
Abstract
Lophotrochozoan species exhibit wide morphological diversity; however, the molecular basis underlying this diversity remains unclear. Here, we explored the evolution of Notch pathway genes across 37 metazoan species via phylogenetic and molecular evolutionary studies with emphasis on the lophotrochozoans. We displayed the components of Notch pathway in metazoans and found that Delta and Hes/Hey-related genes, as well as their functional domains, are duplicated in lophotrochozoans. Comparative transcriptomics analyses allow us to pinpoint sequence divergence of multigene families in the Notch signalling pathway. We identified the duplication mechanism of a mollusc-specific gene, Delta2, and found it displayed complementary expression throughout development. Furthermore, we found the functional diversification not only in expanded genes in the Notch pathway (Delta and Hes/Hey-related genes), but also in evolutionary conservative genes (Notch, Presenilin, and Su(H)). Together, this comprehensive study demonstrates conservation and divergence within the Notch pathway, reveals evolutionary relationships among metazoans, and provides evidence for the occurrence of developmental diversity in lophotrochozoans, as well as a basis for future gene function studies.
Similar content being viewed by others


Introduction
Lophotrochozoa is a monophyletic group of animals that includes Platyhelminthes, bryozoans, brachiopods, annelids, molluscs, and other animals that share a common ancestor. Lophotrochozoan species exhibit a high level of biodiversity; for example, Mollusca is one of the richest groups of animals containing over 100,000 different species1, and over 16,500 species of Annelida have been described worldwide2. Therefore, the superphylum Lophotrochozoa is essential to our understanding of metazoan evolution.
Early developmental pathways have been shown to play a vital role in the evolution of biodiversity, such as pathways that control embryonic development involving transforming growth factor β (TGF-β), wingless/integrated (Wnt), receptor protein tyrosine kinase (RPTK), Janus kinase/signal transducer and activator of tran-ions (Jak/STAT), Hedgehog, retinoic acid signalling (RA), hox cluster (Hox), and Notch3. In particular, the Notch pathway is essential for regulating cellular identity, proliferation, differentiation, and apoptosis via lateral inhibition, lineage decisions, and boundary induction, which all play vital roles in metazoan development4,5.
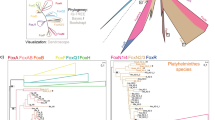
The Notch pathway contains the Notch receptor, the ligands Delta and Jagged (known as Serrate in Drosophila melanogaster), the regulatory factors Fringe, Numb, Deltex, Mastermind, Presenilin, and Nrarp, the transcription factor CSL family (invertebrate Suppressor of Hairless (Su(H)), vertebrate CBF1 (also known as rbpj), and nematode LAG-1; Su(H) is used in this paper), and the Hes (Hairy/enhancer of Split)/Hey (Hairy/Enhancer of Split related with YRPW motif) target gene family5. The Notch gene can be divided into three parts: an extracellular domain (NECD) consisting of 29–36 epidermal growth factor (EGF) repeats and three Lin12/Notch (LNR) repeats, a transmembrane (TM) domain, and an intracellular domain (NICD) that includes several ankyrin (ANK) repeats and a region containing proline, glutamate, serine, and threonine (PEST)5. The Notch ligand Delta contains a Delta/Serrate/Lag (DSL) domain, which is crucial for its interactions with the Notch receptor, whereas its extracellular regions contain several EGF repeats. Some genes that have lost EGF repeats or other motifs are defined as Delta-like (Dll). Jagged, another Notch ligand, contains a von Willebrand factor type C (VWC) domain in addition to the domains in Delta. The vital transcription factor Su(H) has the following functional domains: C-terminal domain (CTD), beta–trefoil domain (BTD), and the N-terminal domain (NTD). Both the BTD and NTD contact DNA, and the BTD and CTD interact with NICD, whereby the CTD binds both the ANK of NICD and Mastermind (MAM)6,7,8. Presenilin encodes a multi-span membrane protein and catalytic subunit that only contains a Presenilin domain5. The terminal genes of the Notch pathway include Hes/Hey-related genes that belong to group E of the basic helix-loop-helix (bHLH) superfamily (including Hes, Hey, Helt (also known as Hesl), and Clockwork orange (Cwo)) and encode a bHLH domain as well as a hairy/orange domain9. The pathway is exhibited in Fig. 1a.

Major genes of the Notch pathway. (a) Diagram of the Notch pathway. (b) Species colours represent different phyla. Dashed lines represent unresolved phylogenetic positions for ctenophores and sponges. Lines indicate none and “*” indicates lack of a proteome. Species abbreviations are described in the “Methods” section.
Previous studies have indicated that the Notch pathway is conserved in metazoans10, and traditional model organisms have revealed the various functions of the Notch pathway, which includes regulation of polarity because its loss results in abnormal anterior–posterior polarity or incorrect left–right asymmetry in somites11,12. The Notch signalling pathway also plays central roles in vertebrate somitogenesis13,14,15 and differentiation of the epidermis and cilia16. Consequently, the function of the Notch pathway in non-model organisms has received considerable attention. In the sponge Amphimedon queenslandica, the Notch pathway is involved in regulation of diverse cell types during development17, and it plays essential roles during nervous system development18 and boundary formation19 in cnidarians. Recent lophotrochozoan studies have associated the Notch pathway with formation of germ layers, neurogenesis, segments, and chaetogenesis20,21,22,23,24; however, it remains unclear how the pathway evolved in these organisms.
The comparative genomic and transcriptomic analysis was widely used in evolutionary developmental biology25,26. Here, we investigated the conservation and divergence of Notch pathway gene components and annotated genes encoding molecules that affect early development of 37 metazoan species. In addition, we performed a comparative genomic study on core components of the Notch pathway (Notch, Delta, Jagged, Presenilin, Su(H), Hes, Hey, Helt, and Cwo) in metazoans and elucidated their evolutionary relationships using phylogenetic analysis. Consequently, this study clarified the molecular mechanisms responsible for evolution of the novel molluscan gene Delta2 and elucidated patterns of Notch pathway gene expression using comparative transcriptome during development, thus providing a basis for further evo-devo research on lophotrochozoans.
Methods
Data collection and Notch pathway gene identification
We selected 37 metazoan species for our study: two Ctenophora, Beroe ovata (Bvo; version 1.0) and Mnemiopsis leidyi (Mle); one Porifera, Amphimedon queenslandica (Aqu; version 1.0); two Cnidaria, Hydra vulgaris (Hvu; version 1.0) and Nematostella vectensis (Nve; version 1.0); one Acoela, Hofstenia miamia (Hmi; version 1.0); 20 Lophotrochozoa, including one Brachiopoda Lingula anatina (Lan; version 2.0), two Platyhelminthes, Schistosoma mansoni (Sma; version 2) and Echinococcus multilocularis (Emu; version EMULTI002), four Annelida, Platynereis dumerilii (Pdu), Helobdella robusta (Hro; version 1.0), Urechis unicinctus (Uun) and Capitella teleta (Cte; version 1.0), thirteen Mollusca, Acanthopleura granulate (Agr), Aplysia californica (Aca; version 3.0), Elysia chlorotica (Ech; version 2.0), Biomphalaria glabrata (Bgl; version 1.0), Lottia gigantea (Lgi; version 1.0), Haliotis discus hannai (Hdh), Crassostrea gigas (Cgi; version oyster_v9), Crassostrea virginica (Cvi; version 3.0), Mizuhopecten yessoensis (Mye; version 2.0), Chlamys farreri (Cfa), Pinctada fucata martensii (Pma), Octopus bimaculoides (Obi; version 2.0), and Octopus vulgaris (Ovu; version 1.0); one Nematoda, Caenorhabditis elegans (Cel; version WBcel235); two Arthropoda, Drosophila melanogaster (Dme; version Release 6 plus ISO1 MT) and Apis mellifera (Ame; version 3.1); two Echinodermata, Strongylocentrotus purpuratus (Spu; version 5.0) and Apostichopus japonicus (Aja, version 1.0); one Hemichordata, Saccoglossus kowalevskii (Sko; version 1.1); and five Chordata, Styela clava (Scl, version 2.0), Branchiostoma belcheri (Bbe; version Haploidv18h27), Danio rerio (Dre; version GRCz11), Mus musculus (Mmu; version GRCm39), and Homo sapiens (Has; GRCh38.p13). All genomes and proteomes were downloaded from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/), except those of P. fucata martensii, which was obtained from GigaDB (http://gigadb.org/dataset/100240)27, C. farreri, which was obtained from CfBase (http://mgb.ouc.edu.cn/cfbase/html/)28, and A. granulata, which was obtained from https://alabama.app.box.com/s/1hsryfff61i01qrljyasrjnu8j7qg2nj. The genomes of M. leidyi, P. dumerilii, U. unicinctus, and H. discus hannai have not been published yet. The genome and protein sequence datasets were searched for each species. Genes were identified using the default protein-to-nucleotide Basic Local Alignment Search Tool (tblastn) in NCBI. Sequences with the lowest E-value were selected for analysis after all matches with unexpected domain architecture had been discarded after being corrected by Gene Wise29.
We defined Notch as a gene containing several EGF domains, three LNR repeats, a TM domain, and several ANK repeats, Delta as a gene containing a DSL domain, an MNLL domain, and EGF domains, and Jagged as a gene having the same domains as Delta, with an additional VWC domain. Similarly, Presenilin and BTD/LAG1-DNAbind domains belonged to Presenilin and Su(H), respectively. The bHLH domains of H. sapiens and D. melanogaster were used as query sequences in tblastn searches for Hes/Hey-related (Hey, Hes, Helt, and Clockwork) members in metazoans.
HMMER3.3 (http://hmmer.org/) was used to screen significant domains in Notch pathway genes with a low cut-off threshold value of E-5 against all datasets. Profile hidden Markov models (profile HMMs) were accessed via Pfam (http://pfam.xfam.org/). All protein domain visualisations were checked by scanning sequences using the NCBI Conserved Domain search30 and Simple Modular Architecture Research Tool (SMART)31. Pfam accession numbers are available in Supplementary Table S1.
Phylogenetic analysis
We constructed phylogenetic trees using 27 species (A. queenslandica and H. vulgaris as outgroups) whose Notch pathway protein sequences were well assembled. Multiple alignments were produced using Clustal W in MEGA X software under default parameters that were manually adjusted32. TrimAL (http://phylemon2.bioinfo.cipf.es/) was used to trim protein sequences under automated1 mode. Since the phylogenetic trees constructed using Bayesian Inference (BI), neighbour-joining (NJ), and maximum likelihood (ML) methods were consistent, only trees of nine core components constructed using the ML method with the best fit model (WAG + CAT) in Fasttree (version 2.1.11, 1000 bootstrap replicates) are shown33. Trees were prepared using FigTree (version 1.4.3), iTOL (https://itol.embl.de/), and EvolView (https://evolgenius.info//evolview-v2/#login). All BLAST gene query sequences were obtained from published papers34,35.
The phylogenetic trees of nine core genes (Notch, Delta, Jagged, Presenilin, Su(H), Hes, Hey, Helt, and Cwo) were constructed using entire protein sequences (IDs listed in Supplementary Tables S2 and S3). The DSL family tree was constructed using the DSL domain of each gene. If a gene contained more than one DSL domain, ‘-’ was used to denote their order in the gene.
Transcriptomic analysis of gene expression
Gene expression levels were measured as reads per kilobase per million (RPKM) or fragments per kilobase million (FPKM). Transcriptomic data (RPKM) from the developmental stages and adult tissues of C. gigas were obtained from NCBI (accession GSE31012) and the supplementary material of a published paper36. Transcriptomic data (RPKM) for P. martensii were obtained from GigaDB and the supplementary material of the associated publication27. The raw data of A. queenslandica, A. japonicus, H. discus hannai, M. yessoensis, M. leidyi, S. clava, and U. unicinctus were obtained from the study by Wang et al.25. The RNA sequencing values for D. melanogaster were retrieved from the FlyBase website (http://flybase.org/). The time-course of D. rerio was derived from a previous report37. For species with no reference genomes, the sratoolkit (version 2.10.8) was used to convert SRR raw data downloaded from NCBI to FASTq format. Transcriptome assembly was performed using Trinity (version 2.2.0) and RSEM (version 1.3.3), which were used to calculate the expression profiles. For species with completely spliced genomes, Hisat2 (version 2.1.0) was used to build index and mapping, SamTools (version 1.11) to format conversion, and Cufflinks (version 2.2.1) to calculate the expression levels. Data were visualised using TBtools (version 1.089)38. Owing to differences in measurement methods of gene expression levels, only expression trends between species were analysed. Abbreviation definitions and divisions of developmental stages are available in Supplementary Table S4.
Results
Identification of Notch pathway genes and related domains
To investigate the diversity of development-related domains in different species, we selected 18 key domains from developmental pathways (including TGF-β, Wnt, Jak/STAT, RPTK, Notch, Hedgehog, RA, Fox, Hox, and ERK) and predicted the number of domains across 34 species whose genomes had been completely sequenced. The EGF, Homeobox, bHLH, and SH2 domains were extensively expanded across metazoans (16,894, 4300, 2259, and 1966, respectively; Table S5), whereas genes encoding ERK-JNK_inhib, TALPID3, HH_signal, RAI16-like, and STAT_bind were relatively conserved (33, 41, 72, 74, and 76, respectively). Overall, EGF was the most expanded domain in lophotrochozoans, and DSL and bHLH also showed high numbers of domains (highlighted in red). Notably, genes encoding the EGF, DSL, and bHLH domains were all related to the Notch pathway. To elucidate the evolution of diversity in Notch pathway components, we identified nine functional domains that serve essential roles in the Notch pathway (Table S6). With the exception of EGF, DSL, and bHLH domain expansions, we found that the Presenilin and BTD/LAG1-DNAbind domains only existed in Presenilin and Su(H), respectively, indicating they were conserved.
To further investigate the origin and evolution of the Notch pathway, we examined the genomes of 37 metazoan species of different evolutionary status, with an emphasis on 20 lophotrochozoans including Platyhelminthes, Brachiopoda, Annelida, and Mollusca. We annotated and compared the numbers of core Notch pathway genes, including the Notch receptor, the Delta and Jagged ligands, the γ-secretase complex component Presenilin, the transcription factor Su(H), and Hes/Hey-related target genes (Fig. 1b). Notch, Presenilin, and Su(H) were identified in the ancestral metazoans Ctenophora (B. ovata and M. leidyi) and Porifera (A. queenslandica), indicating that major Notch pathway components were present before metazoan ancestors; however, neither Delta nor Jagged were identified in Ctenophora. Hes/Hey-related genes were found in Cnidaria, whereas Helt and Cwo first appeared in Lophotrochozoa. Notably, Delta was duplicated in several Mollusca including bivalves, gastropods, and cephalopods (Fig. 1b, red). The ancestral mollusc Polyplacophora A. granulata had Helt, indicating that some gastropods probably lost this gene, such as A. californica, E. chlorotic, H. discus hannai, and B. glabrata. Notch pathway genes also showed expansion in vertebrates resulting from two genome-wide duplications (2R), which provided genetic variation during vertebrate evolution39,40 However, it remains unclear how Delta was duplicated in molluscs.
DSL gene family evolution
The Notch pathway ligands Delta and Jagged have similar domain architectures (MNLL, DSL, and EGF) and both belong to the DSL family. Phylogenetic analysis indicated that Delta and Jagged descended from the same ancestor in early metazoans, such as the sponge A. queenslandica and the hydra H. vulgaris, which concurred with the results of previous studies34 (Fig. 2). In addition, Delta displayed a broadly similar topology to Jagged in eumetazoans, yet Jagged appeared later than Delta. Surprisingly, we found that molluscs, including bivalves, cephalopods, and gastropods, fell into another Delta clade, which we named Delta2 (Fig. 2). Interestingly, Delta2 occurred both independently and appeared later than Delta1, indicating functional gene differentiation. Phylogenetic analyses revealed that Delta2 was absent in the ancestral mollusc Polyplacophora A. granulata but was present in other molluscs, indicating that Delta2 originated in the ancestor of Gastropoda, Cephalopoda, and Bivalvia. Moreover, Delta2 was identified in L. gigantea and H. hannai but lost in gastropods B. glabrata, E. chlorotica, and A. californica.

Phylogeny of Notch ligands. Phylogenetic tree of Delta and Jagged genes in metazoans, as determined using the maximum likelihood method. The tree was constructed using 76 ligand genes from 27 complete metazoan genomes (including 16 lophotrochozoans; Platyhelminthes was not included owing to their specific parasitic lifestyle). Green indicates Delta1 in Lophotrochozoans. Yellow indicates Delta2. Pink indicates Jagged (Serrate in D. melanogaster) in Lophotrochozoans. Fasttree support values are shown at the basal node. Sequence IDs are shown in Supplementary Table S2. The Delta protein sequences are shown in Supplementary Data 1.
Next, we analysed the mechanism of molecular evolution underlying Delta duplication. Although domain architecture was quite well conserved between Delta1 and Delta2, the Delta2 sequences were shorter than those of Delta1 (Fig. 3a). Notably, we found that the early termination in Delta2 that occurred approximately 1–10 amino acids after the arginine (R) of Delta1 downstream may have caused this difference (Fig. 3a, black frame). The terminal patterns are clarified in Fig. 3b. A G base was inserted in the termination sequences of Delta2 of the bivalve C. gigas, which caused the terminal TGA codon and resulted in early termination. In the Gastropoda L. gigantea, early termination occurred owing to a mutation in the second leucine (L) codon (red triangle) in the termination sequences, resulting in the termination codon TTA being corrected to TGA. In M. yessoensis, the C base was deleted before cysteine (C, red triangle) in termination sequences, which led to a frameshift mutation. Similarly, deletion of the second C base in isoleucine (I) in Cephalopoda O. bimaculoides and O. vulgaris resulted in a frameshift mutation in Delta2, likely leads to early termination (Fig. 3b).

Early termination mechanisms in mollusc-specific Delta2. (a) Delta1 and Delta2 gene structures and blast (DNAMAN) results. The protein sequences indicated in the purple frame are TM domains whereas those indicated in the black frame are common termination positions for Delta2. The sequences in the red frame were analysed in detail in (b). (b) Delta2 termination mechanisms in different molluscs. Red solid circles indicate branches containing Delta2. Red hollow circles indicate no Delta2. Both amino acid and nucleotide sequences are displayed here, and the left column includes the termination sequences in lophotrochozoan species whereas the right column denotes sequences after correction. The colours of amino acids represent the functions of codons; amino acids of the same colour have similar functions. The same colour nucleotide represents the same base. The amino acids with a dashed line corresponding to bases in the red frames and triangles represent mutation codon sites in Delta2. “*” means termination codon. MEGA X was used for visualizing mutation sites.
To further investigate the functional differentiation between the ligands in Mollusca, we analysed expression patterns throughout development using transcriptomic data of Mollusca C. gigas, P. martensii, M. yessoensis, H. discus hannai, and Echinodermata A. japonicus (outgroup) (Fig. 4). The figure shows that Delta or Delta1 were generally highly expressed during the blastula and early larva stages, and Delta genes displayed complementary expression throughout development in Mollusca. In M. yessoensis, Delta1 was increased during the gastrula (G), trochophore (T), and D-shaped larva (D) stages, whereas Delta2 was highly expressed during the zygote (Z) and 2–8 cell (C) stages. In C. gigas and P. martensii, Delta2 was increased after the umbone larva stage, unlike Delta1. In H. discus hannai, Delta2 was upregulated in the middle veliger stage (M), whereas Delta1 showed downregulation at this stage. It seemed that Delta2 was a negatively regulated gene of Delta1. Moreover, in C. gigas and M. yessoensis, the expression pattern of Jagged was more similar to Delta2 than Delta1, whereas in P. martensii and H. discus hannai the expression pattern of Jagged was more similar to Delta1. Intriguingly, the expression level of Delta1 was generally higher than that of Delta2 (Fig. 4; Table. S4). This might result from the loss of the TM domain in Delta2, which results in failure of the downstream signal transmission. We also noticed that the expression level of Mye-Delta2, Mye-Jagged, and Hdh-Jagged was very low. As discussed above, the different expression patterns suggest functional divergence between Delta1 and other ligands after duplication. The ligands of the Notch pathway may coordinate with each other and work together.

Expression pattern of Notch ligands. Transcriptomic data of five species throughout development (C. gigas, P. martensii, M. yessoensis, H. discus hannai, and A. japonicus). Heatmaps in red display the expression trends of genes. The X-axis of histogram shows the developmental points and the Y-axis denotes fragments per kilobase million (FPKM) or reads per kilobase per million (RPKM). Sequence IDs are shown in Supplementary Table S2. Developmental point and species name abbreviations are described in Supplementary Table S4.
To explore how DSL expanded, we screened 138 DSL family genes in lophotrochozoan species and constructed an unrooted phylogenetic tree (Fig. S1a). All DSL family genes were divided into four groups (Fig. S1b) and split the tree into two branches: one branch contained genes encoding DSL or DSL tandem repeats but not the EGF domain (purple clades), being present only in bivalves, whereas the other branch contained genes encoding the DSL, EGF, and other domains (blue clades), including Delta and Jagged clades. The tree shows that the expansion of DSL mainly resulted from tandem repeats present among bivalves, and the branch length show that DSL combined with EGF underwent rapid differentiation.
Hes/Hey-related gene family evolution
To investigate divergence within terminal Notch pathway genes, we constructed an unrooted molecular phylogenetic tree of Hes/Hey-related family members (Hes, Hey, Helt, and Cwo) using the bHLH domain (Fig. 5a). Hes, Hey, Helt, and Cwo fell into different clades, whereas Hes was expanded in some species lineages including lophotrochozoans, ecdysozoans, and chordates (genes in red), which was also demonstrated in previous studies35. We selected several species from different expanded lineages of Hes genes for comparative transcriptome analysis (Fig. 5b). The Hes gene clusters of the Lophotrochozoa C. gigas and the Ecdysozoa D. melanogaster were expressed almost during the embryo development stages, implying conserved and specified functions of Hes clusters in expanded lineages. However, in Chordata D. rerio, Hes genes were significantly upregulated during the segmentation, pharyngula, and hatching stages, which displayed that, although the expression pattern of expanded genes of one species was similar, the expression differences and functional differentiations of expanded genes among different specific expanded lineages existed as well. Hey was conserved, whereas Helt and Cwo were novel in the Hes/Hey-related family, and they displayed different expression patterns not only in lophotrochozoans but also in other species (Fig. 5c). These results suggest that the terminal genes of the Notch pathway play different roles in different lineages, which could be responsible for the phenotypic differentiation in larval stages among species.

Phylogeny and transcriptome of Hes/Hey-related gene family across metazoans. (a) A phylogenetic tree was constructed by aligning the bHLH domain of 233 genes. Red labels indicate expanded Hes gene clusters that are analysed in (b). (b) Transcriptomic data of expanded Hes gene clusters in lophotrochozoan, Ecdysozoa, and Chordata. (c) Transcriptomic data of Hey, Helt, and Cwo in nine species. Sequence IDs are shown in Supplementary Table S3. bHLH domain protein sequences are given in Supplementary Data S3. Developmental point and species name abbreviations are described in Supplementary Table S4.
Notch, Presenilin, and Su(H) evolution
The evolutionary relationships of Notch, Presenilin, and Su(H) in Lophotrochozoa from Porifera to Chordata were clarified by phylogenetic analysis. In lophotrochozoans, L. anatine showed a closer relationship with P. dumerilii and C. teleta than with molluscs in phylogenetic trees of Notch and Presenilin (Figs. S2 and S3); however, these relationships differed for Su(H) (Fig. S4). We then analysed gene developmental expression profiles of the three genes or gene families in 11 species for which comprehensive developmental transcriptomic data are available. Transcriptomic analysis revealed that Notch, Presenilin, and Su(H) were highly expressed during the early embryo developmental stages of most species, but we also found lineage-specific developmental expression patterns of these genes in A. queenslandica and U. unicinctus, which indicates gene expression divergence of these conserved genes in a clade-specific mode (Fig. S5).
Discussion
The Notch pathway has been well studied in model animals such as D. melanogaster, C. elegans, and vertebrates4; however, few studies have investigated the Notch pathway in lophotrochozoans. This study significantly expands our knowledge of conservation and divergence within Notch pathway multigene families and elucidates the evolution of core Notch pathway genes in metazoans, with a strong focus on lophotrochozoan species. Interestingly, we found a novel Delta2 gene in molluscs and proposed the molecular mechanisms of its evolution. Moreover, comparative omics-based analyses revealed differences in gene structure and function between species, whereas gene expression patterns were also observed throughout development, particularly for Hes/Hey-related genes. Together, our findings enrich our understanding of the Notch pathway and provide a powerful approach for exploring the evolution of developmental pathway genes.
In this study, we found that Notch pathway gene domains could be traced back to Ctenophora, considered one of the earliest evolving extant species41 (Fig. 1b). Previous studies only verified the Notch pathway genes in Porifera17, but it is still uncertain whether Notch pathway genes are functional in Ctenophora4. A recent study determined that some Notch pathway domains, such as MNLL and DSL, existed not only in animals, but also in choanoflagellate species, suggesting that Notch and Delta appeared much earlier than previously thought42. The expansion of DSL and EGF domains in lophotrochozoans has also been associated with the recombination of Notch ligands and some are considered to be Delta-like genes20 (Fig. S1a), indicating that the functional evolution of ligand genes is probably ongoing, which was consistent with the varying expression patterns of Notch ligands observed in the current study43,44. We also found complementary expression patterns between Delta1 and Delta2 in Mollusca (Fig. 4); one of the possible reasons is both genes shared MNLL and DSL domains and EGF repeats, the functions of which are likely complementary, interchangeable, or antagonistic45,46.
Interestingly, we identified the appearance of Delta2 in Mollusca that may have resulted from early termination near arginine after Delta duplication (Fig. 3) and we speculated the reasons for early termination differed between species probably owing to high selection pressure and long differentiation time, leading to large sequence variation. In general, duplication of evolutionary genes could lead to non-, sub-, neo-, and synfunctionalisation47,48. Because of the different expression pattern (Fig. 4), Delta duplication in molluscs likely resulted in neofunctionalisation, meaning that Delta1 retained its ancestral function and Delta2 acquired new functions. In C. gigas and P. martensii, Delta2 expression was notably upregulated during umbo larva (Fig. 4). As is known, shells, gills, feet, and eye spots are formed at the umbo larva stage36, and thus the observed expression pattern is likely related to specific characteristics that are crucial for phenotype differentiation21,34. Indeed, recent studies have revealed that the Notch pathway plays a key role in shell colour, which is crucial for measuring economic value20,49.
The terminal genes exhibited gene expansion and expression differentiation among metazoans in this study (Fig. 5). The expanded genes, which probably resulted from gene duplication35, showed similar expression patterns associated with embryonic development among C. gigas and D. melanogaster, but the expression pattern was different from that of D. rerio. Unlike the Hes genes clusters (also called E(spl) genes) in D. melanogaster, which had an upregulated expression during the stages of blastoderm and gastrula50, the duplicated terminal genes expressed in D. rerio were consistently highly expressed after the gastrula and segmentation stages (Fig. 5b), likely resulting from periodic activation of the Notch pathway to control somitogenesis51,52. The functional differentiation of other Hes/Hey-related genes were also reported in previous studies. For example, Gazave et al. found that Hes participates in chaetal sac formation in P. dumerilii, whereas Rivera et al. found that Hes affects the segmentation process in H. robusta23,34. In mammals, Hes controls cellular differentiation and leads to neuronal development abnormalities53, whereas the novel gene Helt (Hesl) is reportedly regulated by Notch and plays essential roles in neuronal differentiation54,55; however, the functions of these genes in lophotrochozoans remain unclear. Although Cwo was found to be a transcriptional repressor and novel circadian pacemaker component in D. melanogaster56, it is uncertain whether the Cwo and Notch pathways are closely connected.
Notch, Presenilin, and Su(H) are conserved genes in the Notch pathway, and displayed high expression levels during the early developmental stages of most species (Fig. S5). Although the expression patterns of these genes in species were similar, the functions were diverse as well. It was reported that, during the embryo stage of Cnidarian N. vectensis, Notch broadly crossed several tissues including the pharyngeal, body wall endodermal, and ectoderm18, as well as in another Cnidarian H. vulgaris57. In Mollusca Ilyanassa obsolete, Notch and Su(H) are clearly important for endoderm formation and cell fates20. For Annelida H. robusta and C. teleta, Notch participates in segment formation, as well as in vertebrates13,14,15,22. Furthermore, these genes were also shown to be associated with neurogenesis in N. vectensis, S. purpuratus, and D. melanogaster, but not in P. dumerilii18,34,58,59. The functional differentiation of conserved genes enriched the diversification of species. However, there are few studies on the regulatory factor Presenilin. Moreover, even though the expression level of some developmental stages are very low, they also performed functions, like some Hes genes in D. melanogaster50.
Conclusions
In this study, we demonstrated conservation and divergence within the Notch pathway and revealed the evolution of core Notch pathway genes across metazoan genomes, with a strong focus on lophotrochozoan species. We demonstrated that the Notch pathway can be traced back to Porifera, Ctenophora and even earlier organisms, and that novel genes were differentiated in lophotrochozoans. Comparative transcriptomics revealed similarities and differences in lophotrochozoans compared to other metazoans. In addition, we identified the novel Delta2 gene in molluscs, which may function in specific developmental stages of lophotrochozoans along with Hes/Hey-related genes, and proposed its formation mechanisms. We also discovered the expansion of target genes and the differentiation of the expression pattern of conservative genes in different species. However, future experiments are required to confirm these expression patterns and clarify gene function. Together, our study demonstrates that comparative and evolutionary analyses are essential tools for studying pathway evolution. Exploring the Notch pathway in lophotrochozoans will improve our understanding of their development and phenotype diversity, which could provide a basis for future gene function studies.
References
Guo, X. Use and exchange of genetic resources in molluscan aquaculture. Rev. Aquac. 1, 251–259. https://doi.org/10.1111/j.1753-5131.2009.01014.x (2009).
Zrzavý, J., Říha, P., Piálek, L. & Janouškovec, J. Phylogeny of Annelida (Lophotrochozoa): total-evidence analysis of morphology and six genes. BMC Evol. Biol. 9, 189. https://doi.org/10.1186/1471-2148-9-189 (2009).
Barolo, S. & Posakony, J. W. Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. 16, 1167–1181. https://doi.org/10.1101/gad.976502 (2002).
Gazave, E. et al. Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol. Biol. 9, 249. https://doi.org/10.1186/1471-2148-9-249 (2009).
Bray, S. J. Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689. https://doi.org/10.1038/nrm2009 (2006).
Wilson, J. J. & Kovall, R. A. Crystal structure of the CSL-Notch-mastermind ternary complex bound to DNA. Cell 124, 985–996. https://doi.org/10.1016/j.cell.2006.01.035 (2006).
Nam, Y., Sliz, P., Song, L. Y., Aster, J. C. & Blacklow, S. C. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 124, 973–983. https://doi.org/10.1016/j.cell.2005.12.037 (2006).
Kopan, R. & Ilagan, M. X. G. The canonical notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. https://doi.org/10.1016/j.cell.2009.03.045 (2009).
Gyoja, F. & Satoh, N. Evolutionary aspects of variability in bHLH orthologous families: insights from the pearl oyster Pinctada fucata. Zool. Sci. 30, 868–876. https://doi.org/10.2108/zsj.30.868 (2013).
Artavanis-Tsakonas, S., Rand, M. D. & Lake, R. J. Notch signaling: cell fate control and signal integration in development. Science 284, 770–776. https://doi.org/10.1126/science.284.5415.770 (1999).
Feller, J., Schneider, A., Schuster-Gossler, K. & Gossler, A. Noncyclic Notch activity in the presomitic mesoderm demonstrates uncoupling of somite compartmentalization and boundary formation. Genes Dev. 22, 2166–2171. https://doi.org/10.1101/gad.480408 (2008).
Levin, M. Left–right asymmetry in embryonic development: a comprehensive review. Mech. Dev. 122, 3–25. https://doi.org/10.1016/j.mod.2004.08.006 (2005).
de Angelis, M. H., Mclntyre, J. & Gossler, A. Maintenance of somite borders in mice requires the Delta homologue Dll1. Nature 386, 717–721. https://doi.org/10.1038/386717a0 (1997).
Van Eeden, F. et al. Mutations affecting somite formation and patterning in the zebrafish Danio rerio. Development 123, 153–164. https://doi.org/10.1111/j.1365-2303.1996.tb00550.x (1996).
Huppert, S. S., Ilagan, M. X. G., De Strooper, B. & Kopan, R. Analysis of Notch function in presomitic mesoderm suggests a γ-secretase-independent role for presenilins in somite differentiation. Dev. Cell 8, 677–688. https://doi.org/10.1016/j.devcel.2005.02.019 (2005).
Lowell, S., Jones, P., Le Roux, I., Dunne, J. & Watt, F. M. Stimulation of human epidermal differentiation by Delta-Notch signalling at the boundaries of stem-cell clusters. Curr. Biol. 10, 491–500. https://doi.org/10.1016/s0960-9822(00)00451-6 (2000).
Richards, G. S. & Degnan, B. M. The expression of Delta ligands in the sponge Amphimedon queenslandica suggests an ancient role for Notch signaling in metazoan development. EvoDevo 3, 15. https://doi.org/10.1186/2041-9139-3-15 (2012).
Marlow, H., Roettinger, E., Boekhout, M. & Martindale, M. Q. Functional roles of Notch signaling in the cnidarian Nematostella vectensis. Dev. Biol. 362, 295–308. https://doi.org/10.1016/j.ydbio.2011.11.012 (2012).
Münder, S. et al. Notch signalling defines critical boundary during budding in Hydra. Dev. Biol. 344, 331–345. https://doi.org/10.1016/j.ydbio.2010.05.517 (2010).
Gharbiah, M., Nakamoto, A., Johnson, A. B., Lambert, J. D. & Nagy, L. M. Ilyanassa Notch signaling implicated in dynamic signaling between all three germ layers. Int. J. Dev. Biol. 58, 551–562. https://doi.org/10.1387/ijdb.140149ln (2015).
Thamm, K. & Seaver, E. C. Notch signaling during larval and juvenile development in the polychaete annelid Capitella sp. I. Dev. Biol. 320, 304–318. https://doi.org/10.1016/j.ydbio.2008.04.015 (2008).
Rivera, A. S., Gonsalves, F. C., Song, M. H., Norris, B. J. & Weisblat, D. A. Characterization of Notch-class gene expression in segmentation stem cells and segment founder cells in Helobdella robusta (Lophotrochozoa; Annelida; Clitellata; Hirudinida; Glossiphoniidae). Evol. Dev. 7, 588–599. https://doi.org/10.1111/j.1525-142x.2005.05062.x (2005).
Rivera, A. S. & Weisblat, D. A. And Lophotrochozoa makes three: Notch/Hes signaling in annelid segmentation. Dev. Genes. Evol. 219, 37–43. https://doi.org/10.1007/s00427-008-0264-6 (2009).
Schiemann, S. M. et al. Clustered brachiopod Hox genes are not expressed collinearly and are associated with lophotrochozoan novelties. Proc. Natl. Acad. Sci. 114, E1913–E1922. https://doi.org/10.1073/pnas.1614501114 (2017).
Wang, J. et al. Evolutionary transcriptomics of metazoan biphasic life cycle supports a single intercalation origin of metazoan larvae. Nat. Ecol. Evol. 4, 725–736. https://doi.org/10.1038/s41559-020-1138-1 (2020).
Irie, N. & Kuratani, S. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat. Commun. https://doi.org/10.1038/ncomms1248 (2011).
Du, X. et al. The pearl oyster Pinctada fucata martensii genome and multi-omic analyses provide insights into biomineralization. Gigascience 6, gix0659. https://doi.org/10.1093/gigascience/gix059 (2017).
Li, Y. et al. Scallop genome reveals molecular adaptations to semi-sessile life and neurotoxins. Nat. Commun. 8, 1–11. https://doi.org/10.1038/s41467-017-01927-0 (2017).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucl. Acids Res. 47, W636–W641. https://doi.org/10.1093/nar/gkz268 (2019).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucl. Acids Res. 45, D200–D203. https://doi.org/10.1093/nar/gkw1129 (2017).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucl. Acids Res. 46, D493–D496. https://doi.org/10.1093/nar/gkx922 (2018).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. https://doi.org/10.1093/molbev/msy096 (2018).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE https://doi.org/10.1371/journal.pone.0009490 (2010).
Gazave, E., Lemaître, Q. I. & Balavoine, G. The Notch pathway in the annelid Platynereis: insights into chaetogenesis and neurogenesis processes. Open Biol. 7, 160242. https://doi.org/10.1098/rsob.160242 (2017).
Bao, Y., Xu, F. & Shimeld, S. M. Phylogenetics of lophotrochozoan bHLH genes and the evolution of lineage-specific gene duplicates. Genome Biol. Evol. 9, 869–886. https://doi.org/10.1093/gbe/evx047 (2017).
Zhang, G. et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54. https://doi.org/10.1038/nature11413 (2012).
Domazet-Lošo, T. & Tautz, D. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 468, 815–818. https://doi.org/10.1038/nature09632 (2010).
Chen, C. et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. https://doi.org/10.1016/j.molp.2020.06.009 (2020).
Putnam, N. H. et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071. https://doi.org/10.1038/nature06967 (2008).
Westin, J. & Lardelli, M. Three novel Notch genes in zebrafish: implications for vertebrate Notch gene evolution and function. Dev. Genes. Evol. 207, 51–63. https://doi.org/10.1007/s004270050091 (1997).
Wallberg, A., Thollesson, M., Farris, J. S. & Jondelius, U. The phylogenetic position of the comb jellies (Ctenophora) and the importance of taxonomic sampling. Cladistics 20, 558–578. https://doi.org/10.1111/j.1096-0031.2004.00041.x (2004).
Richter, D. J., Parinaz, F., Eisen, M. B. & Nicole, K. Gene family innovation, conservation and loss on the animal stem lineage. Elife 7, e34226 (2018).
Baladrón, V. et al. dlk acts as a negative regulator of Notch1 activation through interactions with specific EGF-like repeats. Exp. Cell Res. 303, 343–359. https://doi.org/10.1016/j.yexcr.2004.10.001 (2005).
Bray, S. J., Takada, S., Harrison, E., Shen, S.-C. & Ferguson-Smith, A. C. The atypical mammalian ligand Delta-like homologue 1 (Dlk1) can regulate Notch signalling in Drosophila. BMC Dev. Biol. 8, 11. https://doi.org/10.1186/1471-213X-8-11 (2008).
Gu, Y., Hukriede, N. A. & Fleming, R. J. Serrate expression can functionally replace Delta activity during neuroblast segregation in the Drosophila embryo. Development 121, 855–865 (1995).
Sun, X. & Artavanis-Tsakonas, S. Secreted forms of DELTA and SERRATE define antagonists of Notch signaling in Drosophila. Development 124, 3439–3448 (1997).
Gitelman, I. Evolution of the vertebrate twist family and Synfunctionalization: a mechanism for differential gene loss through merging of expression domains. Mol. Biol. Evol. 24, 1912–1925. https://doi.org/10.1093/molbev/msm120 (2007).
Cho, S.-J., Valles, Y., Giani, V. C. Jr., Seaver, E. C. & Weisblat, D. A. Evolutionary dynamics of the wnt gene family: a lophotrochozoan perspective. Mol. Biol. Evol. 27, 1645–1658. https://doi.org/10.1093/molbev/msq052 (2010).
Feng, D., Li, Q., Yu, H., Zhao, X. & Kong, L. Comparative transcriptome analysis of the Pacific oyster Crassostrea gigas characterized by shell colors: identification of genetic bases potentially involved in pigmentation. PLoS ONE https://doi.org/10.1371/journal.pone.0145257 (2015).
Delidakis, C., Monastirioti, M. & Magadi, S. S. in Current Topics in Developmental Biology Vol. 110 (ed Reshma Taneja) 217–262 (Academic Press, 2014).
Jiang, Y.-J. et al. Notch signalling and the synchronization of the somite segmentation clock. Nature 408, 475–479. https://doi.org/10.1038/35044091 (2000).
Soza-Ried, C., Öztürk, E., Ish-Horowicz, D. & Lewis, J. Pulses of Notch activation synchronise oscillating somite cells and entrain the zebrafish segmentation clock. Development 141, 1780–1788. https://doi.org/10.1242/dev.102111 (2014).
Kageyama, R. & Ohtsuka, T. The Notch-Hes pathway in mammalian neural development. Cell Res. 9, 179–188. https://doi.org/10.1038/sj.cr.7290016 (1999).
Nakatani, T., Mizuhara, E., Minaki, Y., Sakamoto, Y. & Ono, Y. Helt, a novel basic-helix-loop-helix transcriptional repressor expressed in the developing central nervous system. J. Biol. Chem. 279, 16356–16367. https://doi.org/10.1074/jbc.M311740200 (2004).
Nakatani, T., Minaki, Y., Kumai, M. & Ono, Y. Helt determines GABAergic over glutamatergic neuronal fate by repressing Ngn genes in the developing mesencephalon. Development 134, 2783–2793. https://doi.org/10.1242/dev.02870 (2007).
Kadener, S., Stoleru, D., McDonald, M., Nawathean, P. & Rosbash, M. Clockwork Orange is a transcriptional repressor and a new Drosophila circadian pacemaker component. Genes Dev. 21, 1675–1686 (2007).
Käsbauer, T. et al. The Notch signaling pathway in the cnidarian Hydra. Dev. Biol. 303, 376–390. https://doi.org/10.1016/j.ydbio.2006.11.022 (2007).
Yaguchi, S. et al. Fez function is required to maintain the size of the animal plate in the sea urchin embryo. Development 138, 4233–4243. https://doi.org/10.1242/dev.069856 (2011).
Artavanis-Tsakonas, S., Muskavitch, M. & Yedvobnick, B. Molecular cloning of Notch, a locus affecting neurogenesis in Drosophila melanogaster. Proc. Natl. Acad. Sci. 80, 1977–1981. https://doi.org/10.1073/pnas.80.7.1977 (1983).
Acknowledgements
We are grateful to Lei Liu and Yue Xu for their help with sampling during our study. We thank Shanshan Yao for assistance in transcriptome assembly.
Funding
This study was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA24030105), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB42000000), the Pilot National Laboratory for Marine Science and Technology (No. YJ2019NO01), Key Development Project of Centre for Ocean Mega-Research of Science, Chinese academy of science (No. COMS2019R01), National Natural Science Foundation of China (No. 41976088), the National Science Foundation of China (No. 31972790 to F.W.), and China Agriculture Research System of MOF and MARA.
Author information
Authors and Affiliations
Contributions
X.H. and F.W. completed all data analysis and wrote the manuscript. F.W. and L.Z. conceived and designed the study. F.W. and L.L. supported the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, X., Wu, F., Zhang, L. et al. Comparative and evolutionary analyses reveal conservation and divergence of the notch pathway in lophotrochozoa. Sci Rep 11, 11378 (2021). https://doi.org/10.1038/s41598-021-90800-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90800-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
