
Abstract
The absence of orthogonal aminoacyl-transfer RNA (tRNA) synthetases that accept non-l-α-amino acids is a primary bottleneck hindering the in vivo translation of sequence-defined hetero-oligomers and biomaterials. Here we report that pyrrolysyl-tRNA synthetase (PylRS) and certain PylRS variants accept α-hydroxy, α-thio and N-formyl-l-α-amino acids, as well as α-carboxy acid monomers that are precursors to polyketide natural products. These monomers are accommodated and accepted by the translation apparatus in vitro; those with reactive nucleophiles are incorporated into proteins in vivo. High-resolution structural analysis of the complex formed between one PylRS enzyme and a m-substituted 2-benzylmalonic acid derivative revealed an active site that discriminates prochiral carboxylates and accommodates the large size and distinct electrostatics of an α-carboxy substituent. This work emphasizes the potential of PylRS-derived enzymes for acylating tRNA with monomers whose α-substituent diverges substantially from the α-amine of proteinogenic amino acids. These enzymes or derivatives thereof could synergize with natural or evolved ribosomes and/or translation factors to generate diverse sequence-defined non-protein heteropolymers.

Similar content being viewed by others

Main
Extant organisms biosynthesize polypeptides in a messenger RNA template-dependent manner using a translation apparatus composed of ribosomes, aminoacyl-transfer RNA (tRNA) synthetases, tRNAs and multiple ancillary factors. Repurposing this translation apparatus for the templated in vivo synthesis of mixed-sequence hetero-oligomers—especially those containing non-l-α-amino acids—would provide a biological route to sequence-defined non-peptide polymers with original, tunable and evolvable properties and protein therapeutics with improved stability and expanded function. Chemical methods support the synthesis of sequence-defined non-peptide polymers1,2, but are limited to a small scale and produce considerable waste. Polymerization methods support the synthesis of sequence-controlled materials, but without rigorous sequence definition. By contrast, cellular methods generate little waste, especially on a large scale, achieve longer chain lengths and are scalable for industrial production.
It has been established over the past decade that many non-l-α-amino acids, including α-hydroxy acids3,4, d-α-5, linear6,7,8 and cyclic9 β-, γ-10,11, and long-chain amino acids12, α-aminoxy and α-hydrazino acids13, α-thio acids14, pyridazinone15, aminobenzoic acids16,17, and even 1,3-dicarbonyl monomers that resemble polyketide precursors17, are accepted by natural Escherichia coli ribosomes in small-scale in vitro reactions when acylated on tRNAs18. The structural and electronic diversity of these monomers emphasizes the important role of proximity in promoting bond-forming reactions within the E. coli peptidyl transferase centre (PTC)19. However, there are scant examples of non-l-α-amino acids having been incorporated into proteins in vivo20,21,22,23,24,25,26,27. The absence of orthogonal aminoacyl-tRNA synthetase (aaRS) enzymes that accept non-l-α-amino acid substrates is a primary bottleneck limiting the production of sequence-defined, non-peptide heteropolymers in vivo using wild-type (WT) or engineered ribosomes.
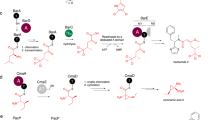
In E. coli, two families of orthogonal aaRS enzymes have been widely employed to introduce hundreds of different non-canonical α-amino acid monomers28,29,30,31,32 into protein. The first includes pyrrolysyl-tRNA synthetase (PylRS) enzymes from methanogenic archaea and bacteria whose natural substrate is pyrrolysine33, an l-α-amino acid found in the active sites of certain enzymes involved in methane metabolism34. The second includes a large family of enzymes derived from Methanocaldococcus jannaschii tyrosyl-tRNA synthetase (MjTyrRS)28,35. These two enzyme classes differ in how they recognize the α-amine of a bound substrate. While MjTyrRS recognizes the substrate α-amine via multiple, direct hydrogen bonds to the side chains of Gln173, Gln155 and Tyr151 (Protein Data Bank (PDB): 1J1U)36, Methanosarcina mazei PylRS (MmPylRS) instead uses water-mediated hydrogen bonds to the Asn346 side chain and Leu301 and Ala302 backbone amides (PDB: 2ZCE; Fig. 1a)37. These differences were exploited to acylate the cognate tRNA of MmPylRS (Mm-tRNAPyl) with a series of conservative Nε-tert-butoxycarbonyl-l-lysine (l-BocK) analogues containing -OH, -H and -NHCH3 in place of the α-amine (Fig. 1b)22.

a, The α-amines of l-α-amino acids are recognized differently by MmPylRS (left)37 and MjTyrRS (right)36. b, Analogues of l-BocK 1 evaluated as substrates for MaPylRS. c, RNAse A assay used to detect the acylation of Ma-tRNAPyl with the BocK analogues shown in b. d, LC–HRMS analysis of Ma-tRNAPyl acylation reactions after RNAse A digestion. The peak masses correspond to adenosine nucleoside 6 acylated on the 2′- or 3′-hydroxy by the indicated monomer. The bottom three traces in each plot are negative controls that lack either the small molecule substrate (∆1, ∆2, or ∆3), enzyme (∆MaPylRS), or tRNA (∆tRNAPyl), while the top trace labeled ‘+’ contains all three necessary components for the acylation reaction. Two isobaric peaks are observed because the sample consists of adenosine acylated on either the 2′- or 3′-hydroxy group. e, Heat map illustrating the relative activities of substrates 1–5 for MaPylRS, as determined by intact tRNA analysis (Supplementary Figs. 3–6) as described in Methods. The yields are reported as percentages based on intact tRNA analysis. The percentage yields were calculated as the ratio of tRNA acylated with the monomer of interest divided by the total tRNA detected by the LC–MS analysis. The black box indicates that no reaction product was detected; the box marked X indicates that the substrate was not investigated.
We hypothesized that the α-amine recognition mode employed by MmPylRS could provide sufficient space for substrates with less conservative substituents than the α-amine if the water molecule in the active site were displaced. Here we report that the tolerance of MmPylRS for substrates with multiple alternative substituents in place of the α-amine extends to Methanomethylophilus alvus PylRS (MaPylRS)38,39 as well as MaPylRS variants (N166A V168L, MaFRS1 and N166A V168K, MaFRS2) that recognize diverse l-phenylalanine derivatives40. MaFRS1 and MaFRS2 also accept phenylalanine derivatives with α-thio, N-formyl-l-α-amino as well as an α-carboxy substituent: 2-benzylmalonic acid. A final variant, MaFRSA (N166A V168A) (ref. 41), shows selectivity for ring-substituted 2-benzylmalonate derivatives over l-Phe.
Malonates contain a 1,3-dicarbonyl unit that represents the defining backbone of polyketide natural products, and after decarboxylation they have the potential to support Claisen-type condensation within the PTC to form a carbon–carbon bond. Structural analysis of MaFRSA complexed with a m-substituted 2-benzylmalonate derivative and a non-hydrolysable ATP analogue revealed how the enzyme uses a unique pattern of hydrogen bonds to differentiate the two prochiral carboxylates in the substrate and accommodate the larger size and distinct electrostatics of an α-carboxy substituent. In vitro translation studies confirmed that tRNAs carrying α-thio, α-carboxy and N-formyl-l-α-amino acid monomers are effectively delivered to and accommodated by E. coli ribosomes. In vivo studies using traditional and engineered E. coli strains confirmed that MaFRSA supports the biosynthesis of model proteins containing internal, aromatic, α-hydroxy acid monomers. This work describes the first aaRSs that accept α-thio acids and α-carboxy acids that could support carbon–carbon bond formation within the ribosome. These activities emphasize the potential of PylRS as a scaffold for evolving new enzymes that act in synergy with natural or evolved ribosomes to generate diverse sequence-defined non-protein heteropolymers.
Results
MaPylRS retains much of the promiscuity of MmPylRS
First, we set out to establish whether the expanded substrate scope of MmPylRS reported for l-BocK analogues (Fig. 1b)22 was retained by MaPylRS, which offers advantages over MmPylRS because it lacks the poorly soluble amino-terminal tRNA-binding domain42 and is easier to express and evaluate in vitro39. The carboxy-terminal catalytic domain of MmPylRS (ref. 37) is 36% identical to MaPylRS and the structures are largely superimposable (Supplementary Fig. 1). To evaluate whether d-BocK as well as l-BocK analogues with -OH or -H in place of the α-amino group were substrates for MaPylRS, we made use of a validated ribonuclease A (RNAse A) liquid chromatography (LC)–high-resolution mass spectrometry (HRMS) assay17,43. This assay exploits RNAse A to cleave the phosphodiester bonds following pyrimidine residues to generate 2′,3′-cyclic phosphate products44. As a result, the residue at the tRNA 3′ terminus is the only mononucleoside product lacking a phosphate (Fig. 1c). The incubation of l-BocK 1 (2 mM) with purified MaPylRS (2.5 µM) and Ma-tRNAPyl (25 µM; Supplementary Fig. 2) at 37 °C for 2 h led to a pair of RNAse A digestion products whose expected mass (496.25142 Da) corresponds to the adenosine nucleoside 6 as a mixture of 2′- and 3′-acylated species (Fig. 1d) Although there is evidence that PylRS from Methanosarcina barkeri and Desulfitobacterium hafniense add Pyl to only the 3′-hydroxy group45, isomerization between the 2′ and 3′ isomers occurs rapidly46 and we were not able to identify which peak corresponds to which isomer. No products with this mass were observed when the reaction mixture lacked Ma-tRNAPyl, l-BocK or MaPylRS, and mass analysis of the intact tRNA product confirmed a 53.8% yield of acylated tRNA (1-tRNAPyl; Supplementary Fig. 3). Under these conditions, l-BocK analogues with either -OH (2) or -H (3) in place of the α-amino group were also substrates for MaPylRS, as judged by RNAse A (Fig. 1d) and intact tRNA mass spectrometry (MS) assays, with acylated tRNA yields of 90.5% (2-tRNAPyl) and 61.6% (3-tRNAPyl; Supplementary Figs. 4 and 5, respectively). No reactivity was detected with d-BocK (Supplementary Fig. 6 and Extended Data Fig. 1), perhaps because of differences in PylRS from M. alvus and M. mazei47. We conclude, therefore, that MaPylRS retains much of the previously reported22 promiscuity of MmPylRS. We note that certain non-natural monomers with relatively high activity, including 2 and 3, led to measurable levels (2.5–13.7%) of diacylated tRNA (Supplementary Table 3). Diacylated tRNAs have been observed as products in cognate reactions of Thermus thermophilus phenylalanyl-tRNA synthetase (PheRS) (ref. 48), and they have been reported to support prokaryotic translation49.
MaPylRS variants retain activity for phenylalanine derivatives
PylRS is a subclass IIc aaRS that evolved from PheRS (ref. 50). MmPylRS variants with substitutions at two positions that alter the architecture of the side-chain-binding pocket (Asn346 and Cys348; Supplementary Fig. 1) accept l-phenylalanine and its derivatives in place of pyrrolysine29,40,50. We integrated the mutations in the two variants that accept unsubstituted l-Phe (MmFRS1 and MmFRS2)40 into the MaPylRS sequence to generate MaFRS1 (N166A and V168L) and MaFRS2 (N166A and V168K) (Supplementary Fig. 2). We then used RNAse A and intact tRNA MS assays to determine whether MaFRS1 or MaFRS2 retained the activity for l-phenylalanine (7) and its analogues in which the l-α-amino group was substituted by -OH (8), -H (9), -NHCH3 (10) or d-NH2 (11; Fig. 2a).

a, Phenylalanine analogues evaluated as substrates for MaFRS1 and MaFRS2. b, Adenosine nucleoside 12 formed during RNAse A digestion of acyl-tRNA. c, LC–HRMS analysis of Ma-tRNAPyl acylation reactions after RNAse A digestion. Adenine nucleoside 12 acylated on the 2′- or 3′-hydroxy of the 3′-terminal ribose of Ma-tRNAPyl could be detected in MaFRS1 and MaFRS2 reactions with l-Phe (7) and substrate 8; substrates 9 and 10 (Z = H, NHCH3) showed more modest reactivity. d, Heat map illustrating the relative yields with substrates 7–11 for MaFRS1 and MaFRS2, as determined by intact tRNA analysis (Supplementary Figs. 8–12 and 20–24) as described in Methods. The reported yields are percentages based on intact tRNA analysis. The black boxes indicate no reaction product was detected.
The incubation of l-Phe (7; 10 mM) with purified MaFRS1 or MaFRS2 (2.5 µM) and Ma-tRNAPyl (25 µM) at 37 °C for 2 h led in both cases to a pair of products whose expected mass (415.17244 Da) corresponds to adenosine nucleoside 12 (Z = l-NH2; Fig. 2b) as the expected mixture of 2′- and 3′-acylated products (Fig. 2c). No product with this mass was observed when the reaction mixture lacked Ma-tRNAPyl, l-Phe, or MaFRS1 or MaFRS2, and mass analysis of the intact tRNA product confirmed yields of 66.1% (MaFRS1) and 65.4% (MaFRS2) of acylated tRNA (7-tRNAPyl; Supplementary Figs. 8 and 20, respectively). l-Phe analogues 8–10 were also substrates for both MaFRS1 and MaFRS2, as judged by both RNAse A (Fig. 2c) and intact tRNA analysis (Supplementary Figs. 8–11 and 20–23), with reactivities decreasing in the order l-α-amino 7 > α-OH 8 ≫ α-H 9 ≈ N-Me-l-α-amino 10 based on intact tRNA yields (Fig. 2d). Mono- and diacylated tRNA products were observed for substrates 7 and 8 (Supplementary Table 3). Despite the fact that the desamino-BocK analogue 3 was a strong substrate for WT MaPylRS, the desamino-Phe analogue 9 showed only modest activity with MaFRS1 and MaFRS2, as observed in the RNAse assay and intact tRNA analysis (Fig. 2c and Supplementary Figs. 10 and 22). Again, no reactivity was detected with d-Phe (Supplementary Figs. 12 and 24 and Extended Data Fig. 1).
MaFRS1 and MaFRS2 process substrates with distinct α-substituents
We then began to explore phenylalanine analogues with larger and electrostatically distinct functional groups at the α-carbon: α-thio acids, N-formyl-l-α-amino acids and α-carboxy acids (malonates; Fig. 3a). α-Thio acids are substrates for extant E. coli ribosomes in analytical-scale in vitro translation reactions with yields as high as 87% of the corresponding α-amino acids14, and thioesters can persist in E. coli for more than 36 h (ref. 51). Peptides and proteins containing thioesters could also act as substrates for polyketide synthase (PKS) modules52, to generate unique keto-peptide natural products, or protein splicing reactions53. Formylation of methionyl-tRNA is important for initiating complex formation, and formylation could enhance initiation with non-methionyl α-amino acids in vivo54,55. Moreover, E. coli ribosomes incorporate monomers containing a 1,3-dicarbonyl moiety at the peptide N terminus to produce keto-peptide hybrids17. To our knowledge, there are currently no aaRS enzymes, orthogonal or otherwise, that accept α-thio, N-formyl-l-α-amino or α-carboxy acid substrates to generate the acylated tRNAs required for in vivo translation (when extant ribosomes are compatible) or ribosome evolution (when extant ribosomes are incompatible).

a, Phenylalanine analogues 13–15 evaluated as substrates for MaFRS1 and MaFRS2. b, LC–HRMS analysis of Ma-tRNAPyl acylation reactions using MaFRS1 or MaFRS2 following RNAse A digestion. Adenosine nucleoside 12 acylated on the 2′- or 3′-hydroxy of the 3′-terminal ribose of Ma-tRNAPyl could be detected in MaFRS1 and MaFRS2 reactions with α-thio acid 13, α-carboxy acid 14 and N-formyl-l-Phe 15. c, LC–MS analysis of intact tRNA products confirms that monomers 13–15 are substrates for MaFRS1 and MaFRS2. An asterisk indicates the peak that corresponds to the indicated acyl-tRNA while a D indicates the peak that corresponds to the decarboxylation of the malonyl-tRNA. d, Heat map illustrating the relative activities of substrates 13–15 with MaFRS1 and MaFRS2, as determined by intact tRNA analysis (Supplementary Figs. 13–16 and 25–30) as described in Methods. The reported yields are percentages based on intact tRNA analysis. The black box indicates that no reaction product was detected. Initially no acyl-tRNA was detected when MaFRS1 was incubated with 13, but when the enzyme concentration was increased fivefold, acyl-tRNA was detected with mono- and diacyl yields of 0.7 and 9.7%, respectively. e, Turnover of MaFRS1 over time with l-Phe (7) and 2-benzylmalonic acid (14) using the malachite green assay. A control with no substrate is shown for comparison. Data from three replicates are shown.
We found that l-Phe analogues in which the α-amino was substituted with an α-thio (substrate 13), α-carboxy (substrate 14) or N-formyl-l-α-amine (substrate 15) moiety were all processed by MaFRS1 and MaFRS2 (Fig. 3a–c). In particular, α-carboxy acid 14 and N-formyl-l-α-amino acid 15 were excellent substrates. The incubation of α-carboxy acid 14 (10 mM) with MaFRS1 or MaFRS2 (2.5 µM) and Ma-tRNAPyl (25 µM) at 37 °C for 2 h, followed by digestion by RNAse A led to the formation of a pair of products whose expected mass (444.15137 Da) corresponds to the adenosine nucleoside 12 (Z = COOH). LC–MS analysis of the intact tRNA products confirmed yields of 24.4% (MaFRS1) and 43.7% (MaFRS2) of acylated tRNA (14-tRNAPyl) (Supplementary Figs. 15 and 26, respectively). Because the α-carbon of substrate 14 is prochiral, mono-acylation of Ma-tRNAPyl can generate two diastereomeric product pairs—one pair in which the 3′-hydroxy group is acylated by the pro-S or pro-R carboxylate and another in which the 2′-hydroxy group is acylated by the pro-S or pro-R carboxylate. These diastereomeric products result from alternative substrate orientations within the enzyme active site (vide infra). We note that intact tRNAs acylated with 2-benzylmalonic acid (14) showed evidence of decarboxylation (indicated by a D in Fig. 3c). No evidence for decarboxylation was observed when the same acyl-tRNAs were evaluated using the RNAse A assay (Extended Data Fig. 2), suggesting that decarboxylation occurs either during work-up or during the LC–MS injection of intact tRNA.
The incubation of N-formyl-l-α-amino acid 15 under identical conditions led to the expected adenosine nucleoside 12 (Z = NHCHO), and LC–MS analysis confirmed yields of 50.0% (MaFRS1) and 31.1% (MaFRS2) of acylated tRNA (15-tRNAPyl; Supplementary Figs. 16 and 27, respectively). The α-thio l-Phe analogue 13 was also a substrate for MaFRS1 and MaFRS2, although higher concentrations of MaFRS1 were necessary to observe the acyl-tRNA product (13-tRNAPyl) using intact tRNA LC–MS (Supplementary Figs. 13, 14 and 25). These results illustrate that the active-site pocket that engages the α-amine in PylRS can accommodate substituents with substantial differences in mass (-NHCHO) and charge (-COO−). Despite these differences, 2-benzylmalonic acid (2-BMA, 14) is an excellent substrate for MaFRS1 and MaFRS2: kinetic analysis using the malachite green assay56 revealed a rate of adenylation by MaFRS1 that was 66% of the rate observed for l-Phe (Fig. 3e). The tolerance for malonate substrates extends to WT MaPylRS itself: the α-carboxylate analogue of l-BocK 16 was also a measurable substrate for WT MaPylRS (Supplementary Fig. 7 and Extended Data Fig. 1).
MaFRSA processes m-2-benzylmalonic acids but not l-Phe
We have demonstrated that MaFRS1 and MaFRS2 have the ability to process substrates with unusual α-substituents, but they can also process l-Phe with comparable efficiency (Fig. 2d), which would interfere with selective charging of the non-l-α-amino acid. Variants of MmPylRS that accept p-, o- and m-substituted l-Phe derivatives have been reported40,41,57,58,59. In particular, MmPylRS containing two active-site mutations (N346A and C348A; henceforth referred to as FRSA) shows high activity for l-Phe analogues with bulky alkyl substituents and low activity towards l-Phe (ref. 41). We expressed and purified an MaPylRS variant containing these mutations (MaFRSA with N166A and V168A; Supplementary Fig. 2) and demonstrated using RNAse A (Fig. 4b) and intact tRNA analysis (Fig. 4c and Supplementary Figs. 31–34) that it shows high activity for derivatives of malonic acid 14 carrying m-CH3 (17), m-CF3 (18) and m-Br (19) substituents and low activity for l-Phe. Again, intact tRNA LC–MS showed evidence of decarboxylation, but this was not observed in the RNAse A assay (Extended Data Fig. 2). Of the m-substituted 2-BMAs, MaFRSA showed the highest activity for m-CF3-2-benzylmalonate (m-CF3-2-BMA, 18). Kinetic analysis56 revealed a rate of adenylation that was 36% of the rate observed for its l-α-amino acid counterpart m-trifluoromethyl-l-phenylalanine (m-CF3-Phe, 20; Fig. 4e). Although derivatives of malonate 14 carrying m-CH3 (17), m-CF3 (18) and m-Br (19) substituents are also excellent substrates for MaFRS1 (Supplementary Figs. 17–19) and MaFRS2 (Supplementary Figs. 28–30), MaFRSA shows substantially lower activity for l-Phe, allowing the selective acylation of tRNA with m-substituted 2-BMAs without interference from l-Phe (Fig. 4d). Indeed, when MaFRSA was incubated with an equal concentration (10 mM) of l-Phe (7) and m-CF3-2-BMA (18), only the malonyl product (18-tRNAPyl) was observed (Supplementary Fig. 35).

a, Phenylalanine analogues 17–19 evaluated as substrates for MaFRSA. b, LC–HRMS analysis of Ma-tRNAPyl acylation products after digestion with RNAse A. c, LC–MS analysis of intact tRNA products confirms that m-substituted 2-BMAs 17–19 are substrates for MaFRSA. An asterisk indicates the peak that corresponds to the indicated acyl-tRNA while a D indicates the decarboxylation product of the malonyl-tRNA. d, Heat map illustrating the relative activities of l-Phe (7) and substrates 17–19 with MaFRS1, MaFRS2 and MaFRSA. The reported yields are percentages based on intact tRNA analysis. e, Turnover of MaFRSA over time with m-CF3-l-Phe (20) and m-CF3-2-BMA (18) using the malachite green assay. A control with no substrate is shown for comparison. Data from three replicates are shown.
Structure of the MaFRSA/m-CF3-2-BMA complex
To better understand how 2-BMA substrates are accommodated by the MaFRSA active site, we solved the crystal structure of MaFRSA bound to both m-CF3-2-BMA and the non-hydrolysable ATP analogue adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP). The structure was refined at 1.8 Å resolution with clear substrate density for m-CF3-2-BMA visible in the electron density map (Extended Data Fig. 3 and Supplementary Table 4). MaFRSA crystallized with two protein chains in the asymmetric unit and an overall architecture resembling previously published PylRS structures (Fig. 5a)37,60,61. The two protein chains in the asymmetric unit are not identical and interact with different orientations of m-CF3-2-BMA (Fig. 5b). One orientation of m-CF3-2-BMA (chain A, light purple) mimics that of l-pyrrolysine (Pyl) bound to MmPylRS37 and would result in adenylation of the pro-R carboxylate (Fig. 5c); the other orientation (chain B, dark purple) would result in adenylation of the pro-S carboxylate (Fig. 5d).

a, Structure of the MaFRSA dimer containing two non-identical chains in the asymmetric unit. b, Alignment of the active sites of chains A (light purple) and B (dark purple) reveals m-CF3-2-BMA (grey) bound in two alternative conformations. c, In chain A, m-CF3-2-BMA is coordinated by an extensive hydrogen bond network (orange dashes) that positions the pro-R carboxylate oxygen for nucleophilic attack (blue dashes). Interatomic distances (in Å) are shown alongside the dashed lines. d, In chain B, m-CF3-2-BMA is coordinated by similar hydrogen bonds, but in this case the pro-S carboxylate is rotated away from AMP-PNP with a loss of the hydrogen bond to Arg150 (red dashes) and a longer distance between the pro-S carboxylate nucleophile and the α-phosphate of AMP-PNP. e, Alignment of active site A with WT MmPylRS bound to Pyl and AMP-PNP (PDB: 2ZCE, blue)37 illustrates the difference between the water-mediated hydrogen bonds (orange dashes) to the α-amine of Pyl in PylRS compared with the direct carboxy-to-backbone hydrogen bonding of m-CF3-2-BMA bound to MaFRSA. f, Comparison of active site B with MmBtaRS (N346G, C348Q) bound to Bta (PDB: 4ZIB, red)62 reveals similar binding modes with hydrogen bonds (orange dashes) between the substrate carboxylate and backbone amide of the enzyme when Asn166/Asn346 (Ma/Mm numbering) is mutated.
Regardless of orientation, discrete networks of hydrogen bonds are used by MaFRSA to discriminate between the pro-R and pro-S carboxylates of m-CF3-2-BMA. In chain A, the pro-S carboxylate accepts a hydrogen bond from the backbone amides of Leu121 and Ala122 and the phenolic -OH of Tyr206 (Fig. 5c and Extended Data Fig. 4a). A hydrogen bond from the Arg150 guanidinium positions the pro-R carboxylate for nucleophilic attack with a distance of 2.7 Å between the carboxy oxygen and the α-phosphorus of AMP-PNP. In chain B, the orientation of m-CF3-2-BMA is flipped relative to the conformation bound to chain A (Fig. 5d and Extended Data Fig. 4b). The pro-R carboxylate accepts a hydrogen bond from the backbone amides of Leu121 and Ala122 and the phenolic -OH of Tyr206, as seen for the pro-S carboxylate in chain A. However, in chain B, the pro-S carboxylate is rotated away from AMP-PNP and towards Tyr206, resulting in loss of the hydrogen bond to Arg150 and a longer distance of 3.9 Å between the carboxy oxygen and the α-phosphorus of AMP-PNP. RNAse A analysis of Ma-tRNAPyl acylation by m-CF3-2-BMA showed more than two peaks of identical mass (Fig. 4) that likely correspond to the two diastereomeric pairs formed from attack of the tRNA 2′- or 3′-hydroxy group on the activated pro-R or pro-S carboxylate. More than two peaks with identical mass were also observed as RNAse A digestion products in Ma-tRNAPyl acylation reactions of other m-substituted 2-BMAs (Fig. 4b). While the m-CF3-2-BMA conformation in chain A seems to be more favourably positioned for catalysis, suggesting that the pro-R carboxylate is acylated preferentially, the appearance of more than two peaks in the RNAse A assay suggests that both conformations are catalytically competent.
As anticipated, the non-reactive, pro-S carboxylate of m-CF3-2-BMA is recognized by MaFRSA chain A using interactions that are distinct from those used by MmPylRS to recognize the Pyl α-amine. In the MmPylRS/Pyl/AMP-PNP complex (PDB: 2ZCE)37, the Pyl α-amine is recognized by water-mediated hydrogen bonds to the backbone amides of Leu301 and Ala302 and the side-chain carbonyl of Asn346 rather than by direct hydrogen bonds to the backbone amides of Leu121 and Ala122, as seen for the recognition of the non-reacting carboxylate of m-CF3-2-BMA by both chains of MaFRSA (Fig. 5e). The interactions used to recognize the non-reacting carboxylate of m-CF3-2-BMA are, however, similar to those used to recognize the single carboxylate of diverse α-amino acids by other PylRS variants with mutations at Asn166/Asn346 (Ma/Mm numbering). For example, the structures of MmPylRS variants with mutations at Asn346, such as MmIFRS and MmBtaRS bound to 3-iodo-l-phenylalanine (3-I-F, PDB: 4TQD)58 and 3-benzothienyl-l-alanine (Bta, PDB: 4ZIB)62, respectively, show the substrate bound with the carboxylate directly hydrogen bonded to the Leu301 and Ala302 backbone amides, as seen for m-CF3-2-BMA bound to MaFRSA (Fig. 5f and Extended Data Fig. 5a). In these cases, the bound water seen in the MmPylRS/Pyl/AMP-PNP complex is either absent or displaced. The mutation of Asn166/Asn346 may destabilize the water-mediated hydrogen bonding between the substrate α-amine and backbone amides seen in WT PylRS and promote alternative direct hydrogen bonding of a substrate carboxylate to backbone amides, as seen in MaFRSA, MmIFRS and MmBtaRS.
The largest differences between MaFRSA and other reported PylRS structures are localized in a six-residue loop that straddles β-strands β5 and β6 and contains the active-site residue Tyr206. In the MmPylRS/Pyl/AMP-PNP (PDB: 2ZCE)37 and WT MaPylRS apo structures (PDB: 6JP2)60, the β5–β6 or corresponding loop is either unstructured or in an open conformation, positioning Tyr206/Tyr384 away from the active site, respectively (Extended Data Fig. 5b). Among WT PylRS structures, the Tyr206/Tyr384-containing loop exists in the closed conformation only in the structure of MmPylRS bound to the reaction product Pyl-adenylate (PDB: 2Q7H)61. In this structure, Tyr384 accepts and donates a hydrogen bond to the Pyl-adenylate α-amine and pyrrole nitrogen, respectively, and forms a hydrophobic lid over the active site. In both chains of substrate-bound MaFRSA, the non-reacting carboxylate of m-CF3-2-BMA forms similar hydrogen bonds with Tyr206. These hydrogen bonds effectively close the β5–β6 loop to form a hydrophobic lid over the active site, which may contribute to the high acylation activity observed for m-CF3-2-BMA with MaFRSA. We note that the β5–β6 loop in chain A exhibits lower B-factors than in chain B, indicating tighter binding and providing further evidence that chain A represents the more active binding mode of m-CF3-2-BMA (Extended Data Fig. 5c,d). The two alternative poses of m-CF3-2-BMA in chains A and B correspond to a rotation of ~120° about the Cα–Cβ bond of the substrate that largely maintains interactions with the m-CF3-2-BMA side chain but alters the placement of the reacting carboxylate. This observation emphasizes the dominant stabilizing role of the main-chain hydrogen bonds provided by the backbone amides of Leu121 and Ala122.
In vitro translation initiation via codon skipping
We performed in vitro translation experiments to verify that the uniquely acylated derivatives of Ma-tRNAPyl produced using the MaPylRS variants are effectively shuttled to and accommodated by the E. coli ribosome. Whereas the E. coli initiator tRNAfMet has been engineered into a substrate for MjTyrRS variants to introduce non-canonical l-α-amino acids at the protein N terminus55, Ma-tRNAPyl lacks the key sequence elements for recognition by E. coli initiation factors, precluding its use for initiation in vivo39,63. It has been reported64 that in the absence of methionine, in vitro translation can begin at the second codon of the mRNA template, a phenomenon we refer to as ‘codon skipping’. We took advantage of codon skipping and a commercial in vitro translation kit to evaluate whether Ma-tRNAPyl enzymatically charged with monomers 13–15 would support translation initiation.
To avoid competition with release factor 1 at UAG codons, the anticodon of Ma-tRNAPyl was mutated to ACC (Ma-tRNAPyl-ACC) to recode a glycine GGT codon at position 2 in the mRNA template. To maximize yields of acyl-tRNA, we increased the aaRS/tRNA ratio to 1:2 (monomers 7, 14 and 15) or 1:1 (monomer 13), extended the incubation time to 3 h and used the most active MaFRSx variant for each monomer. These modifications resulted in acyl-tRNA yields of 79% (7, MaFRS1), 13% (13, MaFRS2), 85% (14, MaFRS2) and 82% (15, MaFRS1). The acylated Ma-tRNAPyl-ACC was added with a DNA template encoding a short MGV-FLAG peptide (MGVDYKDDDDK; Fig. 6a) to a commercial in vitro translation kit (PURExpress Δ (aa, tRNA)). When methionine was excluded from the reaction mixture, translation was initiated at the second position by skipping the start codon to produce peptides with the sequence XVDYKDDDDK (X = 7, 13–15). Following FLAG-tag enrichment, LC–HRMS confirmed the initiation with monomers 7 and 13–15 (Fig. 6b). When the mass corresponding to the m/z = M + 2H (13–15) or m/z = M + 3H (7) charge state of each peptide was extracted from the total ion chromatogram (TIC), a clear peak was observed for each peptide. No such peak was observed in reactions that lacked either the DNA template or acyl-tRNA, confirming templated ribosomal initiation with α-thio acid 13, 2-benzylmalonic acid (14) and N-formyl-l-α-amino acid 15. Two peaks of identical mass were observed in the extracted ion chromatogram (EIC) when translation was initiated with 2-benzylmalonic acid (14), which we assigned to diastereomeric peptides resulting from acylation at either the pro-S or pro-R carboxylate. Combined with the multiple peaks present in the RNAse A assay with 2-benzylmalonic acids 17, 18 and 19 (Fig. 4b and Extended Data Fig. 1), as well as the two m-CF3-2-BMA conformations observed in the structure of MaFRSA (Fig. 5b), the two peptide products of identical mass generated in the in vitro translation reactions imply that MaFRSx enzymes can effectively acylate either of the two prochiral carboxylates of a malonic acid substrate.

a, Workflow for in vitro translation via codon skipping. b, EICs and mass spectra of peptide products obtained using Ma-tRNAPyl-ACC charged with monomers 7 and 13–15 by MaFRS1 (7 and 15) or MaFRS2 (13 and 14). The insets show the mass spectra of the major ions used to generate the EIC of the translated peptide initiated with the indicated monomer. The expected (exp) and observed (obs) m/z peaks in the mass spectra are as follows: l-Phe (7) (M + 3H): exp: 420.51906, obs: 420.52249; α-SH 13 (M + 2H): exp: 638.75554, obs: 638.75614; 2-BMA (14) (M + 2H): exp: 644.76442, obs: 644.76498; N-formyl-l-Phe (N-fPhe) 15 (M + 2H): exp: 644.27242, obs: 644.27167. c, Workflow for the in vivo incorporation of monomers 1, 2, 20 and 21 at position 200 of sfGFP. d, Intact protein mass spectra of sfGFP variants purified from DH10B cells co-expressing MaPylRS (top) or MaFRSA (bottom) in the presence of 1 mM BocK (1), α-OH-BocK (2), m-trifluoromethylphenylalanine (20) or α-OH-m-trifluoromethylphenylalanine (21). e, Fidelity (%) of sfGFP containing the indicated residue at position 200 when expressed in E. coli DH10B (i–vii) or DH10B ∆aspC ∆tyrB (viii and ix) using either terrific broth (i–v, viii and ix) or a defined media lacking glutamate (vi and vii). The fidelity was determined as described in Methods and Supplementary Fig. 39.
In vivo translation of ester-amide heteropolymers
We next sought to evaluate whether the ability of MaPylRS and MaFRSA to acylate tRNA with an expanded subset of monomers in vitro would support the translation of backbone-modified protein heteropolymers in vivo. As the yields of tRNAs acylated in vitro with N-methyl-α-amino acids and α-thio acids were low (Fig. 3), N-formyl-α-amino acids lack an appropriate nucleophile and the requirements for intra-PTC bond formation by α-carboxy (malonic) acids are unknown, we focused on the in vivo incorporation of α-OH-BocK (2) and α-OH-m-trifluoromethyl-l-phenylalanine (α-OH-meta-CF3-Phe, 21).
First, we modified established pUltra plasmids65 for aaRS/tRNA expression by adding a lac operator between the tRNA promoter and coding region to make tRNA expression inducible66. The resulting pMega plasmids were more easily propagated than the pUltra plamids67. Using the resulting pMega or modified pEVOL (ref. 68) plasmids, we performed experiments in E. coli DH10B cells transformed with one of two superfolder green fluorescent protein (sfGFP) reporter plasmids (pET22b-sfGFP-200TAG or pET22b-sfGFP-151TAG; Supplementary Fig. 36) encoding Ma-tRNAPyl and either MaPylRS or MaFRSA. The growth was supplemented with 1 mM BocK (1), α-OH-BocK (2), m-CF3-Phe (20) or α-OH-m-CF3-Phe (21), and the emission at 528 nm, close to the λmax for sfGFP, was assessed after 24 h. In most cases, DH10B cells harbouring a pMega plasmid produced two- to threefold higher levels of sfGFP fluorescence than those harbouring a pEVOL plasmid (Extended Data Fig. 6). In all but one case, the α-OH monomers led to approximately 1.5- to 2-fold lower sfGFP fluorescence than the α-NH2 monomers. The highest levels of sfGFP fluorescence were observed in those cases in which α-NH2 or α-OH monomers were encoded at position 200.
Preparative-scale growth was conducted using pET22b-sfGFP-200TAG and pMega-MaPylRS or pMega-MaFRSA, the top-performing plasmids in the plate reader assay, and the sfGFP products were isolated by metal affinity chromatography (Supplementary Fig. 37). As observed previously with WT MmPylRS, E. coli DH10B cells expressing MaPylRS and grown in the presence of 1 mM BocK (1) or α-OH-BocK (2) expressed an sfGFP variant whose mass corresponded to the presence of a single BocK side chain (Fig. 6d); analogous cells expressing MaFRSA and grown in the presence of 1 mM m-CF3-Phe (20) or α-OH-m-CF3-Phe (21) expressed an sfGFP variant whose mass corresponded to the presence of a single trifluoromethylphenylalanine side chain (Fig. 6d). Although the intact masses of the proteins grown in the presence of BocK (1) and m-CF3-Phe (20) differed from those grown in the presence of α-OH-BocK (2) or α-OH-m-CF3-Phe (21), the resolution was insufficient to unequivocally establish the ester/amide ratio. To characterize the products more rigorously, we digested the isolated sfGFP variants with GluC (Supplementary Fig. 38) and analysed the products by LC with tandem MS (LC–MS/MS; Fig. 6e(i)–(v) and Supplementary Fig. 39). This analysis revealed that the sfGFP produced in the presence of α-OH BocK (2) contained virtually only an ester linkage at position 200, whereas the sfGFP product generated in the presence of α-OH-m-CF3-Phe (21) contained an ester/amide ratio of 11:85 (Fig. 6e and Supplementary Table 5).
Certain α-hydroxy acids can be metabolized in E. coli to α-amino acids via a two-step oxidation–transamination process. Indeed, in classic work21, a DH10B strain lacking the transaminase genes aspC and tyrB was required to detect cytosolic accumulation of the α-OH analogue of tyrosine, 3-(4-hydroxyphenyl)lactic acid21. We thus repeated the preparative-scale growth of DH10B ∆aspC ∆tyrB harbouring pET22b-sfGFP-200TAG and pMega-MaFRSA supplemented with 1 mM m-CF3-Phe (20) or α-OH-m-CF3-Phe (21). For comparison, we also performed growth in DH10B using a defined media lacking glutamate, the amine donor for AspC and TyrB, in an attempt to minimize transaminase activity. The results of intact mass analysis of sfGFP isolated from DH10B ∆aspC ∆tyrB growth supplemented with α-OH-m-CF3-Phe (21) were again consistent with the expected products (Supplementary Figs. 37 and 39). Both strategies improved the fraction of ester product, as judged by GluC digestion of the intact isolated sfGFP; use of the defined media led to a 39:52 ester/amide ratio, whereas use of DH10B ∆aspC ∆tyrB led to an ester/amide ratio of 44:52 (Fig. 6e, Supplementary Fig. 39 and Supplementary Table 5). These studies reveal that while MaFRSA is sufficiently active in vivo to support the biosynthesis of a sequence-defined hetero-oligomer, more complex E. coli strains69,70 or alternative organisms71 may be required to generate ribosomal products containing multiple esters in high yield.
Discussion
Expanding and reprogramming the genetic code for the templated biosynthesis of sequence-defined heteropolymers demands orthogonal aaRS/tRNA pairs that accept non-l-α-amino acid substrates. Over 50 years ago, the PTC of the E. coli ribosome was reported to tolerate an α-hydroxy substituent in place of the α-amine of phenylalanine and biosynthesize a polyester in vitro3. More recent in vitro studies have broadened the scope of WT PTC reactivity to include diverse nucleophilic heteroatoms in place of the α-amine13,14,16,17. However, with the exception of substrates carrying an α-hydroxy substituent21,22,23,24,27, none of these non-l-α-amino acid monomers is a substrate for any known orthogonal aaRS. The challenge is that most aaRSs of known structure simultaneously engage both the substrate α-amine and α-carboxylate moieties via multiple main-chain and side-chain hydrogen bonds to position the α-carboxylate for adenylation and acylation. These well-conserved hydrogen bond networks complicate the engineering of new enzymes that accept substrates with alternative amine and/or carboxylate orientations or conformations (such as β-amino acids or aminobenzoic acids), or those whose α-substituents are large and/or electrostatically distinctive (such as malonates, α-thio acids and N-acyl-α-amino acids).
The water-mediated hydrogen bond network in the active site of MmPylRS has been shown to facilitate the recognition of substrates with conservative substitutions (α-OH, α-H and α-NHCH3) of the α-amino group22. Here we have expanded the scope of monomers recognized and processed by PylRS variants to include α-thio acids and N-formyl-l-α-amino acids, as well as those that carry an α-carboxy functional group in place of the α-amine, that is, prochiral malonic acids with protein-like side chains. Monomers containing α-thio, N-formyl-l-α-amino and α-carboxy substituents in place of the α-amine can be incorporated into polypeptides at the N terminus by the native E. coli translational apparatus; those with an α-hydroxy substituent can be introduced into proteins in vivo, albeit in a side-chain- and position-specific manner. Ester-containing biopolymers produced at scale could generate biomaterials that change shape and self-cleave in a pH- and/or environment-selective manner.
Although thioesters and malonic acids are ubiquitous intermediates in polyketide and fatty acid biosynthesis72,73, as far as we know, aaRS enzymes that act on α-thio or α-carboxy acids were previously unknown, and tRNAs acylated with a polyketide precursor represent distinct chemical species. Such tRNAs could forge a link between ribosomal translation and the assembly-line PKSs74 responsible for protein and polyketide biosynthesis, respectively. Combined with synthetic genomes69,70, ribosomes capable of carbon–carbon bond formation would set the stage for the template-driven biosynthesis of unique hybrid biomaterials and sequence-defined polyketide-peptide oligomers, such as those produced by PKS-non-ribosomal peptide synthetase biosynthetic modules.
Methods
Expression and purification of MaPylRS, MaFRS1, MaFRS2 and MaFRSA for biochemical analysis
The plasmids used to express WT MaPylRS (pET32a-MaPylRS) and MaFRS1 (pET32a-MaFRS1) were constructed by inserting synthetic double-stranded DNA (dsDNA) fragments (Supplementary Table 1) into the NdeI-NdeI cut sites of a pET32a vector using the Gibson method75. pET32a-MaFRS2 and pET32a-MaFRSA were constructed from pET32a-MaFRS1 using a Q5 Site-Directed Mutagenesis Kit (NEB). Primers RF31 and RF32, and RF32 and RF33 (Supplementary Table 1) were used to construct pET32a-MaFRS2 and pET32a-MaFRSA, respectively. The sequences of the plasmids spanning the inserted regions were confirmed via Sanger sequencing at the University of California Berkeley DNA Sequencing Facility using T7 forward and reverse (T7 F and T7 R) primers (Supplementary Table 1), and the complete sequence of each plasmid was confirmed by the Massachusetts General Hospital Center for Computational and Integrative Biology DNA Core.
Chemically competent cells were prepared by following a modified published protocol76. Briefly, 5 ml Luria-Bertani (LB) medium was inoculated using a freezer stock of BL21-Gold (DE3)pLysS cells. The following day, 50 ml LB was inoculated with 0.5 ml of the culture from the previous day and incubated at 37 °C with shaking at 200 r.p.m. until the culture reached an optical density at 600 nm (OD600) in the range of 0.3–0.4. The cells were collected by centrifugation at 4,303g for 20 min at 4 °C. The cell pellet was resuspended in 5 ml of sterile filtered TSS solution (10% (wt/v) polyethylene glycol 8,000, 30 mM MgCl2, 5% (v/v) dimethylsulfoxide in 25 g l−1 LB). The chemically competent cells were portioned into 100 µl aliquots in 1.5 ml microcentrifuge tubes, flash frozen in liquid N2 and stored at −80 °C until use. The following protocol was used to transform plasmids into chemically competent cells: 20 µl KCM solution (500 mM KCl, 150 mM CaCl2 and 250 mM MgCl2) was added to a 100 µl aliquot of cells held on ice along with approximately 200 ng of the requisite plasmid and water to a final volume of 200 µl. The cells were incubated on ice for 30 min and then heat-shocked by placing them for 90 s in a water bath heated to 42 °C. Immediately after heat shock, the cells were placed on ice for 2 min, after which 800 µl LB was added. The cells were then incubated at 37 °C with shaking at 200 r.p.m. for 60 min. The cells were plated onto LB agar plates with the appropriate antibiotic and incubated overnight at 37 °C.
The plasmids used to express WT MaPylRS, MaFRS1, MaFRS2, and MaFRSA were transformed into BL21-Gold (DE3)pLysS chemically competent cells and plated onto LB agar plates supplemented with 100 µg ml−1 carbenicillin. Colonies were picked the following day and used to inoculate 10 ml LB supplemented with 100 µg ml−1 carbenicillin. The cultures were incubated overnight at 37 °C with shaking at 200 r.p.m. The following day, the 10 ml cultures were used to inoculate 1 l LB supplemented with 100 µg ml−1 carbenicillin in 4-l baffled Erlenmeyer flasks. The cultures were incubated at 37 °C with shaking at 200 r.p.m. for 3 h until they reached an OD600 of 0.6–0.8. At this point, isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM and the incubation was continued for 6 h at 30 °C with shaking at 200 r.p.m. The cells were collected by centrifugation at 4,303g for 20 min at 4 °C and the cell pellets stored at −80 °C until the expressed protein was purified as described below.
The following buffers were used for protein purification: wash buffer: 50 mM sodium phosphate (pH 7.4), 500 mM NaCl, 20 mM β-mercaptoethanol, 25 mM imidazole; elution buffer: 50 mM sodium phosphate (pH 7.4), 500 mM NaCl, 20 mM β-mercaptoethanol, 100 mM imidazole; storage buffer: 100 mM HEPES-K (pH 7.2), 100 mM NaCl, 10 mM MgCl2, 4 mM dithiothreitol (DTT), 20% (v/v) glycerol. One cOmplete Mini EDTA-free protease inhibitor tablet was added to the wash and elution buffers immediately before use. To isolate protein, cell pellets were resuspended in wash buffer (5 ml per g cells). The resultant cell paste was lysed at 4 °C by sonication (Branson Sonifier 250) over 10 cycles of 30 s sonication followed by 30 s manual swirling. The lysate was centrifuged at 4,303g for 10 min at 4 °C to separate the soluble and insoluble fractions. The soluble lysate was incubated at 4 °C with 1 ml Ni nitrilotriacetic acid (Ni-NTA) agarose resin (washed with water and equilibrated with wash buffer) for 2 h. The lysate–resin mixture was added to a 65 g RediSep disposable sample load cartridge (Teledyne ISCO) and allowed to drain at room temperature. The protein-bound Ni-NTA agarose resin was then washed three times with 10 ml aliquots of wash buffer. The protein was eluted from the Ni-NTA agarose resin by rinsing the resin three times with 10 ml elution buffer. The elution fractions were pooled and concentrated using a 10 kDa molecular weight cut-off (MWCO) Amicon Ultra-15 centrifugal filter unit (4,303g, 4 °C). The protein was then buffer-exchanged into the storage buffer using the same centrifugal filter unit until the imidazole concentration was less than 5 µM. The protein was dispensed into 20 µl single-use aliquots and stored at −80 °C for up to 8 months. The protein concentration was measured using the Bradford assay77. The yields were between 8 and 12 mg l−1. Proteins were analysed by SDS–PAGE (Supplementary Fig. 2) using Any kD Mini-PROTEAN TGX precast protein gels (BioRad). The gels were run at 200 V for 30 min.
Proteins were analysed by LC–MS to confirm their identity (Supplementary Fig. 2). The samples analysed by MS were resolved using a Poroshell StableBond 300 C8 column (2.1 mm × 75 mm, 5 µm; Agilent, 660750-906) with a 1290 Infinity II ultra-high-performance liquid chromatograph (UHPLC; Agilent, G7120AR). The mobile phases used for separation were 0.1% formic acid in water (mobile phase A) and 100% acetonitrile (mobile phase B), and the flow rate was 0.4 ml min−1. After an initial hold at 5% B for 0.5 min, the proteins were eluted using a linear gradient from 5% to 75% B for 9.5 min, a linear gradient from 75% to 100% B for 1 min, a hold at 100% B for 1 min, a linear gradient from 100% to 5% B for 3.5 min and finally a hold at 5% B for 4.5 min. The protein masses were analysed by LC–HRMS using a 6530 Q-TOF AJS-ESI (Agilent, G6530BAR) instrument. The following parameters were used: gas temperature = 300 °C, drying gas flow = 12 l min−1, nebulizer pressure = 35 psi, sheath gas temperature = 350 °C, sheath gas flow = 11 l min−1, fragmentor voltage = 175 V, skimmer voltage = 65 V, peak-to-peak voltage (Vpp) = 750 V, capillary voltage (Vcap) = 3,500 V, nozzle voltage = 1,000 V and collection rate = 3 spectra s−1.
Analytical size exclusion chromatography (SEC; Supplementary Fig. 2) was performed on an ÄKTA Pure 25 instrument. A flow rate of 0.5 ml min−1 was used for all steps. A Superdex 75 Increase 10/300 GL column (stored and operated at 4 °C) was washed with 1.5 column volumes of degassed, sterile, filtered MilliQ water. The column was equilibrated in 1.5 column volumes of SEC buffer: 100 mM HEPES (pH 7.2), 100 mM NaCl, 10 mM MgCl2, 4 mM DTT. Approximately 800 µg of protein in 250 µl SEC buffer was loaded into a 500 µl capillary loop. The sample loop was washed with 2.0 ml SEC buffer as the sample was injected onto the column. The sample was eluted in 1.5 column volumes of SEC buffer and analysed by UV spectrophotometry at 280 nm.
Transcription and purification of tRNAs
The DNA template used for transcribing Ma-tRNAPyl (ref. 39) was prepared by annealing and extending the single-stranded DNA oligonucleotides Ma-PylT-F and Ma-PylT-R (2 mM; Supplementary Table 1) using OneTaq 2x Master Mix (NEB). The annealing and extension process was performed using the following protocol on a thermocycler (BioRad, C1000 Touch): 94 °C for 30 s, 30 cycles of 94 °C for 20 s, 53 °C for 30 s and 68 °C for 60 s, and finally 68 °C for 300 s. Following the extension, the reaction mixture was supplemented with sodium acetate (pH 5.2) to a final concentration of 300 mM, washed once with 1:1 (v/v) acid phenol–chloroform, twice with chloroform and the dsDNA product precipitated by addition of ethanol to a final concentration of 71%. The pellet was resuspended in water and the concentration of dsDNA determined using a NanoDrop ND-1000 device (Thermo Scientific). The template starts with a single C preceding the T7 promoter, which increases the yields of T7 transcripts78. The penultimate residue of Ma-PylT-R carries a 2′-methoxy modification, which reduces non-templated nucleotide addition by T7 RNA polymerase during in vitro transcription79.
Ma-tRNAPyl was transcribed in vitro using a modified version of a published procedure80. Transcription reactions (25 µl) contained the following components: 40 mM Tris-HCl (pH 8.0), 100 mM NaCl, 20 mM DTT, 2 mM spermidine, 5 mM ATP, 5 mM cytidine triphosphate, 5 mM guanosine triphosphate, 5 mM uridine triphosphate, 20 mM guanosine monophosphate, 0.2 mg ml−1 bovine serum albumin, 20 mM MgCl2, 12.5 ng µl−1 DNA template and 0.025 mg ml−1 T7 RNA polymerase. The reaction mixtures were incubated at 37 °C in a thermocycler for 3 h. Four 25 µl reactions were pooled, and then sodium acetate (pH 5.2) was added to a final concentration of 300 mM in a volume of 200 µl. The transcription reaction mixtures were extracted once with 1:1 (v/v) acid phenol–chloroform, washed twice with chloroform and the tRNA product precipitated by adding ethanol to a final concentration of 71%. After precipitation, the tRNA pellet was resuspended in water and incubated with 8 U of RQ1 RNAse-free DNAse (Promega) at 37 °C for 30 min according to the manufacturer’s protocol. The tRNA was then washed with acid phenol–chloroform and chloroform as described above, precipitated and resuspended in water. To remove small molecules, the tRNA was further purified using a Micro Bio-Spin P-30 gel column, Tris buffer (RNase-free; BioRad) after first exchanging the column buffer with water according to the manufacturer’s protocol. The tRNA was precipitated once more, resuspended in water, quantified using a NanoDrop ND-1000 device, aliquoted and stored at −20 °C.
tRNA was analysed by urea–PAGE (Supplementary Fig. 2) using 10% Mini-PROTEAN Tris-borate-ethylenediaminetetraacetic acid-urea gel (BioRad). The gels were run at 120 V for 30 min and then stained with SYBR Safe gel stain (Thermo Fisher) for 5 min before imaging. Ma-tRNAPyl was analysed by LC–MS to confirm its identity. Samples were resolved on an ACQUITY UPLC BEH C18 column (130 Å, 1.7 µm, 2.1 mm × 50 mm, 60 °C; Waters, 186002350) using an ACQUITY UPLC I-Class PLUS instrument (Waters, 186015082). The mobile phases used were 8 mM triethylamine, 80 mM hexafluoroisopropanol and 5 µM EDTA (free acid) in 100% MilliQ water (mobile phase A) and 4 mM triethylamine, 40 mM hexafluoroisopropanol and 5 µM EDTA (free acid) in 50% MilliQ water–50% methanol (mobile phase B). The analysis was performed at a flow rate of 0.3 ml min−1 and began with mobile phase B at 22%, increasing linearly to 40% B over 10 min, followed by a linear gradient from 40% to 60% B for 1 min, a hold at 60% B for 1 min, a linear gradient from 60% to 22% B over 0.1 min and then a hold at 22% B for 2.9 min. The mass of the RNA was analysed by LC–HRMS with a Xevo G2-XS Tof instrument (Waters, 186010532) in negative ion mode with the following parameters: capillary voltage = 2,000 V, sampling cone = 40, source off-set = 40, source temperature = 140 °C, desolvation temperature = 20 °C, cone gas flow = 10 l h−1, desolvation gas flow = 800 l h−1 and collection rate = 1 spectrum s−1. The expected masses of the oligonucleotide products were calculated using the AAT Bioquest RNA Molecular Weight Calculator. Deconvoluted mass spectra were obtained using the MaxEnt software (Waters).
Procedure for RNAse A assays
The reaction mixtures (25 µl) used to acylate tRNA contained the following components: 100 mM HEPES-K (pH 7.5), 4 mM DTT, 10 mM MgCl2, 10 mM ATP, 0–10 mM substrate, 0.1 U E. coli inorganic pyrophosphatase (NEB), 25 µM Ma-tRNAPyl and 2.5 µM enzyme (MaPylRS, MaFRS1, MaFRS2 or MaFRSA). The reaction mixtures were incubated at 37 °C in a dry air incubator for 2 h. The tRNA samples from the enzymatic acylation reactions were quenched with 27.5 µl RNAse A solution (1.5 U µl−1 RNAse A (MilliporeSigma) and 200 mM sodium acetate, pH 5.2) and incubated for 5 min at room temperature. The proteins were then precipitated by the addition of 50% trichloroacetic acid (Sigma-Aldrich) to a final concentration of 5%. After precipitating protein at −80 °C for 30 min, insoluble material was removed by centrifugation at 21,300g for 10 min at 4 °C. The soluble fraction was then transferred to autosampler vials, kept on ice until immediately before LC–MS analysis and returned to ice immediately afterwards.
The samples analysed by MS were resolved using a Zorbax Eclipse XDB-C18 RRHD column (2.1 mm × 50 mm, 1.8 μm, room temperature; Agilent, 981757-902) fitted with a guard column (Zorbax Eclipse XDB-C18, 2.1 mm ×5 mm, 1.8 µm; Agilent, 821725-903) and a 1290 Infinity II UHPLC (Agilent, G7120AR). The mobile phases used were 0.1% formic acid in water (mobile phase A) and 100% acetonitrile (mobile phase B). The analysis was performed at a flow rate of 0.7 ml min−1 and began with mobile phase B held at 4% for 1.35 min, followed by a linear gradient from 4% to 40% B over 1.25 min, a linear gradient from 40% to 100% B over 0.4 min, a linear gradient from 100% to 4% B over 0.7 min and then finally B held at 4% for 0.8 min. Acylation was confirmed by correctly identifying the exact masses of the 2′- and 3′-acyl-adenosine products corresponding to the substrate tested in the EICs recorded by LC–HRMS using a 6530 Q-TOF AJS-ESI instrument (Agilent, G6530BAR). The following parameters were used: fragmentor voltage = 175 V, gas temperature = 300 °C, gas flow = 12 l min−1, sheath gas temperature = 350 °C, sheath gas flow = 12 l min−1, nebulizer pressure = 35 psi, skimmer voltage =65 V, Vcap = 3,500 V and collection rate = 3 spectra s−1. The expected exact masses of the acyl-adenosine nucleosides (Supplementary Table 2) were calculated using ChemDraw 19.0 and extracted from the TICs (±100 ppm).
Procedure for determining aminoacylation yields using intact tRNA MS
Enzymatic tRNA acylation reactions (25 µl) were performed as described in Procedure for RNAse A assays section. Sodium acetate (pH 5.2) was added to the acylation reactions to a final concentration of 300 mM in a volume of 200 µl. The reaction mixtures were then extracted once with a 1:1 (v/v) mixture of acidic phenol (pH 4.5) and chloroform and washed twice with chloroform. After extraction, the acylated tRNA was precipitated by adding ethanol to a final concentration of 71% and incubation at −80 °C for 30 min, followed by centrifugation at 21,300g for 30 min at 4 °C. After removing the supernatant, acylated tRNA was resuspended in water and kept on ice for analysis.
The tRNA samples from the enzymatic acylation reactions were analysed by LC–MS as described in Transcription and purification of tRNAs section. Because the unacylated tRNA peak in each TIC contained tRNA species that could not be enzymatically acylated (primarily tRNAs that lack the 3′-terminal adenosine81), simple integration of the acylated and non-acylated peaks in the absorbance at 260 nm (A260) chromatogram could not accurately quantify the acylation yield. To accurately quantify the acylation yield, we used the following procedure. For each sample, mass data were collected between m/z = 500 and 2,000. A subset of the mass data collected defined as the raw MS deconvolution range (Supplementary Figs. 3–35) was used to produce the deconvoluted mass spectra (Supplementary Figs. 3–35). The raw MS deconvolution range of each macromolecule species contained multiple peaks corresponding to different charge states of that macromolecule. Within the raw mass spectrum deconvolution range we identified the most abundant charge state peak in the raw mass spectrum of each tRNA species (unacylated, monoacylated and diacylated tRNA), which is identified as the major ion by an asterisk in Supplementary Figs. 3c–35c. To quantify the relative abundance of each species, the exact mass of the major ions (±0.3000 Da) was extracted from the TIC to produce the EICs (Supplementary Figs. 3–35). The EICs were integrated and the areas of the peaks that aligned with the correct peaks in the TIC (as determined from the deconvoluted mass spectrum) were used to quantify the yields (Supplementary Table 3). For the malonic acid substrates, the integrated peak areas of the EICs of both the malonic acid and decarboxylation products were added together to determine the overall acylation yield. Each sample was injected three times; the chromatograms and spectra in Supplementary Figs. 3–35 are representative, and the yields shown in Supplementary Table 3 are averages of the three injections. The expected masses of the oligonucleotide products were calculated using the AAT Bioquest RNA Molecular Weight Calculator, and the molecular masses of the small molecules added to them were calculated using ChemDraw 19.0. All the masses identified in the mass spectra are summarized in Supplementary Data 1.
Malachite green assay to monitor adenylation
Enzymatic adenylation reactions were monitored using malachite green following a previous protocol with modifications56. Each adenylation reaction (60 µl) contained the following components: 200 mM HEPES-K (pH 7.5), 4 mM DTT, 10 mM MgCl2, 0.2 mM ATP, 0–10 mM substrate, 4 U m−1 E. coli inorganic pyrophosphatase (NEB) and 2.5 µM enzyme (MaFRS1 or MaFRSA). The adenylation reactions were incubated at 37 °C in a dry air incubator. Aliquots (10 µl) were withdrawn after 0, 5, 10, 20 and 30 min and quenched by addition to an equal volume of 20 mM EDTA (pH 8.0) on ice. Once all aliquots had been withdrawn, 80 µl malachite green solution (Echelon Biosciences) was added to each aliquot and the mixture incubated at room temperature for 30 min. After shaking for 30 s to remove bubbles, the absorbance at 620 nm was measured on a Synergy HTX plate reader (BioTek). The absorbance was then converted to phosphate concentration using a phosphate calibration curve (0–100 µM) and plotted against time to determine the turnover number.
Structure determination
The following synthetic dsDNA sequence was cloned upstream of MaFRSA (MaPylRS N166A, V168A) into pET32a-MaFRSA by Gibson assembly75 and used for subsequent crystallographic studies: GSS linker-6xHis-SSG linker-thrombin site-MaFRSA (Supplementary Table 1). The sequence of the pET32a-6xHis-thrombin-MaFRSA plasmid was confirmed by Sanger sequencing at Genewiz using the primers T7 F and T7 R (Supplementary Table 1). The procedure used to express and purify MaFRSA for crystallography using pET32a-6xHis-thrombin-MaFRSA was adapted from a protocol used to express and purify WT MaPylRS (ref. 60). BL21-Gold (DE3)pLysS competent cells (Agilent) were transformed with pET32a-6xHis-thrombin-MaFRSA and grown in TB media at 37 °C. Protein expression was induced at an OD600 of 1.2 with 1 mM IPTG. The temperature was lowered to 20 °C and growth was allowed to continue overnight. The cells were pelleted for 1 h at 4,300g and then resuspended in lysis buffer (50 mM potassium phosphate (pH 7.4), 25 mM imidazole, 500 mM sodium chloride, 5 mM β-mercaptoethanol and 1 cOmplete Mini EDTA-free protease inhibitor tablet). The cells were then lysed by homogenization (Avestin Emulsiflex C3). After centrifugation for 1 h at 10,000g, the clarified lysate was bound to TALON metal affinity resin (Takara Bio) for 1 h at 4 °C, washed with additional lysis buffer and eluted with elution buffer (50 mM potassium phosphate (pH 7.4), 500 mM imidazole, 500 mM sodium chloride and 5 mM β-mercaptoethanol). The eluate was dialysed overnight at 4 °C into cleavage buffer (40 mM potassium phosphate (pH 7.4), 100 mM NaCl and 1 mM DTT) and then incubated overnight at room temperature with thrombin protease on a solid agarose support (MilliporeSigma). Following cleavage, the protein was passed over additional TALON resin to remove the 6xHis tag and dialysed overnight at 4 °C into sizing buffer (30 mM potassium phosphate (pH 7.4), 200 mM NaCl and 1 mM DTT). The protein was concentrated and loaded onto a HiLoad 16/600 Superdex 200 pg column (Cytiva Life Sciences) equilibrated with sizing buffer on an ÄKTA Pure 25 fast-liquid chromatograph. Purified MaFRSA was dialysed into storage buffer (10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM MgCl2 and 10 mM β-mercaptoethanol), concentrated to 20 mg ml−1, aliquoted and flash-frozen for crystallography (Supplementary Fig. 2).
The initial crystallization screening conditions were adapted from Seki et al.60. Crystals were grown by hanging-drop vapour diffusion in 24-well plates. Then, 25 µl of 100 mM m-CF3-2-BMA (pH ≈ 7) was added to 1.5 ml microcentrifuge tubes and the water was removed by evaporation. The dried aliquots were then resuspended at a concentration of 100 mM with MaFRSA in storage buffer at three concentrations (6.9, 12.3 and 19.2 mg ml−1) and 10 mM AMP-PNP lithium salt hydrate. The protein–substrate solution (1 µl) was mixed in a 1:1 ratio with the reservoir solution (1 µl) containing 10 mM Tris-HCl (pH 7.4) and 26% polyethylene glycol 3,350, and then incubated above 1 ml of reservoir solution at 18 °C. Crystals with an octahedral shape appeared within 1 week. The crystals were plunged into liquid nitrogen to freeze with no cryoprotectant.
Data were collected at the Advanced Light Source beamline 8.3.1 at 100 K using a wavelength of 1.11583 Å. Data collection and refinement statistics are presented in Supplementary Table 4. The diffraction data were indexed and integrated with XDS82, and then merged and scaled with Pointless83 and Aimless84. The crystals were formed in the space group I4 with unit cell dimensions 108.958, 108.958 and 112.26 Å. The structure was solved by molecular replacement with Phaser85 using a single chain of the WT apo structure of MaPylRS (PDB code: 6JP2)60 as the search model. Two copies of MaFRSA were present in the asymmetric unit. The model was improved with iterative cycles of manual model building in Coot (ref. 86) alternating with refinement in Phenix87,88 using data with a resolution of up to 1.8 Å. Structural analysis and figures were generated using Pymol (version 2.4.2)89.
In vitro translation initiation
The Ma-tRNAPyl-ACC dsDNA template was prepared as described in Transcription and purification of tRNAs section using the primers Ma-PylT-ACC F and Ma-PylT-ACC R (Supplementary Table 1). Ma-tRNAPyl-ACC was also transcribed, purified and analysed as described previously. The enzymatic tRNA acylation reactions (150 µl) were performed as described in Procedure for RNAse A assays section with slight modifications. The enzyme concentration was increased to 12.5 µM (monomers 7, 14 and 15) or 25 µM (monomer 13) and the incubation time was increased to 3 h at 37 °C. Sodium acetate (pH 5.2) was added to the acylation reactions to a final concentration of 300 mM in a volume of 200 µl. The reactions were then extracted once with a 1:1 (v/v) mixture of acidic phenol (pH 4.5) and chloroform and washed twice with chloroform. After extraction, the acylated tRNA was precipitated by adding ethanol to a final concentration of 71% and incubation at −80 °C for 30 min, followed by centrifugation at 21,300g for 30 min at 4 °C. The acylated tRNAs were resuspended in water to a concentration of 307 µM immediately before in vitro translation.
Templates for the expression of MGVDYKDDDDK were prepared by annealing and extending the oligonucleotides MGVflag-1 and MGVflag-2 using Q5 High-Fidelity 2x Master Mix (NEB; Supplementary Table 1). The annealing and extension procedure was performed using the following protocol on a thermocycler (BioRad C1000 Touch): 98 °C for 30 s, 10 cycles of 98 °C for 10 s, 55 °C for 30 s and 72 °C for 45 s, 10 cycles of 98 °C for 10 s, 67 °C for 30 s and 72 °C for 45 s, and finally 72 °C for 300 s. Following the extension, the reaction mixture was supplemented with sodium acetate (pH 5.2) to a final concentration of 300 mM, extracted once with a 1:1 (v/v) mixture of basic phenol (pH 8.0) and chloroform, and washed twice with chloroform. The dsDNA product was precipitated by the addition of ethanol to a final concentration of 71% and incubation at −80 °C for 30 min, followed by centrifugation at 21,300g for 30 min at 4 °C. The dsDNA pellets were washed once with 70% (v/v) ethanol, resuspended in 10 mM Tris-HCl (pH 8.0) to a concentration of 500 ng µl−1 and stored at −20 °C until use in translation.
In vitro transcription/translation by codon skipping of the short FLAG tag-containing peptides X-Val-Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys (XV-Flag, where X = 7, 13, 14 or 15) was carried out using the PURExpress Δ (aa, tRNA) Kit (NEB, E6840S) according to a previous protocol with slight modifications17. The XV-Flag peptides were produced from the following reaction mixtures (12.5 µl): solution A (∆tRNA and ∆aa; 2.5 µl), amino acid stock mix (33 mM l-valine, 33 mM l-aspartic acid, 33 mM l-tyrosine and 33 mM l-lysine; 1.25 µl), tRNA solution (1.25 µl), solution B (3.75 µl), 250 ng dsDNA MGVDYKDDDDK template (0.5 µl) and Ma-tRNAPyl-ACC acylated with 7, 13, 14 or 15 (3.25 µl). The reactions were incubated in a thermocycler (BioRad C1000 Touch) at 37 °C for 2 h and quenched on ice.
The translated peptides were purified from the in vitro translation reaction mixtures by enrichment using anti-FLAG M2 magnetic beads (MilliporeSigma) according to the manufacturer’s protocol with slight modifications. For each peptide, 10 µl of a 50% (v/v) suspension of magnetic beads was used. The supernatant was pipetted from the beads on a magnetic manifold. The beads were then washed twice by incubating with 100 µl TBS buffer (150 mM NaCl and 50 mM Tris-HCl (pH 7.6)) for 10 min at room temperature and then removing the supernatant each time using a magnetic manifold. The in vitro translation reaction mixtures were added to the beads and incubated at room temperature for 30 min with periodic agitation. The beads were washed again three times with 100 µl TBS as described above. The peptides were eluted upon incubation with 12.5 µl of 0.1 M glycine·HCl (pH 2.8) for 10 min. The supernatant was transferred to vials and kept on ice for analysis.
The purified peptides were analysed according to a previously reported protocol17. The supernatant was analysed on an ZORBAX Eclipse XDB-C18 column (1.8 μm, 2.1 mm × 50 mm, room temperature; Agilent) using a 1290 Infinity II UHPLC (Agilent, G7120AR). The following protocol was used for separation: an initial hold at 95% solvent A (0.1% formic acid in water) and 5% solvent B (acetonitrile) for 0.5 min, followed by a linear gradient from 5% to 50% solvent B for 4.5 min at a flow rate of 0.7 ml min−1. The peptides were identified by LC–HRMS using a 6530 Q-TOF AJS-ESI instrument (Agilent, G6230BAR). The following parameters were used: fragmentor voltage = 175 V, gas temperature = 300 °C, gas flow rate = 12 l min−1, sheath gas temperature = 350 °C, sheath gas flow rate = 11 l min−1, nebulizer pressure = 35 psi, skimmer voltage = 5 V, Vcap = 3,500 V and collection rate = 3 spectra s−1. The expected exact masses of the major charge state of each peptide were calculated using ChemDraw 19.0 and extracted from the TICs (±100 ppm).
Plasmids used for in vivo studies
The plasmids used to express WT sfGFP (pET22b-T5/lac-sfGFP) and 151TAG-sfGFP (pET22b-T5/lac-sfGFP-151TAG) in E. coli have been described previously90. pET22b-T5/lac-sfGFP-200TAG was constructed from pET22b-T5/lac-sfGFP using a Q5 Site-Directed Mutagenesis Kit (NEB) with primers CS43 and CS44 (Supplementary Table 1). The synthetase/tRNA plasmid for WT MaPylRS (pMega-MaPylRS) was constructed by inserting a synthetic dsDNA fragment (pMega-MaPylRS) (Supplementary Table 1) into the NotI-XhoI cut sites of a pUltra vector65 by Gibson assembly75 using NEBuilder HiFi DNA Assembly Master Mix (NEB). pMega-MaFRSA was constructed by inserting a synthetic dsDNA fragment (made by annealing primers RF48 and RF49) after inverse PCR of pMega-MaPylRS with primers RF61 and RF62 (Supplementary Table 1) using Gibson assembly75. The sequences of the plasmids spanning the inserted regions were confirmed via Sanger sequencing at the University of California Berkeley DNA Sequencing Facility using primers T7 F and T7 R (Supplementary Table 1), and the complete sequence of each plasmid was confirmed by full-plasmid sequencing at Primordium Labs.
Plate reader analysis of sfGFP expression
E. coli DH10B chemically competent cells were transformed with pET22b-T5/lac-sfGFP-200TAG and either pMega-MaPylRS or pMega-MaFRSA. Colonies were picked and grown overnight in LB with the appropriate antibiotics. The following day, the OD600 of the overnight culture was measured, and all cultures were diluted with LB to an OD600 of 0.10 to generate a seed culture. A monomer cocktail was prepared in LB supplemented with 2 mM IPTG, 2 mM monomer 1, 2, 20 or 21, and the appropriate antibiotics at two times the final concentration (200 µg ml−1 carbenicillin and 100 µg ml−1 spectinomycin). In a 96-well plate (Corning 3904), 100 µl of the seed culture was combined with 100 µl of each monomer cocktail to bring the starting OD600 to 0.05 and half the concentration of the monomer cocktail. The 96-well plate was sealed with a Breathe-Easy sealing membrane (Diversified Biotech) and loaded into a Synergy HTX plate reader (BioTek). The plate was incubated at 37 °C for 24 h with continuous shaking. Two readings were made at 10 min intervals, that is, the absorbance at 600 nm, to measure cell density, and sfGFP fluorescence with excitation at 485 nm and emission at 528 nm.
Expression and purification of sfGFP variants
The plasmids used to express WT sfGFP and sfGFP-200TAG were co-transformed with pMega-MaPylRS or pMega-MaFRSA into DH10B or DH10B ΔaspC ΔtyrB chemically competent cells and plated onto LB agar plates supplemented with 100 µg ml−1 carbenicillin and 100 µg ml−1 spectinomycin. Colonies were picked the following day and used to inoculate 10 ml LB supplemented with 100 µg ml−1 carbenicillin and 100 µg ml−1 spectinomycin. The cultures were incubated overnight at 37 °C with shaking at 200 r.p.m. The following day, 1 ml of each culture was used to inoculate 100 ml TB or defined media (adapted from a published protocol55 with glutamate excluded and 19 other amino acids at 200 µg ml−1) supplemented with 100 µg ml−1 carbenicillin and 100 µg ml−1 spectinomycin in 250-ml baffled Erlenmeyer flasks. The cultures were incubated at 37 °C with shaking at 200 r.p.m. for ~4 h until they reached an OD600 of 1.0–1.2. At this point, IPTG was added to a final concentration of 1 mM and incubation was continued overnight at 37 °C with shaking at 200 r.p.m. The cells were collected by centrifugation at 4,303g for 20 min at 4 °C.
sfGFP variants were purified according to a previously published protocol71. The following buffers were used for protein purification: lysis/wash buffer: 50 mM sodium phosphate (pH 8), 300 mM NaCl and 20 mM imidazole; elution buffer: 50 mM sodium phosphate (pH 8) and 250 mM imidazole; storage buffer: 50 mM sodium phosphate (pH 7), 250 mM NaCl and 1 mM DTT. One cOmplete Mini EDTA-free protease inhibitor tablet was added to the wash and elution buffers immediately before use. To isolate protein, cell pellets were resuspended in 10 ml wash buffer. The resultant cell paste was lysed at 4 °C by homogenization (Avestin Emulsiflex C3) for 5 min at 15,000–20,000 psi. The lysate was centrifuged at 4,303g for 15 min at 4 °C to separate the soluble and insoluble fractions. The soluble lysate was incubated at 4 °C with 1 ml TALON resin (washed with water and equilibrated with wash buffer) for 1 h. The lysate–resin mixture was centrifuged at 4,303g for 5 min to pellet. The supernatant was removed and the protein-bound TALON resin was then washed three times with 5 ml lysis/wash buffer, centrifuging between washes to pellet. The protein was eluted from TALON resin by rinsing the resin five times with 1 ml elution buffer. The elution fractions were pooled and dialysed overnight at 4 °C into storage buffer using 12,000–14,000 MWCO dialysis tubing. The protein concentration was measured using the Pierce 660 nm assay91. Protein samples were concentrated as needed with a 10 kDa MWCO Amicon Ultra-15 centrifugal filter unit (4,303g, 4 °C) to reach a concentration of ≥0.22 mg ml−1 (Supplementary Fig. 37). The protein was stored at 4 °C for later analysis. Yields were between 24 and 324 mg l−1 when expressed in TB, and between 3.6 and 3.7 mg l−1 when expressed in the defined media described above. Proteins were analysed by LC–MS as described above.
Protease digestion and fragment identification by MS
Each isolated sfGFP sample (~10–25 µg) was denatured with 6 M guanidine in 0.15 M Tris buffer (pH 7.5), followed by disulfide reduction with 8 mM DTT at 37 °C for 30 min. The reduced sfGFP was alkylated in the presence of 14 mM iodoacetamide at 25 °C for 25 min, followed by quenching using 6 mM DTT. The reduced and alkylated protein was exchanged into ~40 µl of 0.1 M Tris buffer (pH 7.5) using a Microcon 10 kDa membrane, and then 2.5 µg endoproteinase GluC (in a 0.25 µg µl−1 solution) was added directly to the membrane to achieve an enzyme/substrate ratio of at least 1:10. After 3 h at 37 °C, the digestion was quenched with an equal volume of 0.25 M acetate buffer (pH 4.8) containing 6 M guanidine. The peptide fragments were collected by spinning down through the membrane and subjected to LC–MS/MS analysis.
LC–MS/MS analysis was performed on an Agilent 1290-II HPLC device directly connected to a Thermo Fisher Q Exactive HF high-resolution mass spectrometer. Peptides were separated on a Waters HSS T3 reversed-phase column (2.1 mm × 150 mm) at 50 °C with a 70 min acetonitrile gradient (0.5–35%) in water containing 0.1% formic acid in the mobile phase at a total flow rate of 0.25 ml min−1. The MS data were collected at a resolution of 120,000, followed by data-dependent higher-energy collision dissociation MS/MS at a normalized collision energy of 25%.
Proteolytic peptides were identified and quantified using MassAnalyzer, a program92 developed in house (available in Biopharma Finder, Thermo Fisher). The program performs feature extraction, peptide identification, retention time alignment93 and peak integration in an automated fashion.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The structural model of MaFRSA is available in the Protein Data Bank with PDB code 7U0R. Data for Supplementary Figs. 1–43 are available at Zenodo (https://doi.org/10.5281/zenodo.7754396). Source data are provided with this paper.
References
Lutz, J.-F., Ouchi, M., Liu, D. R. & Sawamoto, M. Sequence-controlled polymers. Science 341, 1238149 (2013).
Barnes, J. et al. Iterative exponential growth of stereo- and sequence-controlled polymers. Nat. Chem. 7, 810–815 (2015).
Fahnestock, S. & Rich, A. Ribosome-catalyzed polyester formation. Science 173, 340–343 (1971).
Ohta, A., Murakami, H., Higashimura, E. & Suga, H. Synthesis of polyester by means of genetic code reprogramming. Chem. Biol. 14, 1315–1322 (2007).
Katoh, T., Iwane, Y. & Suga, H. Logical engineering of D-arm and T-stem of tRNA that enhances d-amino acid incorporation. Nucleic Acids Res. 45, 12601–12610 (2017).
Fujino, T., Goto, Y., Suga, H. & Murakami, H. Ribosomal synthesis of peptides with multiple β-amino acids. J. Am. Chem. Soc. 138, 1962–1969 (2016).
Katoh, T. & Suga, H. Ribosomal incorporation of consecutive β-amino acids. J. Am. Chem. Soc. 140, 12159–12167 (2018).
Adaligil, E., Song, A., Hallenbeck, K. K., Cunningham, C. N. & Fairbrother, W. J. Ribosomal synthesis of macrocyclic peptides with β2- and β2,3-homo-amino acids for the development of natural product-like combinatorial libraries. ACS Chem. Biol. 16, 1011–1018 (2021).
Katoh, T., Sengoku, T., Hirata, K., Ogata, K. & Suga, H. Ribosomal synthesis and de novo discovery of bioactive foldamer peptides containing cyclic β-amino acids. Nat. Chem. 12, 1081–1088 (2020).
Katoh, T. & Suga, H. Ribosomal elongation of cyclic γ-amino acids using a reprogrammed genetic code. J. Am. Chem. Soc. 142, 4965–4969 (2020).
Adaligil, E., Song, A., Cunningham, C. N. & Fairbrother, W. J. Ribosomal synthesis of macrocyclic peptides with linear γ4- and β-hydroxy-γ4-amino acids. ACS Chem. Biol. 16, 1325–1331 (2021).
Lee, J., Schwarz, K. J., Kim, D. S., Moore, J. S. & Jewett, M. C. Ribosome-mediated polymerization of long chain carbon and cyclic amino acids into peptides in vitro. Nat. Commun. 11, 4304 (2020).
Katoh, T. & Suga, H. Consecutive ribosomal incorporation of α-aminoxy/α-hydrazino acids with l/d-configurations into nascent peptide chains. J. Am. Chem. Soc. 143, 18844–18848 (2021).
Takatsuji, R. et al. Ribosomal synthesis of backbone-cyclic peptides compatible with in vitro display. J. Am. Chem. Soc. 141, 2279–2287 (2019).
Lee, J. et al. Ribosome-mediated biosynthesis of pyridazinone oligomers in vitro. Nat. Commun. 13, 6322 (2022).
Katoh, T. & Suga, H. Ribosomal elongation of aminobenzoic acid derivatives. J. Am. Chem. Soc. 142, 16518–16522 (2020).
Ad, O. et al. Translation of diverse aramid- and 1,3-dicarbonyl-peptides by wild type ribosomes in vitro. ACS Cent. Sci. 5, 1289–1294 (2019).
Iskandar, S. E., Haberman, V. A. & Bowers, A. A. Expanding the chemical diversity of genetically encoded libraries. ACS Comb. Sci. 22, 712–733 (2020).
Sievers, A., Beringer, M., Rodnina, M. V. & Wolfenden, R. The ribosome as an entropy trap. Proc. Natl Acad. Sci. USA 101, 7897–7901 (2004).
England, P. M., Zhang, Y., Dougherty, D. A. & Lester, H. A. Backbone mutations in transmembrane domains of a ligand-gated ion channel: implications for the mechanism of gating. Cell 96, 89–98 (1999).
Guo, J., Wang, J., Anderson, J. C. & Schultz, P. G. Addition of an α-hydroxy acid to the genetic code of bacteria. Angew. Chem. Int. Ed. 120, 734–737 (2008).
Kobayashi, T., Yanagisawa, T., Sakamoto, K. & Yokoyama, S. Recognition of non-α-amino substrates by oyrrolysyl-tRNA synthetase. J. Mol. Biol. 385, 1352–1360 (2009).
Li, Y.-M. et al. Ligation of expressed protein α-hydrazides via genetic incorporation of an α-hydroxy acid. ACS Chem. Biol. 7, 1015–1022 (2012).
Bindman, N. A., Bobeica, S. C., Liu, W. R. & van der Donk, W. A. Facile removal of leader peptides from lanthipeptides by incorporation of a hydroxy acid. J. Am. Chem. Soc. 137, 6975–6978 (2015).
Melo Czekster, C., Robertson, W. E., Walker, A. S., Söll, D. & Schepartz, A. In vivo biosynthesis of a β-amino acid-containing protein. J. Am. Chem. Soc. 138, 5194–5197 (2016).
Chen, S., Ji, X., Gao, M., Dedkova, L. M. & Hecht, S. M. In cellulo synthesis of proteins containing a fluorescent oxazole amino acid. J. Am. Chem. Soc. 141, 5597–5601 (2019).
Spinck, M. et al. Genetically programmed cell-based synthesis of non-natural peptide and depsipeptide macrocycles. Nat. Chem. 15, 61–69 (2023).
Liu, C. C. & Schultz, P. G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444 (2010).
Wan, W., Tharp, J. M. & Liu, W. R. Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochim. Biophys. Acta Proteins Proteom. 1844, 1059–1070 (2014).
Vargas-Rodriguez, O., Sevostyanova, A., Söll, D. & Crnković, A. Upgrading aminoacyl-tRNA synthetases for genetic code expansion. Curr. Opin. Chem. Biol. 46, 115–122 (2018).
Italia, J. S. et al. Mutually orthogonal nonsense-suppression systems and conjugation chemistries for precise protein labeling at up to three distinct sites. J. Am. Chem. Soc. 141, 6204–6212 (2019).
Chin, J. W. Expanding and reprogramming the genetic code. Nature 550, 53–60 (2017).
Srinivasan, G., James, C. M. & Krzycki, J. A. Pyrrolysine encoded by UAG in archaea: charging of a UAG-decoding specialized tRNA. Science 296, 1459–1462 (2002).
Ambrogelly, A., Palioura, S. & Soll, D. Natural expansion of the genetic code. Nat. Chem. Biol. 3, 29–35 (2007).
Wang, L., Brock, A., Herberich, B. & Schultz, P. G. Expanding the genetic code of Escherichia coli. Science 292, 498–500 (2001).
Kobayashi, T. et al. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Mol. Biol. 10, 425–432 (2003).
Yanagisawa, T. et al. Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase. J. Mol. Biol. 378, 634–652 (2008).
Borrel, G. et al. Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J. Bacteriol. 194, 6944–6945 (2012).
Willis, J. C. W. & Chin, J. W. Mutually orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs. Nat. Chem. 10, 831–837 (2018).
Wang, Y.-S. et al. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of l-phenylalanine and its derivatives. Mol. Biosyst. 7, 714–717 (2011).
Wang, Y.-S., Fang, X., Wallace, A. L., Wu, B. & Liu, W. R. A rationally designed pyrrolysyl-tRNA synthetase mutant with a broad substrate spectrum. J. Am. Chem. Soc. 134, 2950–2953 (2012).
Herring, S. et al. The amino-terminal domain of pyrrolysyl-tRNA synthetase is dispensable in vitro but required for in vivo activity. FEBS Lett. 581, 3197–3203 (2007).
McMurry, J. L. & Chang, M. C. Y. Fluorothreonyl-tRNA deacylase prevents mistranslation in the organofluorine producer Streptomyces cattleya. Proc. Natl Acad. Sci. USA 114, 11920–11925 (2017).
Findly, D., Herries, D. G., Mathias, A. P., Rabin, B. R. & Ross, C. A. The active site and mechanism of action of bovine pancreatic ribonuclease. Nature 190, 781–784 (1961).
Englert, M. et al. Aminoacylation of tRNA 2′- or 3′-hydroxyl by phosphoseryl- and pyrrolysyl-tRNA synthetases. FEBS Lett. 587, 3360–3364 (2013).
Griffin, B. E., Jarman, M., Reese, C. B., Sulston, J. E. & Trentham, D. R. Some observations relating to acyl mobility in aminoacyl soluble ribonucleic acids. Biochemistry 5, 3638–3649 (1966).
Gottfried-Lee, I., Perona, J. J., Karplus, P. A., Mehl, R. A. & Cooley, R. B. Structures of Methanomethylophilus alvus pyrrolysine tRNA-synthetases support the need for de novo selections when altering the substrate specificity. ACS Chem. Biol. 17, 3470–3477 (2022).
Stepanov, V. G., Moor, N. A., Ankilova, V. N. & Lavrik, O. I. Phenylalanyl-tRNA synthetase from Thermus thermophilus can attach two molecules of phenylalanine to tRNAPhe. FEBS Lett. 311, 192–194 (1992).
Wang, B., Zhou, J., Lodder, M., Anderson, R. D. & Hecht, S. M. Tandemly activated tRNAs as participants in protein synthesis. J. Biol. Chem. 281, 13865–13868 (2006).
Ko, J. et al. Pyrrolysyl-tRNA synthetase variants reveal ancestral aminoacylation function. FEBS Lett. 587, 3243–3248 (2013).
Xuan, W. et al. Site-specific incorporation of a thioester containing amino acid into proteins. ACS Chem. Biol. 13, 578–581 (2018).
Hwang, S., Lee, N., Cho, S., Palsson, B. & Cho, B.-K. Repurposing modular polyketide synthases and non-ribosomal peptide synthetases for novel chemical biosynthesis. Front. Mol. Biosci. 7, 87 (2020).
Muir, T. W., Sondhi, D. & Cole, P. A. Expressed protein ligation: a general method for protein engineering. Proc. Natl Acad. Sci. USA 95, 6705–6710 (1998).
Varshney, U., Lee, C. P., Seong, B. L. & RajBhandary, U. L. Mutants of initiator tRNA that function both as initiators and elongators. J. Biol. Chem. 266, 18018–18024 (1991).
Tharp, J. M. et al. Initiation of protein synthesis with non-canonical amino acids in vivo. Angew. Chem. Int. Ed. 59, 3122–3126 (2020).
Cestari, I. & Stuart, K. A spectrophotometric assay for quantitative measurement of aminoacyl-tRNA synthetase activity. J. Biomol. Screen. 18, 490–497 (2013).
Wang, Y.-S. et al. Genetic incorporation of twelve meta-substituted phenylalanine derivatives using a single pyrrolysyl-tRNA synthetase mutant. ACS Chem. Biol. 8, 405–415 (2013).
Guo, L.-T. et al. Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc. Natl Acad. Sci. USA 111, 16724–16729 (2014).
Tharp, J. M., Wang, Y.-S., Lee, Y.-J., Yang, Y. & Liu, W. R. Genetic incorporation of seven ortho-substituted phenylalanine derivatives. ACS Chem. Biol. 9, 884–890 (2014).
Seki, E., Yanagisawa, T., Kuratani, M., Sakamoto, K. & Yokoyama, S. Fully productive cell-free genetic code expansion by structure-based engineering of Methanomethylophilus alvus pyrrolysyl-tRNA synthetase. ACS Synth. Biol. 9, 718–732 (2020).
Kavran, J. M. et al. Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl Acad. Sci. USA 104, 11268–11273 (2007).
Englert, M. et al. Probing the active site tryptophan of Staphylococcus aureus thioredoxin with an analog. Nucleic Acids Res. 43, 11061–11067 (2015).
Laursen, B. S., Sørensen, H. P., Mortensen, K. K. & Sperling-Petersen, H. U. Initiation of protein synthesis in bacteria. Microbiol. Mol. Biol. Rev. 69, 101–123 (2005).
Lee, J. et al. Expanding the limits of the second genetic code with ribozymes. Nat. Commun. 10, 5097 (2019).
Chatterjee, A., Sun, S. B., Furman, J. L., Xiao, H. & Schultz, P. G. A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52, 1828–1837 (2013).
DeBenedictis, E. A., Söll, D. & Esvelt, K. M. Measuring the tolerance of the genetic code to altered codon size. eLife 11, e76941 (2022).
Goldman, E. & Jakubowski, H. Uncharged tRNA, protein synthesis, and the bacterial stringent response. Mol. Microbiol. 4, 2035–2040 (1990).
Young, T. S., Ahmad, I., Yin, J. A. & Schultz, P. G. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374 (2010).
Ostrov, N. et al. Design, synthesis, and testing toward a 57-codon genome. Science 353, 819–822 (2016).
Fredens, J. et al. Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518 (2019).
González, S. S. et al. Genetic code expansion in the engineered organism Vmax X2: high yield and exceptional fidelity. ACS Cent. Sci. 7, 1500–1507 (2021).
Nivina, A., Yuet, K., Hsu, J. & Khosla, C. Evolution and diversity of assembly-line polyketide synthases. Chem. Rev. 119, 12524–12547 (2019).
Tsai, S. The structural enzymology of iterative aromatic polyketide synthases: a critical comparison with fatty acid synthases. Annu. Rev. Biochem. 87 503–531 (2018).
Walsh, C. T., O’Brien, R. V. & Khosla, C. Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem. Int. Ed. 52, 7098–7124 (2013).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Chung, C. T., Niemela, S. L. & Miller, R. H. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc. Natl Acad. Sci. USA 86, 2172–2175 (1989).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
Baklanov, M. Effect on DNA transcription of nucleotide sequences upstream to T7 promoter. Nucleic Acids Res. 24, 3659–3660 (1996).
Kao, C., Zheng, M. & Rüdisser, S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3′ terminus of RNAs transcribed by T7 RNA polymerase. RNA 5, 1268–1272 (1999).
Seki, E., Yanagisawa, T. & Yokoyama, S. in Noncanonical Amino Acids: Methods and Protocols (ed. Lemke, E. A.) 49–65 (Humana Press, 2018).
Sprinzl, M. & Cramer, F. in Progress in Nucleic Acid Research and Molecular Biology Vol. 22 (ed. Cohn, W. E.) 1–69 (Academic Press, 1979).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Evans, P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D 67, 282–292 (2011).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D 69, 1204–1214 (2013).
Bunkóczi, G. et al. Phaser.MRage: automated molecular replacement. Acta Crystallogr. D 69, 2276–2286 (2013).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
The PyMOL Molecular Graphics System, Version 1.8 (Schrödinger LLC, 2015).
Grasso, K. T. et al. A facile platform to engineer Escherichia coli tyrosyl-tRNA synthetase adds new chemistries to the eukaryotic genetic code, including a phosphotyrosine mimic. ACS Cent. Sci. 8, 483–492 (2022).
Antharavally, B. S., Mallia, K. A., Rangaraj, P., Haney, P. & Bell, P. A. Quantitation of proteins using a dye–metal-based colorimetric protein assay. Anal. Biochem. 385, 342–345 (2009).
Zhang, Z. Large-scale identification and quantification of covalent modifications in therapeutic proteins. Anal. Chem. 81, 8354–8364 (2009).
Zhang, Z. Retention time alignment of LC/MS data by a divide-and-conquer algorithm. J. Am. Soc. Mass Spectrom. 23, 764–772 (2012).
Acknowledgements
We thank H. Celik, A. Lund and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance, J. Kuriyan for generously sharing crystallography equipment, J. Holton and G. Meigs for X-ray diffraction assistance, S. Zheng for advice on synthesis, and I. Knudson for advice on in vitro translation. We are especially grateful to P. Schultz (Scripps Research) for donating the DH10B ∆aspC ∆tyrB deletion strain and to S. Miller and J. Cate for meaningful discussion and feedback. This work was supported by the NSF Center for Genetically Encoded Materials (C-GEM; CHE 2002182). R.F. was supported by Agilent Technologies. C.V.S. was supported by a Berkeley Fellowship for Graduate Study. L.T.R. was supported by the NSF Graduate Research Fellowship Program (grant no. 1752814). Beamline 8.3.1 at the Advanced Light Source is operated by the University of California at San Francisco with generous grants from the National Institutes of Health (R01 GM124149 and P30 GM124169), Plexxikon and the Integrated Diffraction Analysis Technologies programme of the US Department of Energy Office of Biological and Environmental Research. The Advanced Light Source (Berkeley, CA) is a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy Office of Basic Energy Sciences (contract no. DE-AC02-05CH11231). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
R.F., C.V.S., O.A. and A.S. designed the project. R.F. led the acylation experiments and in vivo protein expression and assisted with the crystallography and in vitro translation. C.V.S. led the crystallography, in vitro translation and in vivo protein expression and assisted with the acylation experiments. L.T.R. synthesized the materials and assisted with the crystallography. N.X.H. assisted with the acylation experiments. B.S. and Z.Z. designed and performed the GluC digestions and mass spectrometry analyses. E.F. and A.C. designed the plasmids and developed the protein expression and isolation conditions. S.S. and C.L.G. assisted with the crystallography. R.F., C.V.S. and A.S. prepared the paper.
Corresponding author
Ethics declarations
Competing interests
R.F., C.V.S. and A.S. have submitted a patent application ‘Methods to Generate Novel Acyl-tRNA Species’ (2022, US Patent application no. PCT/US23/63304) related to this work. The application discloses PylRS enzymes that accept α-hydroxy acids, α-thio acids, N-formyl-l-α-amino acids and α-carboxy acid monomers (malonic acids) that are formally precursors to polyketide natural products, and methods of use.
Peer review
Peer review information
Nature Chemistry thanks Jian Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Additional RNAse A assay experiments analyzed by LC-HRMS.
The enzyme and substrate are noted in the top left of each plot. These data provide evidence that MaPylRS accepts 16 as a substrate but not 5. Similarly, MaFRS1 and MaFRS2 accept 17, 18, and 19, but not 11.
Extended Data Fig. 2 RNAse A assay analyzing decarboxylation of malonyl-tRNAs.
a, Substituted malonic acid monomers used in this study. b, Left, 26a-d: malonyl adenosine nucleosides formed when malonyl-tRNA is digested by RNAse A; right, 27a and 27b: decarboxylation products of malonyl-adenosine nucleosides. c, LC-HRMS analysis of Ma-tRNAPyl acylation reactions after RNAse A digestion. Reactions were performed as described in Methods. The EIC for the malonyl product 26 as a mixture of 2’ and 3’ isomers (two pairs of diastereomers) (pink, top) shows the expected peaks whereas the EIC for the decarboxylation product 27, also as a mixture of 2’ and 3’ isomers (black, bottom) shows that the decarboxylation product is absent in all cases except the MaPylRS-catalyzed acylation of Ma-tRNAPyl with monomer 16. These data provide evidence that the decarboxylation of malonates charged to Ma-tRNAPyl observed in the intact tRNA mass analysis (Supplementary Figs. 7, 15, 17-19, 26, 28-30 and 32-35) occurs during either the workup or the LC-MS itself.
Extended Data Fig. 3 Ligand electron densities of m-CF3-2-BMA (18) and AMP-PNP bound to MaFRSA.
Electron density shown from 2F0-Fc map contoured at 1σ for m-CF3-2-BMA bound to chain A (a) and chain B (b) and AMP-PNP bound to chain A (c) and chain B (d).
Extended Data Fig. 4 Expanded view of MaFRSA recognition of m-CF3-2-BMA (18).
Additional residues comprising the active site shown in chain A (light purple, a) and chain B (dark purple, b). Dashed lines indicate hydrogen-bonding interactions (orange), path of nucleophilic attack (blue), or missing hydrogen bonds (red).
Extended Data Fig. 5 Structural recognition of m-CF3-2-BMA (18) by MaFRSA and β7-β8 loop positioning in comparison with published PylRS structures.
a, Alignment of MmIFRS (N346S/C348Q, yellow, PDB: 4TQD) bound to 3-iodo-L-phenylalanine and AMP-PNP and MaFRSA bound to m-CF3-2-BMA (18) and AMP-PNP chain A (light purple) illustrating similar interactions between substrate carboxylate and backbone amides. b, The flexible β7-β8 loop ranges between unstructured, an open conformation, and a closed conformation across PylRS structures. MaFRSA bound to m-CF3-2-BMA and AMP-PNP (light purple), wild-type MaPylRS apo (green, PDB: 6JP2), wild-type MmPylRS bound to pyrrolysine and AMP-PNP (blue, PDB: 2ZCE), and wild-type MmPylRS bound to pyrrolysyl-adenylate (brown, PDB: 2Q7H). Chain A (light purple, c) exhibits lower B-factors in the β5-β6 loop than chain B (dark purple, d) indicated by dark blue and thin ribbon for lower B-factors and dark red and thick ribbon for higher B-factors.
Extended Data Fig. 6 Plate reader analysis of sfGFP expression in DH10B E. coli with n = 4 replicates.
Emission at 528 nm after 24 h sfGFP expression in E. coli DH10B cells harboring pEVOL or pMega plasmids encoding a, MaPylRS and Ma-tRNAPyl in the presence of 0 or 1 mM BocK (1) or α-OH BocK (2) or b, MaFRSA and Ma-tRNAPyl in the presence of 0 or 1 mM m-CF3-L-phenylalanine (20) or α-OH m-CF3- phenylalanine (21).
Supplementary information
Supplementary Information
Materials, Synthesis notes, Supplementary Figs. 1–43, Tables 1–5 and References.
Supplementary Data 1
All masses identified in intact tRNA mass spectra are listed here. N-1 and N-2 refer to the tRNA missing the final nucleotides 1 and 2 at the 3′ end, respectively. P and PPP indicate that the 5′ end of the tRNA has a mono- or triphosphate, respectively. N+G and N+GG refer to tRNA products with non-templated addition of guanosine residues identified in the mass spectrum. Note that for some enzyme/substrate pairs there is evidence that the N+G products are acylated by the synthetase, suggesting that the untemplated guanosine addition does not exclude these tRNA species from activity with the synthetase.
Source data
Source Data Fig. 1
Numerical MS data.
Source Data Fig. 2
Numerical MS data.
Source Data Fig. 3
Numerical MS data.
Source Data Fig. 4
Numerical MS data.
Source Data Fig. 6
Numerical MS data.
Source Data Extended Data Fig. 1
Numerical MS data.
Source Data Extended Data Fig. 2
Numerical MS data.
Source Data Extended Data Fig. 6
Numerical fluorescence emission data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fricke, R., Swenson, C.V., Roe, L.T. et al. Expanding the substrate scope of pyrrolysyl-transfer RNA synthetase enzymes to include non-α-amino acids in vitro and in vivo. Nat. Chem. 15, 960–971 (2023). https://doi.org/10.1038/s41557-023-01224-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41557-023-01224-y
