantino, P. et al. Interstitial lung disease induced by anti-ERBB2 antibody-drug conjugates: a review. JAMA Oncol. 7, 1873–1881 (2021).
Uppal, H. et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin. Cancer Res. 21, 123–133 (2015).
Ansary, A. M. et al. Effect of Ado-trastuzumab emtansine on autologous platelet kinetics and function. JCO Precis. Oncol. 6, e2200237 (2022).
Powell, C. A. et al. Pooled analysis of drug-related interstitial lung disease and/or pneumonitis in nine trastuzumab deruxtecan monotherapy studies. ESMO Open 7, 100554 (2022).
Tang, M. et al. Clinical pharmacology of the antibody-drug conjugate enfortumab vedotin in advanced urothelial carcinoma and other malignant solid tumors. J. Clin. Oncol. 40, 568–568 (2022).
Bardia, A. et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann. Oncol. 32, 746–756 (2021).
Rugo, H. S. et al. Safety analyses from the phase 3 ASCENT trial of sacituzumab govitecan in metastatic triple-negative breast cancer. NPJ Breast Cancer 8, 98 (2022).
Yin, O. et al. Population pharmacokinetics of trastuzumab deruxtecan in patients with HER2-positive breast cancer and other solid tumors. Clin. Pharmacol. Ther. 109, 1314–1325 (2021).
Gibiansky, L. et al. Population pharmacokinetic analysis for tisotumab vedotin in patients with locally advanced and/or metastatic solid tumors. CPT Pharmacomet. Syst. Pharmacol. 11, 1358–1370 (2022).
Moore, K. N. et al. 605P Population pharmacokinetic (PK) analysis of mirvetuximab soravtansine (MIRV) in patients with folate receptor α (FRα)-positive cancer. Ann. Oncol. 33, S822–S823 (2022).
Lu, D. et al. Population pharmacokinetics of trastuzumab emtansine (T-DM1), a HER2-targeted antibody-drug conjugate, in patients with HER2-positive metastatic breast cancer: clinical implications of the effect of covariates. Cancer Chemother. Pharmacol. 74, 399–410 (2014).
Yardley, D. A. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int. J. Breast Cancer 2013, 137414 (2013).
Kan, S. et al. Up-regulation of HER2 by gemcitabine enhances the antitumor effect of combined gemcitabine and trastuzumab emtansine treatment on pancreatic ductal adenocarcinoma cells. BMC Cancer 15, 726 (2015).
Martin, M. et al. Trastuzumab emtansine (T-DM1) plus docetaxel with or without pertuzumab in patients with HER2-positive locally advanced or metastatic breast cancer: results from a phase Ib/IIa study. Ann. Oncol. 27, 1249–1256 (2016).
Cortés, J. et al. Efficacy and safety of trastuzumab emtansine plus capecitabine vs trastuzumab emtansine alone in patients with previously treated ERBB2 (HER2)-positive metastatic breast cancer: a phase 1 and randomized phase 2 trial. JAMA Oncol. 6, 1203–1209 (2020).
Janjigian, Y. Y. et al. Dose-escalation and dose-expansion study of trastuzumab deruxtecan (T-DXd) monotherapy and combinations in patients (pts) with advanced/metastatic HER2+ gastric cancer (GC)/gastroesophageal junction adenocarcinoma (GEJA): DESTINY-Gastric03. J. Clin. Oncol. 40, 295–295 (2022).
Levy, B. et al. MA13.07 TROPION-Lung02: initial results for datopotamab deruxtecan plus pembrolizumab and platinum chemotherapy in advanced NSCLC. J. Thorac. Oncol. 17, S91 (2022).
Moore, K. N. et al. Safety and activity findings from a phase 1b escalation study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with carboplatin in patients with platinum-sensitive ovarian cancer. Gynecol. Oncol. 151, 46–52 (2018).
Risbridger, G. P., Davis, I. D., Birrell, S. N. & Tilley, W. D. Breast and prostate cancer: more similar than different. Nat. Rev. Cancer 10, 205–212 (2010).
Mamounas, E. P. et al. Adjuvant T-DM1 versus trastuzumab in patients with residual invasive disease after neoadjuvant therapy for HER2-positive breast cancer: subgroup analyses from KATHERINE. Ann. Oncol. 32, 1005–1014 (2021).
Hurvitz, S. A. et al. TRIO-US B-12 TALENT: Phase II neoadjuvant trial evaluating trastuzumab deruxtecan with or without anastrozole for HER2-low, HR+ early stage breast cancer. J. Clin. Oncol. 40 (Suppl. 16), TPS623 (2022).
Andre, F. et al. Dose-finding and -expansion studies of trastuzumab deruxtecan in combination with other anti-cancer agents in patients (pts) with advanced/metastatic HER2+ (DESTINY-Breast07 [DB-07]) and HER2-low (DESTINY-Breast08 [DB-08]) breast cancer (BC).J. Clin. Oncol. 40, 3025 (2022).
Galluzzi, L., Humeau, J., Buqué, A., Zitvogel, L. & Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 17, 725–741 (2020).
Bauzon, M. et al. Maytansine-bearing antibody-drug conjugates induce in vitro hallmarks of immunogenic cell death selectively in antigen-positive target cells. OncoImmunology 8, e1565859 (2019).
Cao, A., Heiser, R., Law, C.-L. & Gardai, S. J. Abstract 4914: Auristatin-based antibody drug conjugates activate multiple ER stress response pathways resulting in immunogenic cell death and amplified T-cell responses. Cancer Res. 76, 4914–4914 (2016).
Hasan, M. M., Laws, M., Jin, P. & Rahman, K. M. Factors influencing the choice of monoclonal antibodies for antibody–drug conjugates. Drug Discov. Today 27, 354–361 (2022).
Nicolò, E. et al. Combining antibody-drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives. Cancer Treat. Rev. 106, 102395 (2022).
Spain, L., Diem, S. & Larkin, J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 44, 51–60 (2016).
Emens, L. A. et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomised, double-blind trial. Lancet Oncol. 21, 1283–1295 (2020).
Rosenberg, J. E. et al. LBA73 — Study EV-103 Cohort K: antitumor activity of enfortumab vedotin (EV) monotherapy or in combination with pembrolizumab (P) in previously untreated cisplatin-ineligible patients (pts) with locally advanced or metastatic urothelial cancer (la/mUC). Ann. Oncol. 33 (Suppl. 7), S1441 (2022).
Hamilton, E. et al. Abstract PD3-07: Trastuzumab deruxtecan (T-DXd; DS-8201) with nivolumab in patients with HER2-expressing, advanced breast cancer: a 2-part, phase 1b, multicenter, open-label study. Cancer Res. https://doi.org/10.1158/1538-7445.Sabcs20-pd3-07 (2021).
Galsky, M. D. et al. Primary analysis from DS8201-A-U105: A phase 1b, two-part, open-label study of trastuzumab deruxtecan (T-DXd) with nivolumab (nivo) in patients (pts) with HER2-expressing urothelial carcinoma (UC). J. Clin. Oncol. 40, 438–438 (2022).
Schmid, P., Jung, K. H., Wysocki, P. J., Jassem, J. & Ma, C. 166MO — Datopotamab deruxtecan (Dato-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic triple-negative breast cancer (a/mTNBC): initial results from BEGONIA, a phase 1b/2 study. Ann. Oncol. 33, S199 (2022).
Grivas, P. et al. TROPHY-U-01 Cohort 3: sacituzumab govitecan (SG) in combination with pembrolizumab (Pembro) in patients (pts) with metastatic urothelial cancer (mUC) who progressed after platinum (PLT)-based regimens. J. Clin. Oncol. 40, 434–434 (2022).
Schmid, P. et al. Abstract PD11-08: PD11-08 Trastuzumab deruxtecan (T-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic hormone receptor-negative (HR−), HER2-low breast cancer: updated results from BEGONIA, a phase 1b/2 study. Cancer Res. 83, PD11-08-PD11-08 (2023).
Waks, A. G. et al. Phase Ib study of pembrolizumab in combination with trastuzumab emtansine for metastatic HER2-positive breast cancer. J. Immunother. Cancer https://doi.org/10.1136/jitc-2022-005119 (2022).
Borges, V. F. et al. Tucatinib combined with Ado-trastuzumab emtansine in advanced ERBB2/HER2-positive metastatic breast cancer: a phase 1b clinical trial. JAMA Oncol. 4, 1214–1220 (2018).
Spring, L. M. et al. Phase 1b clinical trial of ado-trastuzumab emtansine and ribociclib for HER2-positive metastatic breast cancer. NPJ Breast Cancer 7, 103 (2021).
Bardia, A. et al. Abstract 2638: Sacituzumab govitecan, combination with PARP inhibitor, talazoparib, in metastatic triple-negative breast cancer (TNBC): translational investigation. Cancer Res. 82, 2638–2638 (2022).
O’Malley, D. M. et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol. Oncol. 157, 379–385 (2020).
Matulonis, U. A. et al. Efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant ovarian cancer with high folate receptor alpha expression: results from the SORAYA study. J. Clin. Oncol. 41, 2436–2445 (2023).
Liao, M. Z. et al. Model-informed therapeutic dose optimization strategies for antibody-drug conjugates in oncology: what can we learn from US Food and Drug Administration-approved antibody-drug conjugates? Clin. Pharmacol. Ther. 110, 1216–1230 (2021).
Hanna, K. S. Clinical overview of enfortumab vedotin in the management of locally advanced or metastatic urothelial carcinoma. Drugs 80, 1–7 (2020).
Moore, K. N. et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 123, 3080–3087 (2017).
Rivera, E. & Cianfrocca, M. Overview of neuropathy associated with taxanes for the treatment of metastatic breast cancer. Cancer Chemother. Pharmacol. 75, 659–670 (2015).
Iveson, T. et al. Prospective pooled analysis of four randomized trials investigating duration of adjuvant (adj) oxaliplatin-based therapy (3 vs 6 months {m}) for patients (pts) with high-risk stage II colorectal cancer (CC). J. Clin. Oncol. 37, 3501–3501 (2019).
Lu, D. et al. Time-to-event analysis of polatuzumab vedotin-induced peripheral neuropathy to assist in the comparison of clinical dosing regimens. CPT Pharmacomet. Syst. Pharmacol. 6, 401–408 (2017).
Deeks, E. D. Polatuzumab vedotin: first global approval. Drugs 79, 1467–1475 (2019).
Norsworthy, K. J. et al. FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist 23, 1103–1108 (2018).
Advani, A. et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: results of a phase I study. J. Clin. Oncol. 28, 2085–2093 (2010).
Kantarjian, H. et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer 119, 2728–2736 (2013).
Kantarjian, H. M. et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 375, 740–753 (2016).
Hurvitz, S. A. et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. Lancet 401, 105–117 (2023).
Tarantino, P. et al. Aiming at a tailored cure for ERBB2-positive metastatic breast cancer: a review. JAMA Oncol. https://doi.org/10.1001/jamaoncol.2021.6597 (2022).
Tamura, K. et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study. Lancet Oncol. 20, 816–826 (2019).
Goto, K. et al. LBA55 Trastuzumab deruxtecan (T-DXd) in patients (Pts) with HER2-mutant metastatic non-small cell lung cancer (NSCLC): interim results from the phase 2 DESTINY-Lung02 trial. Ann. Oncol. 33, S1422 (2022).
Autio, K. A., Boni, V., Humphrey, R. W. & Naing, A. Probody therapeutics: an emerging class of therapies designed to enhance on-target effects with reduced off-tumor toxicity for use in immuno-oncology. Clin. Cancer Res. 26, 984–989 (2020).
Bordeau, B. M., Yang, Y. & Balthasar, J. P. Transient competitive inhibition bypasses the binding site barrier to improve tumor penetration of trastuzumab and enhance T-DM1 efficacy. Cancer Res. 81, 4145–4154 (2021).
Chen, P., Bordeau, B. M., Zhang, Y. & Balthasar, J. P. Transient inhibition of trastuzumab–tumor binding to overcome the “binding-site barrier” and improve the efficacy of a trastuzumab–gelonin immunotoxin. Mol. Cancer Ther. 21, 1573–1582 (2022).
Boni, V. et al. Praluzatamab ravtansine, a CD166-targeting antibody–drug conjugate, in patients with advanced solid tumors: an open-label phase I/II trial. Clin. Cancer Res. 28, 2020–2029 (2022).
Aoyama, M., Tada, M., Yokoo, H., Demizu, Y. & Ishii-Watabe, A. Fcγ receptor-dependent internalization and off-target cytotoxicity of antibody-drug conjugate aggregates. Pharm. Res. 39, 89–103 (2022).
Scribner, J. A. et al. Preclinical evaluation of IMGC936, a next-generation maytansinoid-based antibody–drug conjugate targeting ADAM9-expressing tumors. Mol. Cancer Ther. 21, 1047–1059 (2022).
Mazor, Y. et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs 7, 377–389 (2015).
Sellmann, C. et al. Balancing selectivity and efficacy of bispecific epidermal growth factor receptor (EGFR) × c-MET antibodies and antibody-drug conjugates. J. Biol. Chem. 291, 25106–25119 (2016).
Hamblett, K. J. et al. Abstract 3914: ZW49, a HER2-targeted biparatopic antibody-drug conjugate for the treatment of HER2-expressing cancers. Cancer Res. 78, 3914–3914 (2018).
Antonarelli, G. et al. Research and clinical landscape of bispecific antibodies for the treatment of solid malignancies. Pharmaceuticals https://doi.org/10.3390/ph14090884 (2021).
Jhaveri, K. et al. 460MO Preliminary results from a phase I study using the bispecific, human epidermal growth factor 2 (HER2)-targeting antibody-drug conjugate (ADC) zanidatamab zovodotin (ZW49) in solid cancers. Ann. Oncol. 33, S749–S750 (2022).
Yamaguchi, A. et al. Chemical generation of small molecule-based bispecific antibody-drug conjugates for broadening the target scope. Bioorg. Med. Chem. 32, 116013 (2021).
de Goeij, B. E. et al. Efficient payload delivery by a bispecific antibody-drug conjugate targeting HER2 and CD63. Mol. Cancer Ther. 15, 2688–2697 (2016).
Andreev, J. et al. Bispecific antibodies and antibody-drug conjugates (ADCs) bridging HER2 and prolactin receptor improve efficacy of HER2 ADCs. Mol. Cancer Ther. 16, 681–693 (2017).
Hong, Y., Nam, S.-M. & Moon, A. Antibody–drug conjugates and bispecific antibodies targeting cancers: applications of click chemistry. Arch. Pharm. Res. 46, 131–148 (2023).
Shang, C. et al. Abstract 4256: YH012, a novel bispecific anti-HER2 and TROP2 antibody-drug conjugate, exhibits potent antitumor efficacy. Cancer Res. 82, 4256–4256 (2022).
Coumans, R. G. E. et al. A platform for the generation of site-specific antibody–drug conjugates that allows for selective reduction of engineered cysteines. Bioconjugate Chem. 31, 2136–2146 (2020).
Sussman, D. et al. Engineered cysteine antibodies: an improved antibody-drug conjugate platform with a novel mechanism of drug-linker stability. Protein Eng. Des. Sel. 31, 47–54 (2018).
Zhou, Q. et al. Site-specific antibody conjugation to engineered double cysteine residues. Pharmaceuticals https://doi.org/10.3390/ph14070672 (2021).
Tian, F. et al. A general approach to site-specific antibody drug conjugates. Proc. Natl Acad. Sci. USA 111, 1766–1771 (2014).
Nagaraja Shastri, P. et al. Nonclinical development of next-generation site-specific HER2-targeting antibody-drug conjugate (ARX788) for breast cancer treatment. Mol. Cancer Ther. 19, 1822–1832 (2020).
Zhou, Q. Site-specific antibody conjugation for ADC and beyond. Biomedicines https://doi.org/10.3390/biomedicines5040064 (2017).
Simmons, J. K., Burke, P. J., Cochran, J. H., Pittman, P. G. & Lyon, R. P. Reducing the antigen-independent toxicity of antibody-drug conjugates by minimizing their non-specific clearance through PEGylation. Toxicol. Appl. Pharmacol. 392, 114932 (2020).
Liu, S. et al. Abstract 4263: A novel pegylated biparatopic antibody-drug conjugate (pb-adc) targeting cancers with low HER2+ expression. Cancer Res. 82, 4263–4263 (2022).
Shao, T. et al. Construction of paclitaxel-based antibody–drug conjugates with a PEGylated linker to achieve superior therapeutic index. Signal. Transduct. Target. Ther. 5, 132 (2020).
Walker, J. A. et al. Hydrophilic sequence-defined cross-linkers for antibody–drug conjugates. Bioconjugate Chem. 30, 2982–2988 (2019).
Yamazaki, C. M. et al. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 12, 3528 (2021).
Świderska, K. W., Szlachcic, A., Opaliński, Ł., Zakrzewska, M. & Otlewski, J. FGF2 dual warhead conjugate with monomethyl Auristatin E and α-amanitin displays a cytotoxic effect towards cancer cells overproducing FGF receptor 1. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19072098 (2018).
Nilchan, N. et al. Dual-mechanistic antibody-drug conjugate via site-specific selenocysteine/cysteine conjugation. Antib. Ther. 2, 71–78 (2019).
Kumar, A. et al. Synthesis of a heterotrifunctional linker for the site-specific preparation of antibody-drug conjugates with two distinct warheads. Bioorg. Med. Chem. Lett. 28, 3617–3621 (2018).
Janku, F. et al. Preclinical characterization and phase I study of an anti-HER2-TLR7 immune-stimulator antibody conjugate in patients with HER2+ malignancies. Cancer Immunol. Res. 10, 1441–1461 (2022).
Mersana Therapeutics. Mersana Therapeutics Announces Clinical Hold on XMT-2056 Phase 1 Clinical Trial https://ir.mersana.com/news-releases/news-release-details/mersana-therapeutics-announces-clinical-hold-xmt-2056-phase-1#:~:Text=This%20action%20follows%20the%20company’s,its%20cause%20remain%20under%20investigation (13 March 2023).
Bordeau, B. M., Nguyen, T. D., Polli, J. R., Chen, P. & Balthasar, J. P. Payload-binding Fab fragments increase the therapeutic index of MMAE antibody–drug conjugates. Mol. Cancer Ther. 22, 459–470 (2023).
Toffoli, G. et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J. Clin. Oncol. 24, 3061–3068 (2006).
Liu, X., Cheng, D., Kuang, Q., Liu, G. & Xu, W. Association of UGT1A1*28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: a meta-analysis in Caucasians. Pharmacogenomics J. 14, 120–129 (2014).
Kweekel, D. M. et al. UGT1A1*28 genotype and irinotecan dosage in patients with metastatic colorectal cancer: a Dutch Colorectal Cancer Group study. Br. J. Cancer 99, 275–282 (2008).
Wijsenbeek, M. S. et al. Home monitoring in interstitial lung diseases. Lancet Respir. Med. 11, 97–110 (2023).
Broos, C. E. et al. Daily home spirometry to detect early steroid treatment effects in newly treated pulmonary sarcoidosis. Eur. Respir. J. 51, 1702089 (2018).
Moor, C. C. et al. Diurnal variation in forced vital capacity in patients with fibrotic interstitial lung disease using home spirometry. ERJ Open Res. https://doi.org/10.1183/23120541.00054-2020 (2020).
von Minckwitz, G. et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med. 380, 617–628 (2018).
Pondé, N. F., Zardavas, D. & Piccart, M. Progress in adjuvant systemic therapy for breast cancer. Nat. Rev. Clin. Oncol. 16, 27–44 (2019).
Braun, M. S. & Seymour, M. T. Balancing the efficacy and toxicity of chemotherapy in colorectal cancer. Ther. Adv. Med. Oncol. 3, 43–52 (2011).
Gennari, A. et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 32, 1475–1495 (2021).
Giordano, S. H. et al. Systemic therapy for advanced human epidermal growth factor receptor 2-positive breast cancer: ASCO Guideline update. J. Clin. Oncol. 40, 2612–2635 (2022).
Marmé, F. et al. 58O - Safety interim analysis (SIA) of the phase III postneoadjuvant SASCIA study evaluating sacituzumab govitecan (SG) in patients with primary HER2-negative breast cancer (BC) at high relapse risk after neoadjuvant treatment. Ann. Oncol. 33, S148–S149 (2022).
Hurvitz, S. A. et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 19, 115–126 (2018).
Petrylak, D. P. et al. Study EV-103 cohort H: antitumor activity of neoadjuvant treatment with enfortumab vedotin monotherapy in patients (pts) with muscle invasive bladder cancer (MIBC) who are cisplatin-ineligible. J. Clin. Oncol. 40, 435–435 (2022).
Schmitz, S., Duhoux, F. & Machiels, J. P. Window of opportunity studies: do they fulfil our expectations? Cancer Treat. Rev. 43, 50–57 (2016).
Prat, A. et al. LBA3 Patritumab deruxtecan (HER3-DXd) in early-stage HR+/HER2- breast cancer: final results of the SOLTI TOT-HER3 window of opportunity trial. Ann. Oncol. 33, S164 (2022).
Tarantino, P., et al. Abstract PD18-01: Adjuvant trastuzumab emtansine versus paclitaxel plus trastuzumab for stage I HER2+ breast cancer: 5-year results and correlative analyses from ATEMPT (TBCRC033). Cancer Res. 83, PD18-01-PD18-01 (2023).
Tolaney, S. M. et al. Adjuvant trastuzumab emtansine versus paclitaxel in combination with trastuzumab for stage I HER2-positive breast cancer (ATEMPT): a randomized clinical trial. J. Clin. Oncol. 39, 2375–2385 (2021).
Barroso-Sousa, R. et al. Cardiac outcomes of subjects on adjuvant trastuzumab emtansine vs paclitaxel in combination with trastuzumab for stage I HER2-positive breast cancer (ATEMPT) study (TBCRC033): a randomized controlled trial. NPJ Breast Cancer 8, 18 (2022).
Cortazar, P. et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164–172 (2014).
Hellmann, M. D. et al. Pathological response after neoadjuvant chemotherapy in resectable non-small-cell lung cancers: proposal for the use of major pathological response as a surrogate endpoint. Lancet Oncol. 15, e42–e50 (2014).
Menzies, A. M. et al. Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat. Med. 27, 301–309 (2021).
Meredith, K. L. et al. Pathologic response after neoadjuvant therapy is the major determinant of survival in patients with esophageal cancer. Ann. Surg. Oncol. 17, 1159–1167 (2010).
Chun, Y. S. et al. Significance of pathologic response to preoperative therapy in pancreatic cancer. Ann. Surg. Oncol. 18, 3601–3607 (2011).
Morton, D. et al. Preoperative chemotherapy for operable colon cancer: mature results of an international randomized controlled trial.J. Clin. Oncol. 41, 1541–1552 (2023).
Rugo, H. S. et al. Primary results from TROPiCS-02: a randomized phase 3 study of sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) in patients (Pts) with hormone receptor-positive/HER2-negative (HR+/HER2-) advanced breast cancer. J. Clin. Oncol. 40, LBA1001–LBA1001 (2022).
Tagawa, S. T. et al. TROPHY-U-01: a phase II open-label study of sacituzumab govitecan in patients with metastatic urothelial carcinoma progressing after platinum-based chemotherapy and checkpoint inhibitors. J. Clin. Oncol. 39, 2474–2485 (2021).
Cortés, J. et al. Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N. Engl. J. Med. 386, 1143–1154 (2022).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022).
Shitara, K. et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med. 382, 2419–2430 (2020).
Krop, I. E. et al. Phase 1b/2a study of trastuzumab emtansine (T-DM1), paclitaxel, and pertuzumab in HER2-positive metastatic breast cancer. Breast Cancer Res. 18, 34 (2016).
Santin, A. D. et al. Safety and activity of anti-mesothelin antibody-drug conjugate anetumab ravtansine in combination with pegylated-liposomal doxorubicin in platinum-resistant ovarian cancer: multicenter, phase Ib dose escalation and expansion study. Int. J. Gynecol. Cancer https://doi.org/10.1136/ijgc-2022-003927 (2022).
von Minkwitz, G. et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med. 380, 617–628 (2019).
Harbeck, N. et al. De-escalation strategies in human epidermal growth factor receptor 2 (HER2)–positive early breast cancer (BC): final analysis of the West German Study Group adjuvant dynamic marker-adjusted personalized therapy trial optimizing risk assessment and therapy response prediction in early BC HER2- and hormone receptor-positive phase II randomized trial — efficacy, safety, and predictive markers for 12 weeks of neoadjuvant trastuzumab emtansine with or without endocrine therapy (ET) versus trastuzumab plus ET. J. Clin. Oncol. 35, 3046–3054 (2017).
Hoimes, C. J. et al. Enfortumab vedotin plus pembrolizumab in previously untreated advanced urothelial cancer. J. Clin. Oncol. 41, 22–31 (2023).
Acknowledgements
No funding was provided for this work. We thank V. H. Goldstein of Dana-Farber Cancer Institute for submission assistance. The authors would also like to thank R. Colombo of Zymeworks (Vancouver, Canada) for providing precious insights during the writing of this paper. This article reflects the views of the authors and should not be construed to represent FDA views or policies.
Author information
Authors and Affiliations
Contributions
P.T. and B.R. researched data and wrote this manuscript. All authors made substantial contributions to discussions of content and reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
P.T. has acted as an adviser and/or consultant of AstraZeneca, Daiichi Sankyo, Gilead and Lilly. B.R. reports fees from Regeneron not related to the present article. S.M.T. has acted as a consultant and/or adviser of 4D Pharma, Aadi Bio, ARC Therapeutics, AstraZeneca, Bayer, BeyondSpring Pharmaceuticals, Blueprint Medicines, Bristol Myers Squibb, CytomX Therapeutics, Daiichi Sankyo, Eisai, Eli Lilly, Ellipses Pharma, Genentech/Roche, Gilead, Incyte Corporation, Infinity Therapeutics, Menarini/Stemline, Merck, Myovant, Novartis, Odonate Therapeutics, OncoSec Medical Inc., OncXerna, Pfizer, Reveal Genomics, Sanofi, Seattle Genetics, Umoja Biopharma, Zentalis, Zetagen, and Zymeworks and received research funding from AstraZeneca, Bristol Myers Squibb, Eisai, Exelixis, Gilead, Genentech/Roche, Lilly, Merck, NanoString Technologies, Novartis, OncoPep, Pfizer, Sanofi, and Seattle Genetics. S.M.P. declares no competing interests.
Peer review
Peer reviewer information
Nature Reviews Clinical Oncology thanks H. Burris III and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tarantino, P., Ricciuti, B., Pradhan, S.M. et al. Optimizing the safety of antibody–drug conjugates for patients with solid tumours. Nat Rev Clin Oncol 20, 558–576 (2023). https://doi.org/10.1038/s41571-023-00783-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41571-023-00783-w
This article is cited by
-
Pioneering the Way: The Revolutionary Potential of Antibody–Drug Conjugates in NSCLC
Current Treatment Options in Oncology (2024)
-
Emerging Toxicities of Antibody-Drug Conjugates for Breast Cancer: Clinical Prioritization of Adverse Events from the FDA Adverse Event Reporting System
Targeted Oncology (2024)
