
Abstract
Nemerteans (ribbon worms) and phoronids (horseshoe worms) are closely related lophotrochozoans—a group of animals including leeches, snails and other invertebrates. Lophotrochozoans represent a superphylum that is crucial to our understanding of bilaterian evolution. However, given the inconsistency of molecular and morphological data for these groups, their origins have been unclear. Here, we present draft genomes of the nemertean Notospermus geniculatus and the phoronid Phoronis australis, together with transcriptomes along the adult bodies. Our genome-based phylogenetic analyses place Nemertea sister to the group containing Phoronida and Brachiopoda. We show that lophotrochozoans share many gene families with deuterostomes, suggesting that these two groups retain a core bilaterian gene repertoire that ecdysozoans (for example, flies and nematodes) and platyzoans (for example, flatworms and rotifers) do not. Comparative transcriptomics demonstrates that lophophores of phoronids and brachiopods are similar not only morphologically, but also at the molecular level. Despite dissimilar head structures, lophophores express vertebrate head and neuronal marker genes. This finding suggests a common origin of bilaterian head patterning, although different heads evolved independently in each lineage. Furthermore, we observe lineage-specific expansions of innate immunity and toxin-related genes. Together, our study reveals a dual nature of lophotrochozoans, where conserved and lineage-specific features shape their evolution.
Similar content being viewed by others


Main
Lophotrochozoans represent more than one-third of known marine animals and play important ecological roles1. It is widely accepted that they comprise a major protostome clade, although the clade nomenclature depends on the taxa included. Nevertheless, one classification scheme proposes that protostomes consist of two sister groups—spiralians (most of which exhibit spiral cleavage) and ecdysozoans (that shed their exoskeletons)2. According to the narrow definition, Lophotrochozoa is a subgroup of spiralians and most lophotrochozoans possess either lophophore or trochophore larvae during the planktonic stage. Lophotrochozoans sensu stricto include annelids (for example, leeches and polychaete worms), molluscs (for example, snails and octopuses), nemerteans (ribbon worms), phoronids (horseshoe worms), ectoprocts (bryozoans, otherwise known as moss animals) and brachiopods (lamp shells), although many phylogenetic relationships within the group remain unresolved3,4,5. Molecular phylogenetics suggests that nemerteans and phoronids are closely related3, yet these two phyla have divergent body plans and exhibit no morphological synapomorphic traits. In particular, they have different lifestyles with distinct larval forms and they possess different types of feeding apparatus. For example, nemerteans are unsegmented worms. Mostly predators, they have an eversible proboscis derived from the rhynchocoel (that is, a fluid-filled tubular chamber) for capturing prey and for defence. In contrast, phoronids are sessile filter feeders with ciliated tentacles called lophophores—horseshoe-shaped feeding apparatus that are also shared by ectoprocts and brachiopods. Given the incompatibility of molecular and morphological phylogenies for these groups, the origins of nemerteans and phoronids have remained obscure, although some studies support the close relationship of phoronids and brachiopods.
Our genomic understanding of protostomes is largely based on comparative studies of model ecdysozoans, such as fruit flies and nematodes. Although most developmental genes are shared between protostomes and deuterostomes, some are lost in ecdysozoans, but present in lophotrochozoans. For instance, Nodal, a member of the transforming growth factor-β (TGFβ) superfamily that is required for left–right patterning, has been considered a deuterostome-specific gene, but recently it was found in molluscs6. Similarly, some gene families, such as innate immunity-related genes, are highly reduced in ecdysozoans, but more complex in lophotrochozoans7,8. Recent genomic studies have further shown that annelids and molluscs share various genomic features, such as gene family size and conserved orthologous gene clusters, with invertebrate deuterostomes (for example, amphioxus and sea urchins)9. This observation raises the question of whether lophotrochozoans share some bilaterian ancestral features with invertebrate deuterostomes, which apparently have been lost in ecdysozoans and other lineages during protostome evolution.
Here, we present genomes of the nemertean Notospermus geniculatus and the phoronid Phoronis australis and explore lophotrochozoan evolution using comparative genomics. With both genomic and transcriptomic data, our phylogenetic analyses provide evidence that nemerteans are probably sisters to lophophorates—a clade of animals with horseshoe-shaped lophophores comprising phoronids, ectoprocts and brachiopods, although the position of ectoprocts is questionable under a sensitivity analysis. Our results clearly show that lophotrochozoans have a different evolutionary history than other spiralians (or platyzoans), such as flatworms and rotifers. In particular, lophotrochozoans retain a basic bilaterian gene repertoire, which is probably lost in ecdysozoans and other spiralian lineages. Unexpectedly, genes specifically expressed in lophophores of phoronids and brachiopods are strikingly similar to those employed in vertebrate head formation, although novel genes, expanded gene families and redeployment of developmental genes also contribute to the unique molecular identity of lophophores. Furthermore, we provide examples of lineage-specific genomic features in lophotrochozoans, such as the expansion of innate immunity and toxin-related genes. Taken together, our study reveals the dual nature of lophotrochozoan genomes, showing both conservative and innovative characteristics during their evolution.
Results and discussion
Genome characterization
We sequenced two lophotrochozoan genomes (Supplementary Fig. 1) with at least 220-fold coverage using random shotgun approaches with Illumina MiSeq, HiSeq and Roche 454 platforms (Supplementary Figs. 2–4, Supplementary Tables 1 and 2 and Supplementary Note 1). The haploid genome assembly sizes of the nemertean N. geniculatus and the phoronid P. australis are 859 and 498 Mb, respectively, with N50 lengths of assembled scaffolds of 239 and 655 kb, respectively (Table 1). The genome sizes and assembly quality are comparable to those of other lophotrochozoans, such as the polychaete Capitella teleta (324 Mb)9, Pacific oyster Crassostrea gigas (558 Mb)10 and brachiopod Lingula anatina (406 Mb)11 (Supplementary Table 3). With the support of deep RNA sequencing (RNA-seq) data obtained from 21 libraries, including embryonic stages and adult tissues, we estimated that the Notospermus and Phoronis genomes contain 43,294 and 20,473 protein-coding genes, respectively (Supplementary Fig. 5 and Supplementary Tables 4 and 5). High gene numbers in Notospermus may be related to acquisition of lineage-specific genes and expansions of gene families. Both Notospermus and Phoronis genomes exhibit high heterozygosity (2.4 and 1.2%, respectively) (Supplementary Fig. 6). The abundance of repetitive sequences contributes to the increased size of their genomes (37.5 and 39.4%, respectively). In particular, although the intron–exon structure (8 exons and 7 introns, on average) is similar between Phoronis and Lingula, insertions of transposable elements into introns result in doubling of the Phoronis gene size (14,590 base pair (bp)) compared with that of Lingula (7,725 bp) (Table 1, Supplementary Figs. 7–10 and Supplementary Tables 6–8).
Phylogeny of lophotrochozoans
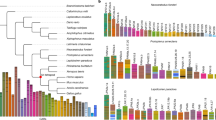
The nomenclature of Lopho-trochozoa varies, depending on whether the sensu stricto or sensu lato definition is considered (Supplementary Figs. 11 and 12 and Supplementary Table 9). To prevent confusion, we used Lophotrochozoa sensu stricto throughout this study. Given that nemerteans possess few morphological features compared with other lophotrochozoans, the phylogenetic position of Nemertea within Lophotrochozoa is highly controversial2,3,12,13,14,15. Some phylogenomic studies place Nemertea as sister to Phoronida and Brachiopoda3,4,5 (Fig. 1a). However, others propose different hypotheses based on various marker sets and substitution models, placing Nemertea in a variety of phylogenetic positions2,13,15,16 (Fig. 1b–d and Supplementary Table 10). To resolve this issue, we applied genome-based phylogenetic analysis (Supplementary Note 2). Using 173 one-to-one orthologous genes from available lophotrochozoan genomes9,10,11,17,18, we showed that Nemertea is close to Phoronida and Brachiopoda (Fig. 1e). Phylogenetic trees based on gene content and transcriptomes also support this relationship (Supplementary Figs. 13 and 14).

a–d, Proposed relationships of the five major clades of lophotrochozoans. a, Kryptrochozoa hypothesis (monophyly of Nemertea, Phoronida and Brachiopoda). b, Nemertea as a sister group to other lophotrochozoans. c, Nemertea as a sister group to Mollusca. d, Nemertea as a sister group to Annelida. e, Phylogeny of lophotrochozoans inferred from 173 one-to-one orthologous genes (62,928 amino acid positions with 92% overall matrix completeness). The maximum-likelihood tree was obtained using LG + Γ, LG + I + Γ, LG4M + Γ and LG4X + Γ models with 1,000 bootstrap replicates. Black circles on nodes indicate 100% bootstrap support from all four models.
Besides the position of Nemertea, several issues about lophotrochozoan phylogeny remain a matter of debate. For example, whether Ectoprocta belongs to the historical superphylum Lophophorata has been contentious2,3,4,5,12,13,14,15 (Supplementary Fig. 15). To test these hypotheses, we retrieved deep RNA-seq reads from 26 taxa, including annelids16, molluscs16, nemerteans7,19,20, phoronids7, ectoprocts5,21 and brachiopods2,7,11. After assembling the transcriptomes de novo, we retained those of high quality (Supplementary Fig. 16 and Supplementary Tables 11–14) and performed phylogenetic analyses with both genomic and transcriptomic data. Our analysis supports monophyly of Brachiopoda, in which Linguliformea and Craniiformea are sisters to Rhynchonelliformea (Supplementary Figs. 17 and 18). Furthermore, Phoronida is probably sister to Ectoprocta. Although the position of Ectoprocta is not certain, our results provide evidence to support the traditional classification of Lophophorata (Phoronida, Ectoprocta and Brachiopoda). Differences between the present results and those of previous studies are possibly due to the selection of different ectoproct gene sets with differing evolutionary rates (Supplementary Figs. 19–21 and Supplementary Table 15), highlighting the importance of careful selection of genes with strong phylogenetic signals22. Further analysis of ectoproct genomes as well as transcriptomes with more complete sampling and higher sequencing coverage will be needed to address its uncertain relationship in lophotrochozoans.
Bilaterian gene repertoire and gene family evolution
To gain insight into bilaterian gene family evolution, we compared lophotrochozoan proteomes with those of other metazoans (Supplementary Table 16 and Supplementary Note 3). The Notospermus genome has experienced a high turnover rate and a recent expansion of gene families compared with Phoronis (Supplementary Fig. 22). Comparing gene families among four lophotrochozoans including Lingula11 and Octopus17, we identified 7,007 lophotrochozoan core gene families, with 1,127 gene families shared only among nemerteans, phoronids and brachiopods, reflecting their relatively close phylogenetic relationships (Fig. 2a). A principle component analysis of gene family size and protein domain showed that lophotrochozoans consistently cluster with invertebrate deuterostomes, such as amphioxus, acorn worms and sea urchins (Fig. 2b and Supplementary Fig. 23). We further determined that lophotrochozoans and deuterostomes share 4,662 gene families that are not found in ecdysozoans or platyzoans, such as flatworms and rotifers. In particular, except for those belonging to eumetazoan genes23, 2,870 gene families are bilaterian-specific. They cannot be found in cnidarians or sponges (Fig. 2c and Supplementary Fig. 24). Many of these gene families carry epidermal growth factor-like, zinc finger and fibronectin domains, which are related to regulation of cell cycle, biological adhesion and immune response (Supplementary Table 17). Thus, our data suggest that an ancestral bilaterian gene repertoire retained in lophotrochozoans and deuterostomes is related to control of homoeostasis and multicellularity24.

a, Venn diagram of shared and unique gene families in four selected lophotrochozoans. Gene families were identified by clustering of orthologous groups using OrthoMCL. b, Principal component (PC) analysis of PANTHER gene family sizes. Invertebrate deuterostomes (bfl, sko and spu) cluster with lophotrochozoans (solid-lined circle). The dashed-lined circle denotes the clustering of vertebrates. c, Matrix of shared gene families among selected metazoans. The cladogram on the left is based on phylogenetic positions inferred from this study. Dashed lines separate the major clades. Note that tunicates (cin) and leeches (hro) share fewer genes with other bilaterians, probably because of their relatively high evolutionary rates and gene loss in each lineage. adi, Acropora digitifera; aqu, Amphimedon queenslandica; ava, Adineta vaga; bfl, Branchiostoma floridae; cel, Caenorhabditis elegans; cgi, C. gigas; cin, Ciona intestinalis; cmi, Callorhynchus milii; cte, C. teleta; dme, Drosophila melanogaster; dpu, Daphnia pulex; dre, Danio rerio; emu, Echinococcus multilocularis; gga, Gallus gallus; hro, Helobdella robusta; hsa, Homo sapiens; lan, L. anatina; lgi, Lottia gigantea; nge, N. geniculatus; nve, Nematostella vectensis; obi, Octopus bimaculoides; ola, Oryzias latipes; pau, P. australis; pfu, Pinctada fucata; sko, Saccoglossus kowalevskii; sma, Schistosoma mansoni; sme, Schmidtea mediterranea; spu, Strongylocentrotus purpuratus; tca, Tribolium castaneum; tru, Takifugu rubripes; xtr, Xenopus tropicalis.
To investigate the evolution of developmental gene content, we annotated transcription factor and signalling pathway-related genes. The Phoronis genome has a smaller number of genes with homeobox and helix-loop-helix binding domains compared with those of other lophotrochozoans (Supplementary Tables 18 and 19). TGFβ and Wnt signalling pathways play important roles in axial patterning, cell specification and control of cell behaviour during embryonic development25,26. Some TGFβ genes modulating Nodal signals, such as Lefty and Univin, are considered deuterostome novelties27. The Notospermus and Phoronis genomes have 15 and 10 TGFβ genes, respectively (Supplementary Table 20). Interestingly, in addition to Nodal, which can be found in the Notospermus, Phoronis and Lingula genomes, we discovered the syntenic linkage of Univin and Bmp2/4 in the Lingula genome, despite its absence in other protostomes. Thus, this finding suggests that the linkage of Univin and Bmp2/4 is a bilaterian ancestral feature that has been lost in some vertebrates and protostomes (Supplementary Fig. 25). Transcriptome analysis shows that Nodal is either not expressed or is expressed at very low levels during early development in Phoronis and Lingula. The Notospermus and Phoronis genomes have 17 and 12 Wnt genes, respectively (Supplementary Table 21). In Notospermus and Phoronis, we identified all Wnt genes (Wnt1, Wnt2, Wnt4–11, Wnt16 and WntA) except Wnt3, which has probably been lost in all protostomes. We failed to find Wnt9 and Wnt10 in Notospermus (Supplementary Figs. 26 and 27). Unlike lophotrochozoans, extensive loss of Wnt genes may be a common feature in Platyhelminthes28 and Pancrustacea29.
Remarkably, we also observed many gene families that are lineage-specific (10–30%) and patchy (~10%; that is, genes retained in certain lineages, but unevenly lost in others) among bilaterians (Supplementary Fig. 28). Together with lineage-specific gene family expansion, these features reflect the dynamics of genome evolution (Supplementary Fig. 29). For instance, the most expanded gene family in Notospermus belongs to retrotransposon-like protein (RTL1). The role of this gene is not clear, but it has been neofunctionalized for developmental processes30. Other expanded gene families in Notospermus are mostly related to toxin metabolism (SLC25A17 and S47A1) and immune response (APAF, IRF5 and IN80C). The most expanded gene families in Phoronis are also related to immunity and programmed cell death (TRI56 and RIPK3) (Supplementary Table 22). Further analysis shows that both Notospermus and Phoronis genomes have more genes with apoptosis-related domains, indicating more complex regulation of cell death programmes (Supplementary Table 23). Notably, gene families related to mucus production, such as mucin-4 (MUC4) and carbohydrate sulfotransferase (CHST) are expanded independently in Phoronis and Lingula and are highly expressed in the lophophores (Supplementary Figs. 30 and 31). This finding indicates possible independent adaptation within each lophophorate lineage, where P. australis may adapt to live with tube-dwelling anemones by protecting themselves with mucus layers. Altogether, our results suggest that both conservation (for example, conserved gene repertoire) and innovation (for example, lineage-specific gene gains and losses and gene family expansion) are fundamental processes shaping the evolution of bilaterian gene families.
Hox genes and conserved bilaterian microsyntenies
Hox genes play essential roles during metazoan development, especially for body patterning and appendage formation31. Notospermus contains 16 Hox genes and two ParaHox genes, although Xlox may have been absent. The Notospermus Hox cluster is disorganized, with Hox genes dispersed in ten different scaffolds (Fig. 3a, Supplementary Figs. 32 and 33 and Supplementary Tables 24–26). In contrast, Phoronis has eight Hox genes in one Hox cluster and three ParaHox genes. We failed to find Scr and Antp in Phoronis. Given that Scr and Antp are expressed in the shell-forming epithelium in brachiopods32, possible gene loss of Scr and Antp in the phoronid lineage may contribute to their shell-less morphology. This may also imply that common lophophorate ancestors had either unmineralized (agglutinated) or mineralized shells that were lost secondarily in crown phoronids33,34. With improved scaffolding, we discovered Lox4 in Lingula, which is linked between Post2 and Antp. Both Notospermus and Phoronis have only one posterior Hox, Post2. Post2 has been identified in polychaetes and brachiopods as a spiralian gene35. Our phylogenetic analysis further shows that Post2 is shared by platyhelminths and all lophotrochozoans. We demonstrated that Post2 has a different evolutionary origin from ecdysozoan AbdB, whereas Post1 may be specific to lophotrochozoans (Supplementary Fig. 34). Interestingly, a recent study shows that rotifers do not have the Post2 gene. Instead, they carry a different posterior Hox gene, MedPost36. This finding suggests different origins of Hox genes among gnathiferans, rouphozoans and lophotrochozoans.

a, Hox clusters in selected protostomes. Arrows indicate transcript directions. Double slashes denote non-continuous linkages between two genes. Black dots signify the end of the scaffolds. b, Matrix of microsyntenic blocks (clustered orthologous genes ≥ 3) among bilaterians. c, An example of neighbouring tightly linked (<20 kb) orthologous genes shared by lophotrochozoans. The numbers on scaffolds indicate genomic distance (kb). ALKB5, alpha-ketoglutarate-dependent dioxygenase alkB homologue 5; COX11, mitochondrial cytochrome c oxidase assembly protein; DRG2, developmentally regulated GTP-binding protein 2; FSCN1, fascin; Sca, scaffold.
Notospermus Hox genes are expressed along the adult anterior–posterior axis with Hox1 and Hox2 expressed anteriorly, Lox2 and Lox4 mid-posteriorly and Post2 posteriorly, but with no strict spatial collinearity. In contrast, Hox gene expression in Phoronis and Lingula does not exhibit apparent spatial polarity (Supplementary Fig. 35). Remarkably, Hox genes are not expressed in the proboscis and head of Notospermus nor in lophophores of Phoronis and Lingula. This anterior Hox-free region is also found in juvenile amphioxus37, hemichordates38, arthropods39, nemerteans40 and annelids41, suggesting that the absence of Hox gene expression at the anterior end is a common adult body plan for all bilaterians.
Unlike the Hox cluster, other conserved gene linkages (‘synteny’) among animals are rarely studied. Conserved microsynteny, such as the pharyngeal gene cluster, is thought to contribute to morphological innovation among deuterostomes, although the regulatory mechanism is still unknown27. We identified ~300–400 conserved microsyntenic blocks (that is, clusters of three or more orthologues with close physical linkages) among lophotrochozoans and amphioxus, indicating a deep bilaterian ancestry of gene linkages (Fig. 3b and Supplementary Note 3). Intriguingly, however, most gene clusters associated with embryonic development, such as Wnt (Wnt9, Wnt1, Wnt6 and Wnt10), ParaHox (Gsx, Xlox and Cdx) and NK (Msxlx, Nkx2.2 and Nkx2.1; Msx, Nkx4, Nkx3, Lbx and Tlx) clusters, are disorganized in Notospermus and Phoronis, although they are retained intact in Lingula (Supplementary Fig. 36). In contrast with the Hox cluster, where transcriptional direction among Hox genes is often the same, neighbouring, tightly linked genes (distance < 20 kb) in the microsyntenic blocks are mostly in opposing directions (Fig. 3c, Supplementary Fig. 37 and Supplementary Table 27). Interestingly, we found that tightly linked genes show significantly lower evolutionary rates, suggesting that they are under strong negative selection. Also, tightly linked genes within microsyntenic blocks tend to be expressed constantly across different species and tissue types (Supplementary Fig. 38 and Supplementary Table 28).
Molecular signature of lophophore and bilaterian head patterning
Traditionally, the lophophore is a feeding apparatus defined as a mesosomal extension with ciliated tentacles that are present in both pterobranch hemichordates and lophophorates. To avoid confusion, here, we apply the term 'lophophore' to the horseshoe-shaped homologous structure shared by brachiopods and phoronids42. Recent immunohistochemical and ultrastructural studies have shown that the lophophore is enriched with neural cells42,43, yet the molecular signature of the lophophore remains unclear. To explore the origin of the lophophore, we applied molecular profiling using an unbiased all-to-all pairwise comparison of different tissues among Notospermus, Phoronis and Lingula using RNA-seq (Fig. 4a–c and Supplementary Note 4). We first conducted comparative transcriptomics by calculating the Spearman’s correlation coefficient (ρ) based on expression levels of 8,650 orthologues shared by all three genomes. The Notospermus proboscis is molecularly distinct from other types of Notospermus tissues (Supplementary Fig. 39) and dissimilar to the Phoronis lophophore (ρ = 0.31) (Fig. 4d). Instead, at the molecular level, the Phoronis lophophore is considerably more similar to the Notospermus head (anterior end and anterior part 1; ρ = 0.46) (Fig. 4a,b,d). Further comparison of Phoronis and Lingula lophophores confirms the shared origin of their feeding apparatus (ρ = 0.61) (Fig. 4b,c,e and Supplementary Fig. 40). Next, to investigate the molecular nature of lophophores, we performed expression profiling based on differentially expressed genes. We identified 2,572 and 1,591 genes that are specifically expressed in the lophophores of Phoronis and Lingula, respectively. Approximately 40% of these genes have no available annotation, reflecting the contribution of a large number of lineage-specific genes to tissue-specific functions (Supplementary Fig. 41).

a–c, Cartoon illustrations of an adult Notospermus (a), Phoronis (b) and Lingula (c; with the dorsal shell removed) with the anterior end facing left. d,e, Analyses of Spearman’s correlation coefficient (ρ) and hierarchical clustering with expression levels of 8,650 orthologous genes from larvae and adult tissues of Notospermus versus Phoronis (d) and Phoronis versus Lingula (e). f, Expression profiles of head patterning-related and neuronal genes in the head of Notospermus and lophophores of Phoronis and Lingula. g,h, Schematic representation of anterior–posterior patterning in bilaterians. g, Simplified phylogeny of bilaterians and regions of anterior positioning heads (highlighted in orange). h, Domain map for conserved signalling components, transcription factors and genes associated with synaptic machinery along the anterior–posterior axis in the last common bilaterian ancestor. A, anterior; AE, anterior end; AM, ampulla; AP1, anterior part 1; AP2, anterior part 2; bHLH, basic helix-loop-helix; CNS, central nervous system; DC, digestive caecum; GT, gut; HMG, high mobility group; LP, lophophore; LV, larva; MB, mid-body; MT, mantle; P, posterior; PB, proboscis; PC, pedicle; PE, posterior end; TK, trunk; TL, trunk with LP.
Many annotated genes in lophophores are related to neural development; for example, those expressed in the Notospermus head (Supplementary Figs. 41 and 42 and Supplementary Table 29). Unexpectedly, we found that vertebrate head markers such as otx, lhx1/5, foxG, pax6 and six3/6 are specifically expressed in both the Notospermus head and the Phoronis lophophore (Fig. 4f, Supplementary Figs. 43 and 44 and Supplementary Tables 30 and 31). Neuronal markers such as soxB2 and achaete-scute (ascl), as well as genes associated with synaptic machinery, such as tyrosine monooxygenase (th) and choline acetyltransferase (chat), are also highly and specifically expressed in lophophores (Fig. 4f and Supplementary Table 32). In addition, we found specific expression of genes for sensory ion channels, such as the cyclic nucleotide-gated olfactory channel (cnga2) and amiloride-sensitive sodium channel subunit beta (scnn1b) in lophophores, suggesting their roles in taste perception and environmental responses (Fig. 4f). These results indicate that lophophores share the molecular nature of the head and anterior centralized nervous system. Interestingly, many of these ‘head/lophophore’ genes overlap with those that are conservatively expressed during the organogenesis stage in vertebrates—the phylotypic period44, including foxG1, pax6, klf2, emx2 and islet1 (Supplementary Tables 29 and 30). Most of these genes are associated with neuronal differentiation, sensory organ development and forebrain development (Supplementary Table 29). Thus, the vertebrate phylotypic period probably reflects the importance of the head patterning step during evolution of bilaterian development.
In bilaterians, the anterior–posterior axis is patterned by a gradient of canonical Wnt signalling through β-catenin45. Along the axis, the bilaterian head develops at the anterior end, characterized by centralization of the nervous system, where Wnt signalling is down-regulated46. Intriguingly, Wnt signalling genes are differentially expressed along the anterior–posterior axis with the Wnt receptor fzd5/8, as well as Wnt antagonists, sfrp1/5 and notum, which are expressed in the head of Notospermus and lophophores of Phoronis and Lingula (Supplementary Fig. 45). Thus, it is tempting to speculate the existence of a conserved anterior–posterior patterning mechanism in which inactivation of Wnt signalling at the anterior end is essential for bilaterian head formation. Superimposed on the conserved patterning system, we found ten homeobox genes (uncx, pou4, six4/5, barx, prox, arx, vsx, alx, msx and nkx1) that are specifically expressed in both Phoronis and Lingula lophophores, but not in the Notospermus head, suggesting a redeployment of developmental genes in patterning lineage-specific structures (Supplementary Fig. 46). Taken together, the lophophore is a structure at the anterior end without Hox gene expression. It expresses Wnt antagonists, head and neuronal markers as well as genes that are associated with synaptic machinery and sensory functions. These features thus resemble the head patterning systems and entities seen in other deuterostomes, ecdysozoans and lophotrochozoans47,48 (Fig. 4g,h). Therefore, despite the lack of morphological similarity, lophophores bear a molecular resemblance to the heads of other bilaterians. Our findings thus suggest a possible common origin of bilaterian head patterning in the bilaterian ancestor of protostomes and deuterostomes, although distinct corresponding structures are formed and evolved independently in different lineages49,50.
Lineage-specific expansion of innate immune genes
Invertebrates defend themselves against infection by viruses, bacteria, fungi or other parasites using innate immune responses that involve pattern recognition and signalling (Fig. 5a). We showed that toll-like receptor (TLR) genes are absent in rotifers, planarians and blood flukes, but are expanded in most lophotrochozoans with numbers of genes comparable to those of deuterostomes (Fig. 5b and Supplementary Note 4). The Notospermus and Phoronis genomes contain 8 and 25 TLR genes, respectively (Supplementary Table 33). Most TLR genes show lineage-specific expansion through tandem duplications (Fig. 5c,d). Although TLR genes are mostly intronless, we found several that carry introns (Fig. 5d). In humans, TLR genes with low numbers (<10) of leucine-rich repeats, such as TLR1, TLR2 and TLR6, recognize glycolipids or lipopeptides, whereas those with high numbers (10–18) of leucine-rich repeats usually target nucleic acids51. Expanded Notospermus and Phoronis TLR genes are mostly long and have low numbers of leucine-rich repeats (Fig. 5e and Supplementary Fig. 47). Some TLR genes are specifically expressed in Phoronis and Lingula lophophores, whereas many of them have low expression across tissues, indicating that they may be triggered by infection8 (Supplementary Fig. 48).

a, Schematic representation of components in the MyD88-dependent pathway of TLR and interleukin-1 receptor (IL-1R) signalling. AP1, activator protein 1; Ig, immunoglobulin; IKK, inhibitor of κB kinase; IRAK, interleukin-1 receptor-associated kinase; JNK, c-Jun N-terminal kinases; LRR, leucine-rich repeat; MKK, mitogen-activated protein kinase kinase; MyD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor-κB; TAB1, TAK1-binding protein 1; TAK1, TGF-beta-activated kinase 1; TIR, toll/interleukin-1 receptor; TRAF, TNF receptor-associated factor. b, Distribution of TLR/IL-1R signalling components among selected metazoans. c, Phylogenetic analysis of TLR genes among Notospermus (green), Phoronis (blue) and Lingula (red), with TIR domains using the neighbour-joining method with the Jones-Taylor-Thornton (JTT) model (49 genes, 133 amino acids, 1,000 bootstrap replicates). Open circles on nodes denote bootstrap support > 60%. d, Genomic organization of TLR genes. Arrows show the direction of transcription. Rectangles indicate exons. e, Gene structure of TLR genes.
Nemertean toxins
Nemerteans produce peptide toxins to capture prey and for defense19. To investigate the origins of nemertean toxins, we annotated 63 putative toxin genes in the Notospermus genome. Of these, 15 genes, such as metalloproteases and phospholipases, are shared with other lophotrochozoans that have no reported toxic proteins, suggesting that those genes may have other roles in metabolism and may have been co-opted for toxic functions (Supplementary Table 34). We focused on 32 putative toxin genes that are specifically present in Notospermus, and found 26 of these differentially expressed in eggs and tissues. Many of these genes, such as C-type lectins (SL27) and serine protease inhibitors (VKT6) expressed in the proboscis are associated with inhibition of platelet aggregation and haemolysis (Supplementary Fig. 49 and Supplementary Table 35). Among these toxin genes, we also found several genes that have high sequence similarities to the stonefish toxin, stonustoxin. Stonustoxin is a pore-forming protein of the membrane attack complex-perforin/cholesterol-dependent cytolysin superfamily, which is widely distributed among eukaryotes52. Wide distribution of this gene in non-toxic taxa suggests that it may play a broader role than envenomation. For known nemertean-specific toxin genes, we could not find neurotoxin B-II or neurotoxin B-IV in the Notospermus genome, indicating they may be lineage-specific in Cerebratulus lacteus. Instead, we found the cytolytic protein cytotoxin A-III, which is expanded in Notospermus (Supplementary Fig. 49). Cytotoxin A-III is a polypeptide cytotoxin that was first isolated from C. lacteus mucus and has also been found in other heteronemerteans53. Notospermus cytotoxin A-III genes are expanded through tandem duplication and expressed throughout the body or specifically in the proboscis and eggs.
Biomineralization
Although phoronids are closely related to brachiopods, they have no mineralized tissues. Chitin synthase genes, which are required for biomineralization, are reduced in Phoronis (6) compared with Lingula (31)11 (Supplementary Fig. 50). Some chitin synthase genes present in molluscs and brachiopods with close orthology cannot be found in Phoronis. This probably indicates loss of these genes in the phoronid lineage, although we cannot exclude the possibility of misannotation. To explore the origin of mineralized tissues in lophophorates, we compared biomineralization-related genes among phoronids and brachiopods, including the mantle transcriptome of the brachiopod Magellania venosa54. We found only five shell matrix protein genes that are shared by Phoronis, Lingula and Magellania (Supplementary Fig. 51). These genes include peroxidasin (PXDN), mucin-5B (MUC5B), serine protease 42 (PRS42), SVEP1 and hemicentin-1 (HMCN1). Notably, most of these genes can also be found in other metazoans with functions other than biomineralization. We failed to find brachiopod-specific shell matrix proteins in the Phoronis genome (Supplementary Table 36)11,54. Thus, our findings suggest that lineage-specific gene expansions, acquisition of novel genes and redeployment of extracellular matrix genes are involved in the evolution of lophophorate biomineralization.
Conclusion
Despite being phylogenetically closely related, nemerteans, phoronids and brachiopods diverged early, perhaps before the Cambrian explosion55. During more than 540 million years of evolution, they have evolved many lineage-specific features and yet retained unexpected elements in terms of the bilaterian gene repertoire and head patterning system. One remarkable finding is that the same developmental head marker genes are expressed in the adult anterior structure, which may highlight their roles in maintaining tissue identity and homoeostasis in all bilaterians. We argue that the molecular basis of morphological features is the combination of the conserved gene repertoire and patterning system, together with lineage-specific gene family expansions and novel genes10,11,17. However, co-option and redeployment of developmental and structural genes in different lineages also contribute to specialization and functions of body structures56. Although our phylogenetic analysis based on transcriptomic data suggests the possible monophyly of lophophorates, an ectoproct genome will be needed for a comprehensive understanding of lophophorate evolution. Given Xenacoelomorpha as the earliest branching bilaterians2, the origins of the bilaterian gene repertoire and heads will be further clarified with the available genomes from Acoela, Nemertodermatida and Xenoturbella. The draft Notospermus and Phoronis genomes presented here, together with our comparative genomics and transcriptomics, provide insight into the conservation and dynamics of lophotrochozoan evolution.
Methods
Biological materials
Adult nemerteans (N. geniculatus) were collected at the Ushimado Marine Institute, Okayama University, Japan. Adult phoronids (P. australis) were collected at Kuroshima Island, near Ushimado town, Okayama, Japan (Supplementary Fig. 1). After starvation, genomic DNA was extracted from intact adults using the phenol/chloroform method.
Genome sequencing and assembly
The Notospermus and Phoronis genomes were sequenced using Illumina MiSeq, HiSeq 2500 and Roche 454 GS FLX + platforms (Supplementary Figs. 2 and 3). Paired-end libraries (286–1,100 bp) were prepared using the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs). Paired-end reads were sequenced to obtain 127 and 71 Gb of data from Notospermus and Phoronis samples, respectively, using Illumina MiSeq (read length 250–400 bp) (Supplementary Tables 1 and 2). A mate pair library from 3 kb DNA fragments was prepared using the Cre-Lox recombination approach. Other mate pair libraries generated from 1.5 to 20 kb DNA fragments were size selected with the automated electrophoresis platforms SageELF or BluePippin (Sage Science) and prepared using the Nextera Mate Pair Sample Prep Kit. Mate pair libraries were sequenced to obtain 100 and 38 Gb of data from Notospermus and Phoronis samples, respectively, using Illumina HiSeq 2500 and MiSeq platforms.
After quality control checks with FastQC (v0.10.1), Illumina reads were quality filtered (Q score ≥ 20) and trimmed with Trimmomatic (v0.33). Roche 454 reads were filtered with PRINSEQ (v0.20.3) to remove duplicated and low-complexity sequences. Mate pair reads prepared from Cre-LoxP and Nextera were filtered with DeLoxer (http://genomes.sdsc.edu/downloads/deloxer/) and NextClip (v0.8), respectively. To overcome high heterozygosity, genomes were assembled using a de Bruijn graph-based assembler, Platanus (v1.2.4)57. Scaffolding was conducted by mapping Illumina paired-end and mate pair reads to contigs using SSPACE (v3.0)58. For the Phoronis genome, a set of long 454 reads (750 bp) with 3 Gb of data was used for scaffolding with SSPACE-LongRead (v1-1)59. Gaps in the scaffolds were filled with GapCloser (v1.12-r6). Redundant allele scaffolds were removed with HaploMerger (2_20151106)60. Genome assembly quality was assessed with N(X) graphs using QUAST (v3.1) (Supplementary Fig. 4). Mitochondrial genomes and high GC scaffolds possibly derived from bacterial contamination were removed using custom Perl scripts. Genome sizes and heterozygosity rates were estimated by k-mer analysis using SOAPec (v2.01) and GCE (v1.0.0), as well as JELLYFISH (v2.0.0)61 and a custom Perl script. Genome assembly completeness was assessed with CEGMA (v2.5)62 (Supplementary Table 3).
Transcriptome sequencing and assembly
RNA-seq of adult tissues and embryonic stages was performed using the Illumina HiSeq 2500 platform. In total, 435 and 174 million RNA-seq read pairs from 15 Notospermus and 6 Phoronis samples, respectively, were generated (read length 100–300 bp) (Supplementary Tables 4 and 5). After quality checking and trimming of raw sequencing reads, transcripts were assembled de novo with Trinity (v2.1.0)63. Transcript isoforms with high similarity (≥ 95%) were removed with CD-HIT-EST. Transcript abundance was estimated with Bowtie (v2.1.0)64 and RSEM (v1.2.26)65 by mapping reads back to the transcript assembly. The trimmed mean of M-values-normalized expression values in fragments per kilobase of transcript per million mapped reads (FPKM) were used to estimate relative expression levels across samples. To reduce data complexity, functional filtering with TransDecoder (v2.0.1)63 was applied with the following three criteria: (1) open reading frames larger than 70 amino acids; (2) sequences with HMMER (v3.1b2) hits against the Pfam database (Pfam-A 29.0; 16,295 families); and (3) sequences with BLASTP (v2.2.29+) hits against the Swiss-Prot database (20160122; 550,299 sequences). Expression filtering was applied with two criteria: (1) expression levels ≥ 1 FPKM in at least one sample; and (2) transcript isoforms with abundances > 5% (Supplementary Figs. 2 and 3).
Repeat analysis
Regions of repetitive sequences in the genomes were identified with RepeatScout (v1.0.5)66 using default settings (that is, a sequence length larger than 50 bp and occurring > 10 times). Repetitive sequences were masked with RepeatMasker (http://www.repeatmasker.org/; v4.0.6). Transposable elements were annotated with TBLASTX and BLASTN searches against Repbase for RepeatMasker (v20150807). Repeat landscape (Kimura genetic distance) was calculated with the Perl script RepeatLandscape.pl bundled within RepeatMasker (v4.0.5+).
Gene prediction and annotation
Non-exon (that is, repeat) hints were generated with RepeatScout and RepeatMasker. Intron hints from spliced alignments of RNA-seq reads were generated using TopHat (v2.0.9) and Bowtie (v2.1.0)64 with the two-step method: (1) genome assembly mapping and (2) exon–exon junction mapping. Exon hints were generated from spliced alignments of transcriptome assemblies using BLAT (v.35). Gene structure was annotated by extraction of open reading frames with PASA (v2.0.2). Gene models were predicted with trained AUGUSTUS (v3.2.1)67 with repeat, intron and exon hints on the soft-masked genome assemblies. KEGG orthology was assigned using the KEGG Automatic Annotation Server. Gene models were annotated with protein identity and domain composition by BLASTP and HMMER searches against the Swiss-Prot and Pfam databases, respectively (Supplementary Fig. 5).
Gene family analysis
After all-to-all BLASTP searches against 31 selected metazoan proteomes (Supplementary Table 13), orthologous groups were identified with OrthoMCL (v2.0.9)68 using a default inflation number (I = 1.5). Venn diagrams were plotted with jvenn. Gene ontology annotation was performed with PANTHER (v10.0) using the PANTHER HMM scoring tool (pantherScore.pl). Gene ontology enrichment analysis was conducted with DAVID (v6.8). Gene family gain-and-loss was estimated using CAFE (v3.1)69. Principal component analysis was performed using the R package, prcomp.
Phylogenetic analysis
Genome-based orthologues with one-to-one relationships were selected with custom Perl scripts from OrthoMCL orthologous groups. Orthologues identified from transcriptomic data with many-to-many relationships were selected with HaMStR (v13.2.3)70. Paralogy screening was conducted with TreSpEx (v1.1)71. Sequence alignments were performed with MAFFT (v7.271)72. Unaligned regions were trimmed with TrimAl (v1.2rev59)73. Species trees were constructed with RAxML (v8.2.4)74 using the maximum-likelihood method with the LG, LG4M and LG4X models. Bayesian analyses were performed with PhyloBayes (v3.3 f)75 using the CAT + GTR model with the first 1,000 trees as a burn-in. For sensitivity analyses, four major factors that may cause systematic errors were assessed as follows: (1) branch length heterogeneity, as measured by the standard deviation of the average pairwise distance between taxa; (2) evolutionary rate, as estimated by the average patristic distance; (3) topological robustness, as defined by the average bootstrap support; and (4) compositional heterogeneity, as measured by relative composition frequency variability. Branch length heterogeneity, average patristic distance and average bootstrap support values were calculated with TreSpEx71. Relative composition frequency variability values were calculated with BaCoCa (v1.1)76.
Microsynteny analysis
At least three orthologues on the same scaffold shared between two species were considered as microsyntenic blocks, as previously described11. In brief, after assigning orthologues with a universal orthologous group identifier using OrthoMCL, the genomic locations of orthologues among different species were compared. All-to-all pairwise comparison was conducted with genome GFF (general feature format) files and OrthoMCL outputs using custom Perl scripts. Detailed step-by-step methods and Perl scripts are available on our genome project website (http://marinegenomics.oist.jp/).
Transcriptome analysis
To identify transcriptomic similarities between tissues, orthologues were identified among species using the bidirectional best hits (that is, reciprocal BLAST) approach. Spearman’s and Pearson’s correlation coefficients were calculated as previously described11. Differential expression analysis was conducted with a Trinity bundled Perl script (run_DE_analysis.pl). Heat maps and clustered matrices were created using R (v3.2.4) with the package Bioconductor (v3.0) and pheatmap (v1.0.8).
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability
This genome project has been registered at NCBI under the BioProject accession PRJNA393252. Genome assemblies have been deposited at DDBJ/ENA/GenBank under accession numbers NMRB00000000 (N. geniculatus) and NMRA00000000 (P. australis). Transcriptome assemblies have been deposited in the NCBI Transcriptome Shotgun Assembly Sequence Database under accession numbers GFRY00000000 (N. geniculatus) and GFSC00000000 (P. australis). Sequencing reads of the genomes and transcriptomes have been deposited in the NCBI Sequence Read Archive under the study accession SRP111350. The updated L. anatina genome (v2.0) has been deposited under the accession number LFEI00000000. Genome browsers, genome assemblies, gene models and transcriptomes, together with annotation files, are available at http://marinegenomics.oist.jp/.
References
Appeltans, W. et al. The magnitude of global marine species diversity. Curr. Biol. 22, 2189–2202 (2012).
Cannon, J. T. et al. Xenacoelomorpha is the sister group to Nephrozoa. Nature 530, 89–93 (2016).
Dunn, C. W. et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749 (2008).
Nesnidal, M. P. et al. New phylogenomic data support the monophyly of Lophophorata and an ectoproct–phoronid clade and indicate that Polyzoa and Kryptrochozoa are caused by systematic bias. BMC Evol. Biol. 13, 253 (2013).
Laumer, C. E. et al. Spiralian phylogeny informs the evolution of microscopic lineages. Curr. Biol. 25, 2000–2006 (2015).
Grande, C. & Patel, N. H. Nodal signalling is involved in left–right asymmetry in snails. Nature 457, 1007–1011 (2009).
Halanych, K. M. & Kocot, K. M. Repurposed transcriptomic data facilitate discovery of innate immunity toll-like receptor (TLR) genes across Lophotrochozoa. Biol. Bull. 227, 201–209 (2014).
Zhang, L. et al. Massive expansion and functional divergence of innate immune genes in a protostome. Sci. Rep. 5, 8693 (2015).
Simakov, O. et al. Insights into bilaterian evolution from three spiralian genomes. Nature 493, 526–531 (2013).
Zhang, G. et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54 (2012).
Luo, Y. J. et al. The Lingula genome provides insights into brachiopod evolution and the origin of phosphate biomineralization. Nat. Commun. 6, 8301 (2015).
Helmkampf, M., Bruchhaus, I. & Hausdorf, B. Phylogenomic analyses of lophophorates (brachiopods, phoronids and bryozoans) confirm the Lophotrochozoa concept. Proc. Biol. Sci. 275, 1927–1933 (2008).
Paps, J., Baguna, J. & Riutort, M. Bilaterian phylogeny: a broad sampling of 13 nuclear genes provides a new Lophotrochozoa phylogeny and supports a paraphyletic basal acoelomorpha. Mol. Biol. Evol. 26, 2397–2406 (2009).
Hausdorf, B., Helmkampf, M., Nesnidal, M. P. & Bruchhaus, I. Phylogenetic relationships within the lophophorate lineages (Ectoprocta, Brachiopoda and Phoronida). Mol. Phylogenet. Evol. 55, 1121–1127 (2010).
Weigert, A. et al. Illuminating the base of the annelid tree using transcriptomics. Mol. Biol. Evol. 31, 1391–1401 (2014).
Kocot, K. M. et al. Phylogenomics of Lophotrochozoa with consideration of systematic error. Syst. Biol. 66, 256–282 (2017).
Albertin, C. B. et al. The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 524, 220–224 (2015).
Takeuchi, T. et al. Bivalve-specific gene expansion in the pearl oyster genome: implications of adaptation to a sessile lifestyle. Zool. Lett. 2, 3 (2016).
Whelan, N. V., Kocot, K. M., Santos, S. R. & Halanych, K. M. Nemertean toxin genes revealed through transcriptome sequencing. Genome Biol. Evol. 6, 3314–3325 (2014).
Egger, B. et al. A transcriptomic–phylogenomic analysis of the evolutionary relationships of flatworms. Curr. Biol. 25, 1347–1353 (2015).
Wong, Y. H. et al. Transcriptome analysis elucidates key developmental components of bryozoan lophophore development. Sci. Rep. 4, 6534 (2014).
Salichos, L. & Rokas, A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497, 327–331 (2013).
Putnam, N. H. et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94 (2007).
Srivastava, M. et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 466, 720–726 (2010).
Massague, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012).
Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 13, 767–779 (2012).
Simakov, O. et al. Hemichordate genomes and deuterostome origins. Nature 527, 459–465 (2015).
Riddiford, N. & Olson, P. D. Wnt gene loss in flatworms. Dev. Genes Evol. 221, 187–197 (2011).
Kao, D. et al. The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. eLife 5, e20062 (2016).
Sekita, Y. et al. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat. Genet. 40, 243–248 (2008).
Pearson, J. C., Lemons, D. & McGinnis, W. Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 6, 893–904 (2005).
Schiemann, S. M. et al. Clustered brachiopod Hox genes are not expressed collinearly and are associated with lophotrochozoan novelties. Proc. Natl Acad. Sci. USA 114, E1913–E1922 (2017).
Moysiuk, J., Smith, M. R. & Caron, J.-B. Hyoliths are Palaeozoic lophophorates. Nature 541, 394–397 (2017).
Zhang, Z. F. et al. An early Cambrian agglutinated tubular lophophorate with brachiopod characters. Sci. Rep. 4, 4682 (2014).
De Rosa, R. et al. Hox genes in brachiopods and priapulids and protostome evolution. Nature 399, 772–776 (1999).
Fröbius, A. C. & Funch, P. Rotiferan Hox genes give new insights into the evolution of metazoan bodyplans. Nat. Commun. 8, 9 (2017).
Schubert, M., Holland, N. D., Laudet, V. & Holland, L. Z. A retinoic acid-Hox hierarchy controls both anterior/posterior patterning and neuronal specification in the developing central nervous system of the cephalochordate amphioxus. Dev. Biol. 296, 190–202 (2006).
Gonzalez, P., Uhlinger, K. R. & Lowe, C. J. The adult body plan of indirect developing hemichordates develops by adding a Hox-patterned trunk to an anterior larval territory. Curr. Biol. 27, 87–95 (2016).
Hughes, C. L. & Kaufman, T. C. Exploring the myriapod body plan: expression patterns of the ten Hox genes in a centipede. Development 129, 1225–1238 (2002).
Hiebert, L. S. & Maslakova, S. A. Hox genes pattern the anterior–posterior axis of the juvenile but not the larva in a maximally indirect developing invertebrate, Micrura alaskensis (Nemertea). BMC Biol. 13, 23 (2015).
Fröbius, A. C., Matus, D. Q. & Seaver, E. C. Genomic organization and expression demonstrate spatial and temporal Hox gene colinearity in the lophotrochozoan Capitella sp. I. PLoS ONE 3, e4004 (2008).
Temereva, E. N. & Kosevich, I. A. The nervous system of the lophophore in the ctenostome Amathia gracilis provides insight into the morphology of ancestral ectoprocts and the monophyly of the lophophorates. BMC Evol. Biol. 16, 181 (2016).
Temereva, E. N. & Tsitrin, E. B. Modern data on the innervation of the lophophore in Lingula anatina (Brachiopoda) support the monophyly of the lophophorates. PLoS ONE 10, e0123040 (2015).
Irie, N. & Kuratani, S. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat. Commun. 2, 248 (2011).
Petersen, C. P. & Reddien, P. W. Wnt signaling and the polarity of the primary body axis. Cell 139, 1056–1068 (2009).
Glinka, A., Wu, W., Onichtchouk, D., Blumenstock, C. & Niehrs, C. Head induction by simultaneous repression of Bmp and Wnt signalling in Xenopus. Nature 389, 517–519 (1997).
Steinmetz, P. R. H. et al. Six3 demarcates the anterior-most developing brain region in bilaterian animals. EvoDevo 1, 14 (2010).
Lowe, C. J., Clarke, D. N., Medeiros, D. M., Rokhsar, D. S. & Gerhart, J. The deuterostome context of chordate origins. Nature 520, 456–465 (2015).
Pani, A. M. et al. Ancient deuterostome origins of vertebrate brain signalling centres. Nature 483, 289–294 (2012).
Santagata, S., Resh, C., Hejnol, A., Martindale, M. Q. & Passamaneck, Y. J. Development of the larval anterior neurogenic domains of Terebratalia transversa (Brachiopoda) provides insights into the diversification of larval apical organs and the spiralian nervous system. EvoDevo 3, 3 (2012).
Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1, 135–145 (2001).
Ellisdon, A. M. et al. Stonefish toxin defines an ancient branch of the perforin-like superfamily. Proc. Natl Acad. Sci. USA 112, 15360–15365 (2015).
Kem, W. R. & Blumenthal, K. M. Purification and characterization of the cytotoxic Cerebratulus A toxins. J. Biol. Chem. 253, 5752–5757 (1978).
Jackson, D. J. et al. The Magellania venosa biomineralizing proteome: a window into brachiopod shell evolution. Genome Biol. Evol. 7, 1349–1362 (2015).
Erwin, D. H. et al. The Cambrian conundrum: early divergence and later ecological success in the early history of animals. Science 334, 1091–1097 (2011).
Jandzik, D. et al. Evolution of the new vertebrate head by co-option of an ancient chordate skeletal tissue. Nature 518, 534–537 (2015).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395 (2014).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Boetzer, M. & Pirovano, W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15, 211 (2014).
Huang, S. et al. HaploMerger: reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res. 22, 1581–1588 (2012).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Fischer, S. et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinformatics 35, 6.12.1-6.12.19 (2011).
De Bie, T., Cristianini, N., Demuth, J. P. & Hahn, M. W. CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006).
Ebersberger, I., Strauss, S. & von Haeseler, A. HaMStR: profile hidden markov model based search for orthologs in ESTs. BMC Evol. Biol. 9, 157 (2009).
Struck, T. H. TreSpEx-detection of misleading signal in phylogenetic reconstructions based on tree information. Evol. Bioinform. Online 10, 51–67 (2014).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Lartillot, N., Lepage, T. & Blanquart, S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288 (2009).
Kuck, P. & Struck, T. H. BaCoCa—a heuristic software tool for the parallel assessment of sequence biases in hundreds of gene and taxon partitions. Mol. Phylogenet. Evol. 70, 94–98 (2014).
Acknowledgements
This study was supported by internal funding from the Okinawa Institute of Science and Technology Graduate University and a Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (B) (16H04824) to N.S. Y.-J.L. was supported by a JSPS Research Fellowship for Young Scientists (DC1) and a JSPS Grant-in-Aid for JSPS Fellows (15J01101). We thank P. W. H. Holland, Y. Yasuoka and E. Shoguchi, as well as all members of the Marine Genomics Unit for helpful discussions. We also thank S. D. Aird for editing the paper.
Author information
Authors and Affiliations
Contributions
Y.-J.L. and N.S. conceived the project. T.A., H.S. and T.S. collected specimens and identified species. M.K., R.K. and Y.-J.L. prepared genomic DNA and RNA samples. M.K. and R.K. carried out DNA and RNA sequencing. Y.-J.L. assembled genomes and transcriptomes, generated gene models and analysed data. K.H. prepared the genome browser. Y.-J.L. wrote the paper and prepared the Supplementary Information with input from other authors. N.S. provided supervision and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figures 1–52, Supplementary tables 1–36, Supplementary Notes 1–4
Supplementary Data 1
List of gene IDs and annotations in Fig. 2a
Supplementary Data 2
List of gene IDs and annotations in Supplementary Figs. 24a,b and 28a
Supplementary Data 3
List of gene IDs and annotated gene ontology enrichment terms in Supplementary Figure 24c–e
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, YJ., Kanda, M., Koyanagi, R. et al. Nemertean and phoronid genomes reveal lophotrochozoan evolution and the origin of bilaterian heads. Nat Ecol Evol 2, 141–151 (2018). https://doi.org/10.1038/s41559-017-0389-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41559-017-0389-y
This article is cited by
-
Diversification of von Willebrand Factor A and Chitin-Binding Domains in Pif/BMSPs Among Mollusks
Journal of Molecular Evolution (2024)
-
Genomic and transcriptomic survey of bryozoan Hox and ParaHox genes with emphasis on phylactolaemate bryozoans
BMC Genomics (2023)
-
The localization of Toll and Imd pathway and complement system components and their response to Vibrio infection in the nemertean Lineus ruber
BMC Biology (2023)
-
Spatial compartmentalisation of bacteria in phoronid microbiomes
Scientific Reports (2023)
-
Brachiopod and mollusc biomineralisation is a conserved process that was lost in the phoronid–bryozoan stem lineage
EvoDevo (2022)
