
Abstract
Increasing evidence points towards the key role of the epithelium in the systemic and over-activated immune response to viral infection, including SARS-CoV-2 infection. Yet, how viral infection alters epithelial–immune cell interactions regulating inflammatory responses, is not well known. Available experimental approaches are insufficient to properly analyse this complex system, and computational predictions and targeted data integration are needed as an alternative approach. In this work, we propose an integrated computational biology framework that models how infection alters intracellular signalling of epithelial cells and how this change impacts the systemic immune response through modified interactions between epithelial cells and local immune cell populations. As a proof-of-concept, we focused on the role of intestinal and upper-airway epithelial infection. To characterise the modified epithelial–immune interactome, we integrated intra- and intercellular networks with single-cell RNA-seq data from SARS-CoV-2 infected human ileal and colonic organoids as well as from infected airway ciliated epithelial cells. This integrated methodology has proven useful to point out specific epithelial–immune interactions driving inflammation during disease response, and propose relevant molecular targets to guide focused experimental analysis.
Similar content being viewed by others


Introduction
Specialised epithelial cells lining the surface of the mammalian gastrointestinal tract form the primary line of defense against external stimuli, working in cohort with resident immune cells to maintain homeostasis and defend the body from infections. Although the role of both the epithelium and the immune system during infection have been assessed in previous studies, these components have often been investigated separately. While this knowledge has been instrumental in advancing medical research, the recent COVID-19 pandemic has pointed out the need for large-scale, integrative models to address key questions that cannot yet be solved with available experimental models. One example is: what is the role of an infected cell in inducing systemic inflammatory responses by communicating to resident immune cells? To address this critical question, existing yet often disconnected datasets and computational approaches can be leveraged to develop a complex but easily interpretable map of how viral molecules are able affect different cell populations in the gut, and how infected cells can in turn modulate local and systemic immune and inflammatory responses.
The recent COVID-19 pandemic is caused by infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). While SARS-CoV-2 mainly targets the lung and upper airways1,2,3, other organs can be infected too, including the heart, kidney, brain, and the intestine4. In addition to directly infecting key organs, the main hurdle of SARS-CoV-2 infection is the excessive inflammatory response mediated by both the innate and adaptive immune systems1,5. The overactivated inflammatory response, also known as cytokine release syndrome (CRS) or cytokine storm, is the result of high levels of circulating cytokines and chemokines, and it is thought to be responsible for the severe COVID-19 symptoms some patients experience6. Yet, there is no clear understanding of which particular inflammatory pathways and cell types are responsible for driving this process, and whether some organs are more important than others in the initiation and maintenance of this syndrome7. The causal role of SARS-CoV-2 on intestinal damage and the role of the small intestine in contributing to CRS was recently highlighted8,9.
Human intestinal organoids have been used as a tool to study SARS-CoV-2 infection in the gut and the inflammatory responses of specific intestinal epithelial cell types10,11,12,13. These studies provided evidence that SARS-CoV-2 is able to infect and actively replicate in human intestinal cells, in particular in enterocytes10,13. These studies also revealed that, contrary to the limited type I and type III interferon (IFN) immune response observed in the lungs14,15, the response to SARS-CoV-2 infection in the gut is characterised by a cell-type specific inflammatory response that is important in the development of systemic reactions11. Examination of human intestinal samples has also shown that infection of gut epithelial cells results in the activation of local immune populations16. Yet, the exact effects of viral infection in the gut and the role of epithelial cell–immune cell interaction in mediating the inflammatory response of the body are not known. This information could ultimately aid the development of treatments and strategies to optimize the level and type of immune response as we would understand better the viral strategies that dysregulate our immune system. Due to the lack of adequate and complex experimental systems, to the best of our knowledge, no study has been carried out so far to analyse epithelial–immune crosstalk in the gastrointestinal tract upon SARS-CoV-2 infection.
Here, we introduce a computational framework to map epithelial–immune interactions, improve our understanding in the gut or in other organs by interpreting existing data better, and importantly, provide a short list of key molecules for targeted experimental validations. To achieve this, we integrated two previously developed intracellular modelling tools (ViralLink and CARNIVAL) with intercellular network approaches (from OmniPath)17,18,19. We present two proof of concept studies on intestinal organoids and upper airways ciliated cells. We used available SARS-CoV-2–human mRNA/protein–protein interaction predictions to model the effect of viral infection on intracellular signalling networks in host intestinal and ciliated cells, and applied published single-cell datasets to create cell-type, organ and context-specific epithelial–immune interaction maps. We demonstrated the importance and usefulness of this map with integrated analyses, which provided an improved understanding of the effect of viral infection on ileal, colonic and airway epithelial cells, and the role of epithelial–immune cell crosstalk during SARS-CoV-2 infection. Ultimately, this framework may help to find key intercellular inflammatory pathways involved in these crosstalks that could pave the way for potential successful strategies against the cytokine release syndrome associated-symptoms observed in severe cases of COVID-19. Importantly, the presented integrated framework will allow investigating other infections and conditions for which our analytical toolkits can be repurposed.
Results
Reconstructing the intestinal epithelial–immune interactome
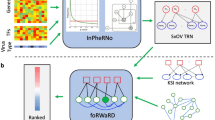
In our previous work, we identified a subpopulation of enterocytes as the prime target of SARS-CoV-2 (BavPat1/2020 strain), with directly infected cells showing a high pro-inflammatory response and little to no interferon-mediated response as the result of a SARS-CoV-2-mediated inhibition of interferon signalling13. These findings highlighted the key role of the gut as a pro-inflammatory reservoir, which primes for further investigation to be able to fully understand SARS-CoV-2 pathogenesis. Building on this study, we created an integrated bioinformatics framework that enables the investigation of the infected epithelial cell–immune cell crosstalk in a cell-type specific manner (Fig. 1). To do so, we exploited a priori knowledge on ligand–receptor interactions [18] to construct intercellular networks connecting epithelial cells and resident immune cells using our previously generated single cell RNA-seq epithelial cell dataset of ileal and colonic organoids infected with SARS-CoV-2 (BavPat1/2020 variant)13 and a separate lamina propria immune cell one20,21.

Schematic workflow illustrating the different analytical steps used to construct the intracellular and intercellular signalling networks between epithelial cells in SARS-CoV-2 infected intestinal organoids (ileal and colonic organoids, 24 h infection) or moderate COVID-19 upper airway ciliated epithelial and immune cell types.
First, we looked at the epithelial cell population with the highest number of ligands among the differentially expressed genes upon infection to assess which epithelial cell type could drive the response to SARS-CoV-2 infection. Our previous findings13 had highlighted immature enterocytes (originally known as “immature enterocytes 2”, an enterocyte subpopulation characterised by MMP7+, MUC1+, CXCL1+) as the epithelial population characterised by the highest number of differentially expressed genes upon infection (Supplementary Fig. 1). In accordance with this, we found that this population was also characterised by the highest number of differentially expressed ligands. Hence, differentially expressed ligands of colonic and ileal immature enterocytes were used to build epithelial–immune intercellular networks by connecting ligands to their binding receptors on immune cells (Fig. 1 and Methods).
To identify the main epithelial and immune cell types involved in intercellular crosstalk, the putative number of ligand–receptor interactions between each epithelial–immune cell type pair was computed. Specifically, all possible interactions between each set of up or downregulated epithelial ligands and each of the receptors expressed by the specific immune cell type (from20 and21) were identified (Fig. 1 and Methods). While both bystander and infected cell populations were affected by viral infection, directly infected intestinal cell populations had a higher number of predicted interactions with immune cells compared to bystander cell populations in both colon and ileum, supporting a role for direct viral infection in altering intercellular signalling in the gut (Fig. 2a). In the colon, the higher number of epithelial–immune interactions was identified between downregulated ligands of infected immature enterocytes and plasma cells, as well as CD4+/CD8+ T cells, macrophages and dendritic cells (DCs) to a lesser extent (Fig. 2a). Conversely, in the ileum, the highest number of interactions was identified between upregulated ligands of infected immature enterocytes and IgA plasma cells, T resident memory (Trm) cells, dendritic cells and resident macrophages (Fig. 2a). Notably, the higher number of interactions in the ileum was not a result of a higher number of upregulated ligands (20), as this was similar to the number of downregulated ones (24) (Fig. 2a, b). Instead, the higher number of interactions was driven by upregulated ligands binding to multiple receptors on each immune cell targeted. A more detailed explanation can be found in the Supplementary Results.

a Heatmap showing the number of interactions between immature enterocytes and resident immune cells. Interactions driven by upregulated and downregulated ligands (ligand direction) are shown separately for infected and bystander cells (status), and for ileum and colonic organoids. The intensity of the colour indicates the number of interactions with the immune cell types whose receptor is targeted by the epithelial cells ligands. The numbers on the ligand direction row refer to the number of upregulated or downregulated ligands driving the indicated interactions with immune cells for the different groups/conditions. Abbreviations: Ileum: inf_macrophage infected macrophage, mast mast cell, CD8_Trm_cyto Resident memory cytotoxic T cell, DC2 dendritic cell 2, Trm Tissue-resident memory T cell, gd_Tcell Gamma delta (γδ) T cells, ILC Innate lymphoid cell, mem_Bcell memory B cell, naive_Bcell naive B cell, TFH_like T follicular helper cells, Trm_Th17 Tissue-resident memory Th17 cells, Treg Regulatory T cell, Tcyto Cytotoxic T cell, Tmem Memory T cells. Colon: ILC Innate lymphoid cell, CD8_IL17 IL-17+ CD8+ T cells, DC dendritic cells, GC_Bcell Germinal center B cells, CD4_PD1 mast mast cell, Treg Regulatory T cell, NK Natural Killer cell, CD4_MThi high mitochondrial CD4+ T cell, CD4_memory CD4+ Memory T cell, CD4_activ_fos_high activated CD4+ T cells (high/low c-fos), CD8_LP CD8+ lymphocyte-predominant cells, CD8_IEL CD8+ intraepithelial lymphocytes. b Venn diagrams showing the number of ligands of the infected immature enterocytes–immune cells intercellular network that are unique or shared between the ileum and colon. Upregulated and downregulated ligands are shown separately. The full list of ligands is available as Table 1.
With this integrated network reconstruction, we have shown the value of our framework in enabling the study of mechanistic details of the effect of SARS-CoV-2, or other viruses, on the human immune system.
The infected epithelial signalling network drives the epithelial–immune interactome
To further understand how SARS-CoV-2 infection drives altered ligand expression in infected intestinal epithelial cells, we used our integrated network model to reconstruct the altered intracellular signalling in directly infected immature enterocytes population driven by SARS-CoV-2. Within our framework, two separate bioinformatics tools, ViralLink and CARNIVAL, were used to construct an intracellular causal network linking perturbed human proteins interacting with SARS-CoV-2 viral proteins or miRNAs to activated transcription factors (TFs) regulating the differentially expressed ligands upon infection, through altered intracellular protein-protein signalling cascades (Fig. 1). By integrating tissue-specific epithelial data, we have constructed two separate causal networks for infected immature enterocytes of the ileum and colon, thus enabling us to distinguish tissue specific differences in infection response (Fig. 1). Detailed analysis of the colon and ileal intracellular network features can be found in the Supplementary Results. Furthermore, using multiple complementary methods of network analysis, we have highlighted the most likely signalling pathways affected upon infection. Finally, by integrating a priori information on SARS-CoV-2 miRNAs/proteins - human protein interactions, we have built separate sublayers of the networks representing altered signalling stemming from upstream perturbations caused by SARS-CoV-2 miRNAs, proteins or both. These networks allowed us to assess the contribution of each of these viral factors in altering the intracellular signalling cascade (Fig. 1 and Methods).
Functional analysis of the tissue-specific intracellular networks is useful to understand how SARS-CoV-2 infection in immature enterocytes affects their function through the modulation of intracellular signalling, and whether any differences in response exist between colon and ileum. Here, a functional overrepresentation analysis (Gene Ontology (GO) and Reactome) of the protein-protein interaction (“PPI”) layer22,23,24,25 of each intracellular causal sub-network (stemming from viral proteins, miRNAs, or both) was integrated in our framework to assess the contributions of SARS-CoV-2 miRNAs or proteins to the changes observed22,23,24,25 (Fig. 1 and Methods). Functional analysis of the PPI layer of the intracellular networks built with ViralLink revealed an overrepresentation of pathways related to inflammation and chemotaxis (Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-kB) signalling, interleukin signalling, chemokine signalling) in both ileum and colon (Fig. 3). Additionally, we found the overrepresentation of functions related to interferon (IFN) signalling and Mitogen-Activated Protein Kinase (MAPK) signalling being overrepresented uniquely in the ileum in both viral protein and miRNA intracellular networks (Fig. 3b). An overrepresentation of laminin-driven interaction pathways, which we observed uniquely for viral miRNA intracellular network in both ileum and colon, could be indicative of an increased recruitment and adhesion of immune cells following infection (Fig. 3). Furthermore, an overrepresentation of pathways related to negative regulation of apoptosis, cell cycle, cell proliferation and growth was found in both ileal and colonic networks, suggesting an effect of SARS-CoV-2 on epithelial cell tissue renewal (Fig. 3). Interestingly, an overrepresentation of WNT signalling pathway, which is key for stem cell renewal, was found uniquely in the viral protein sub-network, in both tissues, while pathways related to the establishment of cell and tissue polarity were found uniquely in the colon, indicating an attempt for tissue healing following viral infection (Fig. 3).

a, b Overview of intracellular and intercellular signalling upon SARS-CoV-2 infection in colonic (a) and ileal (b) infected immature enterocytes and immune cell populations. From left to right: signalling cascade going from SARS-CoV-2 molecules (proteins or miRNAs) to differentially expressed ligands on immature enterocytes and binding receptor groups on immune cells. Intracellular network: SARS-COV-2 molecules are grouped separately if they are viral proteins (bottom) or miRNAs (top). Differentially expressed ligands for which no upstream signalling was identified, but downstream intercellular connections were predicted are excluded from this figure. Differentially expressed ligands are grouped based on the direction of regulation, which is indicated with blue when downregulated (bottom) and red when upregulated (top) when comparing SARS-CoV-2 infected vs uninfected conditions. Colours of the nodes and of the functional analysis indicate if the original network was a miRNA only (yellow), viral protein only (black) or both viral protein and miRNA (grey). Functional overrepresentation analysis was carried out for the “PPI layer” of the intracellular network which includes human binding proteins, intermediary signalling proteins and TFs (adj p value < 0.05, n > 3). Intercellular network: Size of the receptor node represents the sum of receptors within the group targeted by each incoming ligand. Functional analysis is indicated for ligand–receptor groups. Receptor groups layout is based on whether they contributed to the functional analysis of upregulated interactions (red) or downregulated interactions (blue). Receptor groups not contributing to any functions are indicated in light grey.
Functional overrepresentation analysis of the PPI layer of the intracellular networks built with CARNIVAL confirmed similar affected functions upon infection as those found in the ViralLink networks, suggesting functional overlap between networks obtained using these two methods such as senescence or inflammation (Fig. 3 and Supplementary Figs. 3 and 4). Within the outlined framework, the choice of which tool (ViralLink or CARNIVAL) to be used to build the intracellular network for a particular study should be driven by the specific study aims and biological questions. Further information about similarities and differences between the two tools is available in the Methods and Supplementary Results.
Further analysis of these networks can help predict key transcription factors responsible for the upstream regulation of altered ligands upon infection. Here, we found that both colonic and ileal networks generated with CARNIVAL shared similar transcription factors, including ATF2/3, FOS, JUN, STAT1, and NFKB1, which were all upregulated in both tissues upon infection (Supplementary Figs. 3 and 4). These transcription factors play a role in interferon response (STAT126), and inflammation (NFKB127), anti-apoptosis and cell growth (ATF2/328), cell proliferation and differentiation (JUN, FOS29), suggesting an increase in these functions upon SARS-CoV-2 infection in both colon and ileum. Interestingly, viral miRNAs were predicted to target different intracellular signalling processes between colon and ileum (miR_10,11,16,18 in the colon and miR_4,5,6,18 in the ileum). Additionally, by analysing these networks, we observed that NOTCH1 and SMAD4, seem to be central to the intracellular signalling cascade in the colon, by receiving several signals driven by viral miRNAs and viral proteins, respectively (Supplementary Fig. 3). Interestingly, both the Notch and TGF-β SMAD-dependant signalling pathways are involved in intestinal epithelial cell homeostasis, including stem cell maintenance, progenitor cell proliferation30 and maintenance of cell differentiation31, suggesting a modulation of these pathways upon infection. In the ileal network, JAK2 and CREB1, as well as SMAD2, SMAD3 and ERK2 (MAPK1) seem to play a central role in the intracellular PPI signalling driven by viral miRNAs and viral proteins, respectively, and JAK2 and both SMAD2 and SMAD3 were also upregulated upon infection (Supplementary Fig. 4). These transcription factors play a key role in the regulation of immunity (JAK2, CREB1;32,33, cell proliferation and differentiation (MAPK1) and plasticity (SMAD2/3)34,35, suggesting a positive regulation of these functions uniquely in the ileum upon infection.
Reconstruction and analysis of perturbed intracellular signalling in infected enterocytes using two complementary methods highlighted key pathways through which SARS-CoV-2 affects the infected cells, and pointed out transcription factors playing a major role during SARS-CoV-2 infection response.
Altered epithelial-derived ligands drive differential epithelial–immune crosstalk upon infection
To understand the functional impact of epithelial infection on the epithelial–immune interactome, we created intercellular networks by connecting upregulated and downregulated epithelial ligands of colonic and ileal infected immature enterocytes upon infection to their binding receptors on immune cells (Fig. 1 and Methods). Next, for each set of up and downregulated intercellular interactions, we looked at which ligands, receptors and immune cell types were involved in these intercellular interactions, assessing any potential similarities or differences between the colon and ileum (Fig. 1 and Methods). Here, we present the analysis relative to infected immature enterocytes–immune cell interaction, while the analysis relative to bystander immature enterocytes is available as Supplementary Results.
Upregulated ligands of infected immature enterocytes upon infection as well as binding receptors on immune cells were mainly shared among colon and ileum, resulting in most epithelial–immune interactions driven by upregulated ligands being similar in both tissues (one unique to colon, 219 unique to ileum, 66 shared) (Figs. 2b, 4a, b). A full list and detailed description of differences and similarities in ligands, receptors and upregulated intercellular interactions between colon and ileum can be found as Tables 1–3 and as Supplementary Results.

a Venn diagrams showing the number of ligand–receptor interactions in the infected immature enterocytes–immune cells intercellular networks that are unique or shared between the ileum and colon. Intercellular interactions driven by upregulated and downregulated ligands are shown separately. The full list of ligand–receptor interactions is available as Table 3. b Venn diagrams showing the number of receptors in the infected immature enterocytes–immune cells intercellular networks that are unique or shared between the ileal and colonic networks. Receptors targeted by upregulated ligands and downregulated ligands are shown separately. The full list of receptors is available as Table 2.
To understand which epithelial ligands and immune cell receptors were driving most epithelial–immune cell interactions, we scored ligands and receptors based on the number of interactions they were involved in (Fig. 1 and Methods). In both tissues, chemokines (CXCLs) and tumour necrosis factor alpha (TNF-α) were among epithelial ligands (Fig. 5a, b), and chemokine receptors (CXCR 3,4,5,6 and CCR 1,2,5,7,9,10) among the receptors on immune cells driving the highest numbers of upregulated interactions (Fig. 6), overall pointing towards an increased immune cell recruitment upon infection36. The high number of upregulated interactions driven by chemokines could be attributable to the widespread presence of several different chemokine receptors on immune cells (Supplementary Fig. 8).

a, b Bar plot showing the upregulated and downregulated ligands in the colonic (a) and ileal (b) infected immature enterocytes–immune cell network scored by number of interactions (height of the bar plot) and number of immune cells targeted (black dots). Upregulated ligands are shown in red and downregulated ligands in blue.

a, b Bar plot showing the immune receptors targeted by upregulated (top graph) and downregulated (bottom graph) ligands in colonic (a) and ileal (b) infected immature enterocytes, scored by number of interactions (height of the bar plot) and number of immune cells targeted (black dots). The colour of the bar plots indicates the number of ligands targeting each of the receptors indicated. This plot only shows the top 25 receptors by number of interactions, and the full plot is available as Supplementary Fig. 6.
To decipher which specific epithelial ligands and immune receptors were driving the strongest epithelial–immune cell interactions, ligands and receptors participating in epithelial–immune interactions can be scored based on the “sum of receptor expression” value, which takes into account the number of interacting receptors and the level of receptor expression in each immune cell type (Fig. 1 and Methods). In the colon, the strongest upregulated interactions involved the epithelial TNF-α binding to B cells, T cells (CD4/CD8+), NK cells, macrophages and DCs, as well as epithelial chemokines (CXCL2,3, 10) binding to T cells (CD4/CD8+) and NK cells (Fig. 7a). Similarly, in the ileum the strongest upregulated interactions involved epithelial chemokines binding to T cells (Treg, Tcyto, Tmem, CD8 Trm cyto) as well as TNF-α and colony stimulating factor 1 (CSF1) binding to macrophages and DCs (Fig. 7b). Receptors driving the strongest upregulated interactions were mainly chemokine receptors (CXCRs, CCRs) in both colon and ileum, and Receptor Interacting Serine/Threonine Kinase 1 (RIPK1) in the colon only (Fig. 8).

a, b Heatmap showing the upregulated and downregulated interactions in the colon (a) and ileum (b) between intestinal epithelial ligands and resident immune cells upon infection of immature enterocytes with SARS-CoV-2. The strength of the interaction is expressed by accounting for the number of interactions between epithelial ligands and immune receptors and the level of receptor expression of immune cells. The strength of the interaction, named “sum of expression values”, is visualised using a colour gradient from white (weakeast interactions) to purple (strongest interactions). Abbreviations: Ileum: inf_macrophage infected macrophage, mast mast cell, CD8_Trm_cyto Resident memory cytotoxic T cell, DC2 dendritic cell 2, Trm Tissue-resident memory T cell, gd_Tcell Gamma delta (γδ) T cells, ILC Innate lymphoid cell, mem_Bcell memory B cell, naive_Bcell naive B cell, TFH_like T follicular helper cells, Trm_Th17 Tissue-resident memory Th17 cells, Treg Regulatory T cell, Tcyto Cytotoxic T cell, Tmem Memory T cells. Colon: ILC Innate lymphoid cell, CD8_IL17 IL-17+ CD8+ T cells, DC dendritic cells, GC_Bcell Germinal center B cells, CD4_PD1 mast mast cell, Treg Regulatory T cell, NK Natural Killer cell, CD4_MThi high mitochondrial CD4+ T cell, CD4_memory CD4+ Memory T cell, CD4_activ_fos_high activated CD4+ T cells (high/low c-fos), CD8_LP CD8+ lymphocyte-predominant cells, CD8_IEL CD8+ intraepithelial lymphocytes.

a, b Heatmap showing the upregulated and downregulated interactions in the colon (a) and ileum (b) between receptors and resident immune cell types upon infection of immature enterocytes with SARS-CoV-2. The number of interactions in which each receiving receptor on immune cell types is involved is visualised using a colour gradient from blue (weakest interactions) to red (strongest interactions). Abbreviations: Ileum: inf_macrophage infected macrophage, mast mast cell, CD8_Trm_cyto Resident memory cytotoxic T cell, DC2 dendritic cell 2, Trm Tissue-resident memory T cell, gd_Tcell Gamma delta (γδ) T cells, ILC Innate lymphoid cell, mem_Bcell memory B cell, naive_Bcell naive B cell, TFH_like T follicular helper cells, Trm_Th17 Tissue-resident memory Th17 cells, Treg Regulatory T cell, Tcyto Cytotoxic T cell, Tmem Memory T cells. Colon: ILC Innate lymphoid cell, CD8_IL17 IL-17+ CD8+ T cells, DC dendritic cells, GC_Bcell Germinal center B cells, CD4_PD1 mast mast cell, Treg Regulatory T cell, NK Natural Killer cell, CD4_MThi high mitochondrial CD4+ T cell, CD4_memory CD4+ Memory T cell, CD4_activ_fos_high activated CD4+ T cells (high/low c-fos), CD8_LP CD8+ lymphocyte-predominant cells, CD8_IEL CD8+ intraepithelial lymphocytes.
Finally, functional overrepresentation analysis of the participating upregulated epithelial ligands and receiving receptors on immune cells can help to understand the role of each of these epithelial–immune interactions driven by infection (Fig. 1). In line with the extensive overlap in upregulated intercellular interactions (Fig. 4a and Supplementary Fig. 8), most functions were shared between colon and ileum, and included chemotaxis (GPCR signalling, chemokine signalling), immunity (interleukin signalling), apoptosis (caspase activation) and angiogenesis (VEGFA-VEGFR2 pathway) (Supplementary Fig. 9). One colonic-specific function was related to pro-inflammatory responses (TNF signalling) (Supplementary Fig. 9a) and one ileal-specific function was related to stem cell renewal (BMP signalling) (Supplementary Fig. 9b).
Downregulated ligands in infected immature enterocytes upon infection as well as targeted receptors on immune cells were tissue-specific to a large extent, resulting in a large proportion of downregulated interactions being tissue-specific (73 unique to ileum, 125 to colon) (Figs. 2b, 4a, b). A detailed description of differences and similarities in ligands, receptors and downregulated intercellular interactions between colon and ileum can be found as Supplementary Results.
In both tissues, a high number of downregulated interactions was driven by the epithelial ligands human leukocyte antigens (HLA-A/B/C), beta-2-microglobulin (B2M) and calmodulin (CALM1/2) (Fig. 5a, b), and integrins (ITGs), KLRCs and LDL Receptor Related Protein 1 (LRP1) in both colon and ileum (Figs. 4a and 6). Additionally, uniquely in the colon, the highest number of downregulated interactions was driven by two epithelial-derived laminins (LAMC2, LAMB3) (Fig. 2c), and by AKT1 (Protein kinase B, PKB) present on immune cell types (Figs. 4a and 6).
When investigating the strength of these intercellular interactions, we found that HLA-s (HLA-A, B, C) and B2M targeting T cells (colon: CD4/CD8+, Tregs; ileum: Trm, Tregs, cytotoxic T cells), NK cells (colon only), ILCs and macrophages (ileum only) represented the strongest downregulated interactions in both colon and ileum (Fig. 7). Additionally, uniquely in the colon, laminins (LAMB3, LAMC2) targeting T cells and macrophages represented the strongest downregulated interactions (Fig. 7). Receptors driving the strongest downregulated interactions were AKT1 uniquely in the colon (Fig. 8a) as well as integrins, KLRCs and LRP1 in both colon and ileum (Fig. 8a, b)
Functional overrepresentation analysis of the participating downregulated epithelial ligands and receiving receptors on immune cells (Fig. 1 and Methods) revealed shared functions related to antigen processing and cross-presentation (MHC class I–mediated), phagocytosis (endoplasmic reticulum (ER) phagosome pathway, signalling by RHO GTPases) and cell–cell communication (immunoregulatory interactions between a lymphoid and non-lymphoid cell) in both tissues, possibly suggesting decreased epithelial–immune cell crosstalk functions related to the activation of the innate and adaptive immune response37 (Supplementary Fig. 9). Furthermore, several colon-specific functions were related to the extracellular matrix (ECM) organisation and integrin cell surface interactions, which play an important role in processes critical to inflammation, infection, and angiogenesis, thereby suggesting a negative regulation of these vital interactions uniquely in the colon38 (Supplementary Fig. 9a). The only function uniquely overrepresented in the ileum was transcriptional regulation by MECP2 (Supplementary Fig. 9b), whose expression has been shown to play a role in intestinal morphology and function39.
In conclusion, using our framework, we pinpointed tissue-specific and shared epithelial ligands and immune cell receptors participating in the intercellular signalling through which SARS-CoV-2 infected epithelial cells can affect the inflammatory responses of various immune cell types during infection (Fig. 9).

SARS-CoV-2 directly infects colonic and ileal immature enterocytes. Upon infection, transcription factors ATF2/3, JUN, FOS, STAT1 and NFKB1 are modulated, resulting in altered intracellular signalling pathways and altered ligands expression, including upregulation of chemokines (CXCLs) and human leukocyte antigens (HLAs), and downregulation of laminins (LAMB/Cs). Altered ligands expression drives differential intercellular interactions between epithelial ligands and immune cell receptors (chemokine receptors (CXCRs), killer cell lectin-like receptors (KLRs) and integrins (ITGs)) expressed on immune cells.
Implication of epithelial ligands in the inflammatory process
The analysis of our integrated framework of intracellular and intercellular signalling networks in intestinal epithelial cells upon infection has helped point out several differentially expressed ligands participating in epithelial–immune interactions potentially playing a role in driving the inflammatory process upon SARS-CoV-2 infection. To validate their importance during immune reactions, we integrated these predictions with independent data from three previously published studies (Fig. 1).
First, by comparing the differentially expressed ligands upon SARS-CoV-2 infection to DEGs in human colonic organoids exposed to inflammatory cytokines40, we identified 24 ligands whose expression change is regulated by cytokines during intestinal inflammation (Table 4). These ligands are likely to contribute to the inflammatory responses upon infection. Next, by comparing ileal and colonic ligands to data from ImmunoGlobe, a manually curated intercellular immune interaction network41 and ImmunoeXpresso, a collection of cell–cytokine interactions generated through text mining42, we identified 12 ligands previously known to influence immune cell populations (Table 4). The full list of affected immune cell types for each epithelial ligand is available in Table 4. Finally, to understand which ileal and colonic ligands could explain blood cytokine level changes of COVID-19 patients via direct immune cell regulation, we used data from5, and identified six ligands capable to create the detected blood cytokine levels during infection (Table 4).
Using this assessment, we were able to rank the differentially expressed ligands for their importance in the inflammatory process, and subsequently listed the 18 highest ranked ligands, for which there is strong evidence of their role in epithelial–immune cell interactions during the inflammatory SARS-CoV-2 disease response (Table 4). These ligands included CSF1, various chemokines (CXCL10, CXCL11, CXCL2, CXCL3, CCL5, CX3CL1, CXCL8), TNFa and TNFSF13b, and ICAM1 among the upregulated ones; and various laminins (LAMC2, LAMB3), AREG, B2M), human leukocyte antigens (HLAs) (HLA-A, HLA-B) and IL32 among the downregulated ones.
The intracellular and intercellular signalling are also altered in the upper airways in patients with moderate COVID-19
The presented integrated framework can also be applied to infected epithelial cell data from other organs to reveal the effects of viral infection on that specific organ’s function. To show the ease of applicability of our framework to other infected tissues, we have employed it to analyse the effect of SARS-CoV-2 on the intracellular and intercellular signalling of upper airway ciliated epithelial cells during moderate COVID-19 cases (Methods and Fig. 1). Functional analysis of the PPI layer of the intracellular signalling network of infected ciliated cells upon moderate COVID-19 revealed an alteration of pathways related to cell motility and migration, cell adhesion mediated by the ECM (laminin, non-integrins interactions), pro-inflammatory signalling pathways (Interleukin, MAPK, PI3K, NF-kB signalling), cell cycle arrest and intestinal homeostasis (Supplementary Fig. 12).
Intercellular interactions between upper airways ciliated epithelial cells and resident immune cells were mainly driven by upregulated ligands, particularly chemokines (CXCL1/3/6) and HLAs (HLA-A/B/C) binding to chemokine receptors (CCRs, CX(3)CRs) and CD3D/G, KLRs and KIRs (expressed on T-cells and NK cells), respectively (Supplementary Fig. 12 and Supplementary Fig. 13a, b). Interestingly, the strongest upregulated interactions were driven by chemokines and HLAs targeting T-cells (cytotoxic and regulatory), macrophages (resident and non-resident) and B cells, with a second cluster of slightly weaker interactions including NK, dendritic and mast cells (Supplementary Fig. 13c). Together, these results point towards increased recruitment and antigen presentation to these immune cell types. Functional overrepresentation analysis showed that upregulated interactions were related to chemokines/cytokines signalling, antigen processing and presentation, activation of the innate and adaptive immune system and general defense response (Supplementary Fig. 12).
Conversely, downregulated interactions were driven by epidermal growth factor receptor (EGFR) binding to several different receptors, and the ECM protein Tenascin C (TNC) binding to integrins (ITGs) on immune cells (Supplementary Fig. 12 and Supplementary Fig. 13a, b). Downregulated interactions involved most immune cells, with the strongest interactions targeting non-resident macrophages and regulatory T cells (Supplementary Fig. 13c). Functional overrepresentation analysis showed that downregulated interactions were involved in haemostasis and cell adhesion processes mediated by the ECM components (integrin, laminin, syndecan) (Supplementary Fig. 12).
Discussion
In this work, we have developed an integrated framework to model how altered intracellular signalling in epithelial cells drives a different epithelial–immune interactome upon infection. As a proof of concept study, we first applied this model to highlight the putative role of the gut during the immune response following SARS-CoV-2 infection, showing how several intracellular and intercellular mechanisms are affected, with key differences between colon and ileum. A visual schematic of our key findings can be found in Fig. 9. Additionally, we proved the applicability of this framework to other tissues of interest by analysing intra and intercellular interactions of the upper airway epithelium in moderate COVID-19 patients, confirming many of the findings highlighted in the literature, and pointing out key cell–cell interactions of interest.
SARS-CoV-2 has been shown to actively infect and reproduce in the human gut and in human gastrointestinal organoids10,11,13. However, the exact effect of intestinal inflammation and the role of epithelial–immune interactions in the hyperinflammatory immune response (“cytokine storm”) characterising many COVID-19 patients are not known5,6. Accurately modelling these interactions could help identify potential targets that are key to selectively disrupt such cell–cell interactions underlying extreme inflammatory conditions during SARS-CoV-2 infection. This would be extremely important given the failure of most randomised control trials associated with pro-inflammatory drug candidates for COVID-1943.
The altered epithelial–immune cell crosstalk during SARS-CoV-2 infection has been explored within the nasopharynx and lungs using scRNA seq data44. This study found stronger epithelial–immune cell interactions in critically ill patients based on ligand–receptor expression profiles, highlighting the importance of the crosstalk between infected cells and local immune cells in the disease course. However, to our knowledge, no prior study has been carried out so far to investigate the effect of viral infection in host intestinal cells, and the role and contribution of intestinal epithelial cell–immune cell crosstalk during SARS-CoV-2 infection.
In this work, we developed an integrated pipeline to model the effect of intracellular signalling perturbation in epithelial cells on the epithelial–immune interactome in the gut. As a proof-of-concept, we exploited our previously published data on SARS-CoV-2 (BavPat1/2020) infection in intestinal organoids13 to investigate the effect of SARS-CoV-2 proteins and potential miRNAs on ileal and colonic epithelial cell intracellular signalling and function. We added in a distinguishable way the analysis of these potential miRNAs encoded by SARS-CoV-2, as previous studies highlighted the regulatory role of similar miRNAs produced by RNA viruses and their ability to downregulate host genes and affecting host functions45,46,47. Furthermore, we modelled how specific epithelial ligands, whose expression was altered upon infection, were driving specific epithelial–immune interactions via their altered binding to receptors expressed on resident immune cell populations20,21.
While our previous data pointed towards immature enterocytes as the prime target of SARS-CoV-2 infection, the application of our integrated pipeline allowed us to model how this epithelial population, when directly infected, also drives the majority of interactions with gut resident immune cells stemming from their differentially regulated ligands by SARS-CoV-2 (Fig. 2a). Upon infection of immature enterocytes, intracellular signalling pathways were altered, with a direct effect on pathways of inflammation, apoptosis, cell survival and cell death (Fig. 3). Pathways related to cell cycle (negative regulation of G2/M transition) and cell proliferation were also altered upon infection (Fig. 3), in line with a previous phosphoproteomics study finding a correlation with cell cycle arrest upon SARS-CoV-2 infection48. Finally, pathways involved in cell differentiation, cell migration and epithelial polarisation were also modulated upon infection (Fig. 3), which to our knowledge no other study had highlighted before.
By using available ligand–receptor interaction data, we aimed to understand how infected gut epithelial cells recruit resident immune cell populations to find key interactions driving the immune response during infection. Our analysis revealed that IgA plasma cells were the immune cell population with the highest number of cell–cell interactions upon infection, with the highest number of epithelial–immune interactions driven by downregulated epithelial ligands (29) in the colon, and upregulated epithelial ligands (20) in the ileum (Fig. 2a). A possible explanation for these observed tissue-specific differences and on the role of IgA plasma cells can be found in the Supplementary Discussion section.
By further analysing the specific ligand–receptor interactions driving epithelial–immune crosstalk upon SARS-CoV-2 infection, we could observe that strong upregulated interactions upon infection were mostly shared by both colon and ileum, and were represented by chemokine and TNF-α driven interactions, possibly reflecting a general effect of the inflammation process (Figs. 6 and 7). Functional analysis highlighted a relation to proinflammatory signalling pathways, including TNF-α signalling, interleukin signalling and chemotaxis via GPCR signalling, overall suggesting an increasing recruitment and cell adhesion of these immune cell populations upon infection (Supplementary Fig. 9). Notably, four chemokine receptors identified by our study (CXCR6 in the ileum, CCR1/2 and CCR9 in both ileum and colon) are coded in a genomic region found to be a COVID-19 risk locus on chromosome 3, further validating our predictions49.
Conversely, we could observe that strong downregulated interactions were driven by epithelial HLAs (HLA-A, B, C) and B2M, a subcomponent of the major histocompatibility complex I (MHC I) in both tissues (Fig. 6). According to our analysis, these ligands were mainly binding to KLR receptors, which are mainly presented on NK cells (Fig. 6, Supplementary Fig. 8). Downregulation of HLA-KLR interactions may represent an immune evasion mechanism50 that a recent study proposed as a way SARS-CoV-2 protein ORF8 uses to escape host immune surveillance51.
Uniquely in the colon, strong downregulated interactions were driven by epithelial laminins (LAMB3 and LAMC2) and integrins, with T cells and macrophages as the main immune cell types targeted upon infection (Figs. 6 and 7). Laminin–integrin binding contributes to focal adhesion of immune cells to the inflamed tissue52, and downregulation of laminins could represent an additional strategy for immune evasion following viral infection uniquely in the colon. Furthermore, laminins are known to play a role in shaping the architecture of intestinal mucosa, and an altered expression has been observed in Crohn’s disease, a type of IBD, driven by pro-inflammatory cytokines TNF-α and IFN-γ53,54,55. Finally, an additional mechanism that SARS-CoV-2 may use to evade the immune response via the downregulation of calmodulin-phosphodiesterases interactions is further discussed in the Supplementary Discussion section.
With our integrated framework, we provided a key tool to study the effect of intracellular signalling perturbation in gut epithelial cells driving differential epithelial–immune interactions. By applying this workflow on SARS-CoV-2 infected organoids scRNA seq data, we confirmed many of the previous findings about SARS-CoV-2 infection, including the induced pro-inflammatory responses driven by chemokines and the role played by T cells (Fig. 9). Additionally, we uncovered mechanisms by which SARS-CoV-2 may evade the immune responses by interfering with epithelial–immune cell connections. Such mechanisms include downregulation of antigen presentation mediated by HLAs-KLR interactions and of focal adhesion pathways mediated by laminin–integrins interactions (Fig. 9).
In this work, we highlighted a set of intestinal epithelial ligands and immune cell populations implicated in altered epithelial–immune interactions during SARS-CoV-2 infection, which could potentially drive the excessive inflammatory processes seen in severe COVID-19 patients (Table 4). Further experimental validation of bioinformatics predictions is key to validate these processes and the main molecules and cell types involved. To this end, intestinal organoids represent an excellent in vitro model to enable such validations56. Currently, introduction of immune cells to an organoid system is a challenging task. Yet, a recent study where human intestinal CD4+ T cells have been co-cultured with human intestinal organoids57, may represent a promising set-up for future studies to investigate epithelial–immune cell interactions during SARS-CoV-2 induced inflammation in the gut. As reviewed recently by58,59, such co-culture systems could be excellent to study intestinal host-microbe interactions, including the detailed experimental analysis of SARS-CoV-2 infection.
Nonetheless, our integrated workflow presents some limitations. When constructing the intracellular causal network, the effect of SARS-CoV-2 proteins towards human binding partners was always considered as inhibitory. However, this is not always the case. In the future, with increasingly available data, a more refined model could be generated. Furthermore, two different single cell transcriptomics datasets were used for colonic and ileal immune cell populations, due to the unavailability of both datasets from the same experiment. Similarly, IBD uninflamed data and healthy data were used for the ileum and colon respectively, as healthy control scRNAseq immune cell data for both tissues was not available at the time of the analysis. Finally, the a priori resources used to infer the intracellular and intercellular interaction networks may have some intrinsic limitations associated with them60. Specific tools such as LIANA (LIgand–receptor ANalysis frAmework; https://github.com/saezlab/liana) could be used in the future to compare across several resources available, helping to choose the one(s) providing the best overall prediction.
With our integrated workflow, we established a computational method to evaluate the effect of viral infection on host intestinal epithelial cell functions and how this consequently modulates the epithelial–immune crosstalk and immune activation. To demonstrate its applicability to other tissues, we analysed the intracellular and intercellular signalling of upper airway epithelial data of moderate COVID-19 cases. Although no specific information about the infecting SARS-CoV-2 strain was available, we were able to confirm several general findings related to COVID-19 infection previously highlighted in the literature. Following infection of ciliated cells, pro-inflammatory signalling pathways (interleukin, MAPK and NF-kB signalling) were altered, indicating an activated immune status, as previously reported48,61,62,63. Upregulated interactions were driven by chemokines binding to T cells (cytotoxic and regulatory) and macrophages, in line with the known role of chemokines in driving inflammation and immune cell recruitment14,64 and the role played by T cells macrophages in the innate and adaptive immune response to SARS-CoV-265,66. Additionally, upregulated interactions were also driven by HLAs, which are part of the MHC I complex, whose association with symptom severity has been previously highlighted67. Interestingly, the upregulation of intercellular interactions driven by HLAs is the opposite of the effect found in the intestine, which may represent a key difference in the response to SARS-CoV-2 between these two tissues. Finally, we found strong downregulated interactions between Tenascin C (TNC) and integrins, which were related to cell adhesion processes mediated by the ECM components. This is in line with a study finding that proteins associated with focal adhesion and the ECM receptors were decreased in COVID-19 lung tissue, which could indicate a dysregulation of the extracellular microenvironment in this tissue, revealing a possible mechanism of SARS-CoV-2-related lung damage68.
To conclude, we demonstrated that this workflow is not limited to the gut, but it can be easily applied to other organs and cell types (e.g. lung, kidney, heart), provided the right input data is available. The workflow is translatable and both the epithelial and the immune component are replaceable. Furthermore, the presented workflow is transferable to understand any past or future infectious disease, when transcriptomic data of infected and control tissue and viral interactors are available. In this way, our workflow could potentially and efficiently be used in any other infection studies to shed light on the potential intervention points between immune cells and infected cells.
Methods
Intercellular analysis
Input data
Intestinal epithelial cells
Single cell transcriptomics data of colonoids and enteroids infected with SARS-CoV-2 (BavPat1/2020 strain) was obtained from69. Single cell transcriptomics data of upper airway epithelial cells from moderate COVID-19 patients were obtained from44. Strain-level information about the infecting SARS-CoV-2 variant was not available in this study. The R packages ‘Mast’ and ‘Seurat’ were used to identify differentially expressed genes upon infection with SARS-CoV-2 for each epithelial cell type70,71. Specifically, directly infected or bystander cells from intestinal organoids treated with SARS-CoV-2 for 24 h were compared with the equivalent cell type from uninfected organoids. For the airway analysis, ciliated airway epithelial cells from moderate COVID-19 patients were compared with the equivalent cell type in control patients. This cell type was chosen as it is the most prevalent and most affected by SARS-CoV-2 infection together with secretory cell types44. Any genes with adjusted p value ≤0.05 and |log2 fold change (FC)| ≥ 0.5 were considered significantly differentially expressed. Differential expression could only be calculated for cell types within a condition where data was available from ≥3 individual cells.
Intestinal resident immune cells
Single cell expression data from ileal and colonic resident immune cells was obtained from20 and21, respectively. For the analyses, data from healthy samples and uninflamed Crohn’s disease samples was used for colonic and ileal immune cell populations, respectively. Single cell expression data of upper airway immune cells from moderate COVID-19 patients was obtained from44.
Immune cell populations were identified through annotated clustering from20,21,44. Cell type labels were maintained as originally published. Following removal of all genes with count = 0, normalised log2 counts across all samples (separately for each cell type) were fitted to a gaussian kernel72. All genes with expression values above mean minus three standard deviations were considered as expressed genes for the given cell type in the given intestinal location. For the intercellular ligand–receptor predictions in the colon and ileum, a representative collection of immune cells relevant in gut inflammation and SARS-CoV-2 infection based on previous literature was selected, which included all macrophages, T cells, B cells, plasma cells, Innate Lymphoid Cells (ILCs), mast cells and a representative group of dendritic cells (DCs)20,21,49,66,73. For the ligand–receptor predictions of the upper respiratory tract, all immune cell types for which information was available were used in the analysis.
Defining ligand–receptor interactions between cell types
A list of ligand–receptor interactions was obtained from OmniPath on 23 September 2020 using the ‘OmnipathR’ R package18. Source databases used to retrieve the ligand–receptor interactions through OmnipathR included six independent resources (CellPhoneDB, HPMR, Ramilowski 2015, Guide2Pharma, Kirouac 2010, Gene Ontology)25,74,75,76,77,78. No weighing was performed on ligand–receptor interactions, and protein complexes were dealt with by including each of their individual proteins in the list.
Ligand–receptor interactions (intercellular interactions; full list available at https://github.com/korcsmarosgroup/gut-COVID) were predicted between epithelial cells types and resident immune cells according to the following conditions:
-
1.
The ligand is differentially expressed in the epithelial cell (upon SARS-CoV-2 infection—in directly infected or bystander cells of the colon and ileum).
-
2.
The receptor is expressed in the immune cell in the relevant dataset (ie, ileal or colonic immune cells).
-
3.
The ligand–receptor interaction is present in OmniPath.
For the gut analysis, intercellular interactions were defined separately for directly infected epithelial cells and bystander epithelial cell populations in the ileum and in the colon. Enteroid epithelial data was paired with ileal immune cell data20, while colonoid epithelial data was paired with colonic immune cell data21. For the upper airway analysis, ciliated cell data of moderate COVID-19 samples was paired with the same cell type of control samples44. Intercellular interactions were defined between every possible pair of epithelial cells and immune cells for each condition. Interactions derived from upregulated ligands (“upregulated interactions”) were evaluated separately from interactions derived from downregulated ligands (“downregulated interactions”).
Scoring of ligands, receptors and immune cell types involved in ligand–receptor interactions
To assess the importance of specific ligands, receptors and immune cell types, additional parameters were computed using the ligand–receptor network. First, the number of interactions between each epithelial and immune cell type was computed by summing up all the possible interactions between each differentially expressed epithelial ligand and each of the receptors expressed by the specific immune cell type. Second, the number of immune cell types involved in each ligand–receptor pair was computed by counting the number of different immune cell types which were expressing the receiving receptor. Third, for each ligand, a “sum of receptor expression value” was computed for each interacting immune cell type, based on the number of interacting receptors and the mean expression level of the interacting receptors.
Data visualisation
Venn diagrams were generated using the ‘gplots’ R package. Heatmaps were generated using the ‘ggplot2’ and ‘pheatmap’ packages79,80. Barplots were generated with the ‘ggplot2’ package. Network visualisations were done using Cytoscape (version 3.8.2) (Shannon et al. 2003; Su et al. 2014). All scripts used to generate these plots are available on the Github repository of the project (https://github.com/korcsmarosgroup/gut-COVID).
Intracellular analysis
Two previously established tools were employed to predict the effect of SARS-CoV-2 infection on epithelial cells: ViralLink and CARNIVAL17,19. Both tools, using related but distinct methods, infer causal molecular interaction networks. These networks link perturbed human proteins predicted to interact with SARS-CoV-2 viral proteins or miRNAs, to transcription factors known to regulate the observed differentially expressed ligands in infected epithelial cells.
Input data
To reconstruct the intracellular causal networks, three different a priori interactions datasets were used. Information on viral proteins and their interacting human binding partners was obtained from the SARS-CoV-2 collection of the IntAct database on 1st October 202081,82. Predicted SARS-CoV-2 miRNAs and their putative human binding partners were obtained from45. Intermediary signalling protein interactions known to occur in humans were obtained from the core protein-protein interaction (PPI) layer of the OmniPath collection using the ‘OmnipathR’ R package on 7th October 202083. Only directed and signed interactions were included. Interactions between human transcription factors (TFs) and their target genes (TG) were obtained from the DoRothEA collection using the DoRothEA R package on 7th October 202084. Only signed interactions of the top three confidence levels (A, B, C) were included.
Normalised transcript counts and differentially expressed ligands were obtained from single cell transcriptomics data of (i) colonoids and enteroids infected with SARS-CoV-2 obtained from13, or (ii) upper airways samples of moderate COVID-19 patients from44, as previously described.
ViralLink pipeline
Intracellular causal networks were inferred using the ViralLink pipeline, as described in17. Briefly, a list of expressed genes in infected immature enterocytes (originally known as “immature enterocytes 2” (MMP7+, MUC1+, CXCL1+)) from SARS-CoV-2-infected ileal and colonic organoids13 or ciliated epithelial cells from moderate COVID-19 samples44 was generated from a normalised count table by fitting a gaussian kernel72. The list of expressed genes in the infected immature enterocytes population or COVID-19 ciliated epithelial cells was subsequently used to filter the a priori molecular interactions from OmniPath and DoRothEA, to obtain PPI and TF-TG sub-networks where both interacting molecules are expressed (described as “contextualised” networks). Transcription factors regulating the differentially expressed ligands were predicted using the contextualised DoRothEA TF—TG interactions and scored as described in17. Human binding proteins of viral proteins and viral miRNAs obtained from the IntAct database81,82 and45, respectively, were connected to the listed TFs through the contextualised PPIs using a network diffusion approach called Tied Diffusion Through Interacting Events (TieDIE)85. In this model, all viral protein—human binding protein interactions were assumed to be inhibitory in action, based on likely biological function, and given a lack of clear literature evidence of proven action. All viral miRNA—human binding protein interactions were set as inhibitory based on biological action of miRNAs86. The final reconstructed network contains “nodes”, which refers to the interacting partners, and “edges”, which refers to the interaction between nodes. Nodes include viral proteins and miRNAs, human binding proteins, intermediary signalling proteins, TFs and differentially expressed ligands. Edges include activatory or inhibitory interactions.
For ileal, colonic and upper airways data, separate networks were generated using the viral miRNA and viral protein as perturbations, and subsequently joined using the “Merge” function within Cytoscape to generate the final intracellular network. Nodes and edges were annotated according to their involvement in networks downstream of viral miRNAs or proteins. Further analyses were performed separately on the different layers of the network: miRNA specific, protein specific or shared nodes.
CARNIVAL pipeline
Intracellular causal networks were inferred using CARNIVAL and associated tools for analyses of expression data as described in19. For simplicity, we refer to the pipeline as described in19 as the CARNIVAL pipeline. Briefly, PROGENy is used to infer pathway activity from the log2 FC of the infected immature enterocytes 2 gene expression data87. Next, using the TF-TGs interactions (from DoRothEA) and the differential expression data from infected organoids, VIPER was used to score TF activity based on enriched regulon analysis88. Here, only the top 25 TFs regulating at least 15 target genes were taken forward, and a correction for pleiotropic regulation was included. Finally, CARNIVAL applied an integer linear programming approach to identify the most likely paths between human interaction partners of viral proteins or miRNAs and the selected TFs, through PPIs from OmniPath, considering the activity scores from PROGENy and VIPER. Viral protein—human binding protein interactions signs were specified to CARNIVAL as ‘inhibitory’, based on likely biological function, and given a lack of clear literature evidence of proven action. All viral miRNA—human binding protein interactions were also set as inhibitory based on biological action of miRNAs86.
Network functional analysis
Functional overrepresentation analysis was performed on the networks constructed as above-mentioned using the R packages ‘ClusterProfiler’ and ‘ReactomePA’, for Gene Ontology (GO)25) and for Reactome22,23,24 annotations, respectively. For the intercellular network, the analysis was carried out separately for ligand–receptor intercellular interactions driven by either upregulated or downregulated ligands. A complete list of ligand–receptor interactions is available in the GitHub repository of the project (https://github.com/korcsmarosgroup/gut-COVID). For the upregulated interactions, a list of upregulated ligands and their connecting immune receptors was used as the test. For the downregulated interactions, a list of downregulated ligands and their connecting immune receptors was used. Where a list of ligands plus receptors contained <5 genes, it was excluded from the analysis. All ligands and receptors from the original ligand–receptor network used as prior knowledge input for the intercellular analysis was used as the statistical background.
For the intracellular network, the analysis has been done separately for each of the sub-networks (viral protein specific, viral miRNA specific, or shared). For each sub-network, a set of genes that were human binding proteins, intermediary proteins and TFs in the network (“PPI layer”) was used as a test list, and a set of all nodes from the original OmniPath PPI interaction network used as prior knowledge input for the intracellular analysis was used as the statistical background. For the Reactome pathway enrichment analysis the IDs were converted to Entrez Gene ID within the ‘ReactomePA’ package. Functional categories with adjusted p value ≤ 0.05 and with gene count >3 were considered significantly overrepresented.
Selection of ligands involved in the inflammatory process
To show how our approach could help point out specific epithelial-derived ligands driving the inflammatory process upon SARS-CoV-2 infection, the list of differentially expressed ligands in infected immature enterocytes in both colon and ileum was validated using independent data from three previously published studies. To identify ligands whose expression was induced by cytokines, ligands were compared to DEGs in human colonic organoids exposed to cytokines from40. To identify ligands already known to influence immune cell population, ligands were compared to two databases: ImmunoGlobe, a manually curated intercellular immune interaction network41, and ImmunoeXpresso, a collection of cell–cytokine interactions generated through text mining42. Finally, to identify ligands that could directly explain blood cytokine level changes in COVID-19 patients via direct immune cell regulation, ligands were compared to the data from a large dataset we recently manually compiled using COVID-19 patient publications5.
Data availability
The necessary input data for the workflow and the full ligand–receptor interaction tables are available in the GitHub repository of the project (https://github.com/korcsmarosgroup/gut-COVID). All other relevant data is in the main text and in Supplementary files.
Code availability
The code of the entire workflow is available in the GitHub repository of the project (https://github.com/korcsmarosgroup/gut-COVID).
References
Tay, M. Z., Poh, C. M., Rénia, L., MacAry, P. A. & Ng, L. F. P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 20, 363–374 (2020).
Mick, E. et al. Upper airway gene expression reveals suppressed immune responses to SARS-CoV-2 compared with other respiratory viruses. Nat. Commun. 11, 5854 (2020).
Peacock, T. P. et al. The SARS-CoV-2 variant, Omicron, shows rapid replication in human primary nasal epithelial cultures and efficiently uses the endosomal route of entry. Preprint at https://www.biorxiv.org/content/10.1101/2021.12.31.474653v1 (2022).
Gupta, A. et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 26, 1017–1032 (2020).
Olbei, M. et al. SARS-CoV-2 causes a different cytokine response compared to other cytokine storm-causing respiratory viruses in severely ill patients. Front. Immunol. 12, 629193 (2021).
Arunachalam, P. S. et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369, 1210–1220 (2020).
Stone, J. H. et al. Efficacy of tocilizumab in patients hospitalized with Covid-19. N. Engl. J. Med. 383, 2333–2344 (2020).
Kale, S. D. et al. “Small” intestinal immunopathology plays a “Big” role in lethal cytokine release syndrome, and its modulation by interferon-γ, IL-17A, and a Janus kinase inhibitor. Front. Immunol. 11, 1311 (2020).
Martin-Cardona, A. et al. SARS-CoV-2 identified by transmission electron microscopy in lymphoproliferative and ischaemic intestinal lesions of COVID-19 patients with acute abdominal pain: two case reports. BMC Gastroenterol. 21, 334 (2021).
Lamers, M. M. et al. SARS-CoV-2 productively infects human gut enterocytes. Science 369, 50–54 (2020).
Stanifer, M. L. et al. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep. 32, 107863 (2020).
Zang, R. et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 5, 47 (2020).
Triana, S. et al. Single-cell analyses reveal SARS-CoV-2 interference with intrinsic immune response in the human gut. Mol. Syst. Biol. 17, e10232 (2021).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045.e9 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Guo, M., Tao, W., Flavell, R. A. & Zhu, S. Potential intestinal infection and faecal–oral transmission of SARS-CoV-2. Nat. Rev. Gastroenterol. & Hepatol. 18, 269–283 (2021).
Treveil, A. et al. ViralLink: an integrated workflow to investigate the effect of SARS-CoV-2 on intracellular signalling and regulatory pathways. PLoS Comput. Biol. 17, e1008685 (2021).
Türei, D. et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 17, e9923 (2021).
Liu, A. et al. From expression footprints to causal pathways: contextualizing large signaling networks with CARNIVAL. NPJ Syst. Biol. Appl. 5, 40 (2019).
Martin, J. C. et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 178, 1493–1508.e20 (2019).
Smillie, C. S. et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730.e22 (2019).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Yu, G. & He, Q.-Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 12, 477–479 (2016).
Fabregat, A. et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 46, D649–D655 (2018).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
Cartwright, T., Perkins, N. D. & Wilson, L. C. NFKB1: a suppressor of inflammation, ageing and cancer. FEBS J. 283, 1812–1822 (2016).
Persengiev, S. P. & Green, M. R. The role of ATF/CREB family members in cell growth, survival and apoptosis. Apoptosis 8, 225–228 (2003).
Mehic, D., Bakiri, L., Ghannadan, M., Wagner, E. F. & Tschachler, E. Fos and jun proteins are specifically expressed during differentiation of human keratinocytes. J. Invest. Dermatol. 124, 212–220 (2005).
Carulli, A. J. et al. Notch receptor regulation of intestinal stem cell homeostasis and crypt regeneration. Dev. Biol. 402, 98–108 (2015).
Yamada, Y. et al. Functional roles of TGF-β1 in intestinal epithelial cells through Smad-dependent and non-Smad pathways. Dig. Dis. Sci. 58, 1207–1217 (2013).
Shuai, K. & Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3, 900–911 (2003).
Wen, A. Y., Sakamoto, K. M. & Miller, L. S. The role of the transcription factor CREB in immune function. J. Immunol. 185, 6413–6419 (2010).
Valcourt, U., Kowanetz, M., Niimi, H., Heldin, C.-H. & Moustakas, A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 16, 1987–2002 (2005).
Osaki, L. H. & Gama, P. MAPKs and signal transduction in the control of gastrointestinal epithelial cell proliferation and differentiation. Int. J. Mol. Sci. 14, 10143–10161 (2013).
Alejo, A. et al. Chemokines cooperate with TNF to provide protective anti-viral immunity and to enhance inflammation. Nat. Commun. 9, 1790 (2018).
Mantegazza, A. R., Magalhaes, J. G., Amigorena, S. & Marks, M. S. Presentation of phagocytosed antigens by MHC class I and II. Traffic 14, 135–152 (2013).
Mezu-Ndubuisi, O. J. & Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr. Res. 89, 1619–1626 (2021).
Millar-Büchner, P. et al. Severe changes in colon epithelium in the Mecp2-null mouse model of Rett syndrome. Mol. Cell. Pediatr. 3, 37 (2016).
Pavlidis, P. et al. Cytokine responsive networks in human colonic epithelial organoids unveil a novel molecular stratification of inflammatory bowel disease. SSRN J. https://doi.org/10.2139/ssrn.3823433 (2021).
Atallah, M. B. et al. ImmunoGlobe: enabling systems immunology with a manually curated intercellular immune interaction network. BMC Bioinforma. 21, 346 (2020).
Kveler, K. et al. Immune-centric network of cytokines and cells in disease context identified by computational mining of PubMed. Nat. Biotechnol. 36, 651–659 (2018).
Abubakar, A. R. et al. Systematic review on the therapeutic options for COVID-19: clinical evidence of drug efficacy and implications. Infect. Drug Resist. 13, 4673–4695 (2020).
Chua, R. L. et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020).
Saçar Demirci, M. D. & Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. PeerJ 8, e9369 (2020).
Griffiths-Jones, S., Saini, H. K., van Dongen, S. & Enright, A. J. miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–8 (2008).
Bruscella, P. et al. Viruses and miRNAs: more friends than foes. Front. Microbiol. 8, 824 (2017).
Bouhaddou, M. et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182, 685–712.e19 (2020).
Schultze, J. L. & Aschenbrenner, A. C. COVID-19 and the human innate immune system. Cell 184, 1671–1692 (2021).
Koutsakos, M. et al. Downregulation of MHC class I expression by influenza A and B viruses. Front. Immunol. 10, 1158 (2019).
Park, M. D. Immune evasion via SARS-CoV-2 ORF8 protein? Nat. Rev. Immunol. 20, 408 (2020).
Simon, T. & Bromberg, J. S. Regulation of the immune system by laminins. Trends Immunol. 38, 858–871 (2017).
Mahoney, Z. X., Stappenbeck, T. S. & Miner, J. H. Laminin alpha 5 influences the architecture of the mouse small intestine mucosa. J. Cell Sci. 121, 2493–2502 (2008).
Bouatrouss, Y., Herring-Gillam, F. E., Gosselin, J., Poisson, J. & Beaulieu, J. F. Altered expression of laminins in Crohn’s disease small intestinal mucosa. Am. J. Pathol. 156, 45–50 (2000).
Francoeur, C., Escaffit, F., Vachon, P. H. & Beaulieu, J.-F. Proinflammatory cytokines TNF-alpha and IFN-gamma alter laminin expression under an apoptosis-independent mechanism in human intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G592–8 (2004).
Kim, J., Koo, B.-K. & Knoblich, J. A. Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 21, 571–584 (2020).
Schreurs, R. R. C. E., Baumdick, M. E., Drewniak, A. & Bunders, M. J. In vitro co-culture of human intestinal organoids and lamina propria-derived CD4 + T cells. STAR Protoc. 2, 100519 (2021).
Poletti, M., Arnauts, K., Ferrante, M. & Korcsmaros, T. Organoid-based models to study the role of host-microbiota interactions in IBD. J. Crohns Colitis 15, 1222–1235 (2021).
Min, S., Kim, S. & Cho, S.-W. Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp. Mol. Med. 52, 227–237 (2020).
Dimitrov, D. et al. Comparison of Resources and Methods to infer Cell–Cell Communication from Single-cell RNA Data. Preprint at https://www.biorxiv.org/content/10.1101/2021.05.21.445160v1 (2021).
Su, C.-M., Wang, L. & Yoo, D. Activation of NF-κB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 11, 13464 (2021).
Mardi, A., Meidaninikjeh, S., Nikfarjam, S., Majidi Zolbanin, N. & Jafari, R. Interleukin-1 in COVID-19 Infection: Immunopathogenesis and Possible Therapeutic Perspective. Viral Immunol. 34, 679–688 (2021).
Vaz de Paula, C. B. et al. IL-4/IL-13 remodeling pathway of COVID-19 lung injury. Sci. Rep. 10, 18689 (2020).
Koenig, L. M. et al. Blocking inflammation on the way: Rationale for CXCR2 antagonists for the treatment of COVID-19. J. Exp. Med. 217, 9 (2020).
Yang, D. et al. Attenuated interferon and proinflammatory response in SARS-CoV-2-infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J. Infect. Dis. 222, 734–745 (2020).
Sette, A. & Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 184, 861–880 (2021).
Castelli, E. C. et al. MHC variants associated with symptomatic versus asymptomatic SARS-CoV-2 infection in highly exposed individuals. Front. Immunol. 12, 742881 (2021).
Leng, L. et al. Pathological features of COVID-19-associated lung injury: a preliminary proteomics report based on clinical samples. Signal Transduct. Target. Ther. 5, 240 (2020).
Triana, S., Stanifer, M. L., Boulant, S. & Alexandrov, T. COVID19_July.rda. Figshare https://doi.org/10.6084/m9.figshare.13703752.v1 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
McDavid, A., Finak, G. & Yajima, M. MAST: Tools and methods for analysis of single cell assay data in R. R package version 1.18.0 https://github.com/RGLab/MAST/ (2021).
Beal, J. Biochemical complexity drives log-normal variation in genetic expression. Eng. biol. 1, 55–60 (2017).
Filbin, M. R. et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell–cell interactions. Cell Rep. Med. 2, 100287 (2021).
Ben-Shlomo, I., Yu Hsu, S., Rauch, R., Kowalski, H. W. & Hsueh, A. J. W. Signaling receptome: a genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci. STKE 2003, RE9 (2003).
Ramilowski, J. A. et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 6, 7866 (2015).
Pawson, A. J. et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 42, D1098–106 (2014).
Vento-Tormo, R. et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018).
Kirouac, D. C. et al. Dynamic interaction networks in a hierarchically organized tissue. Mol. Syst. Biol. 6, 417 (2010).
ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag New York, 2016).
Kolde, R. Pheatmap: pretty heatmaps. R. package version 1. 2 1, 726 (2012).
Hermjakob, H. et al. IntAct: an open source molecular interaction database. Nucleic Acids Res. 32, D452–5 (2004).
Orchard, S. et al. The MIntAct project - IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–63 (2014).
Türei, D., Korcsmáros, T. & Saez-Rodriguez, J. OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 13, 966–967 (2016).
Garcia-Alonso, L., Holland, C. H., Ibrahim, M. M., Turei, D. & Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 29, 1363–1375 (2019).
Paull, E. O. et al. Discovering causal pathways linking genomic events to transcriptional states using Tied Diffusion Through Interacting Events (TieDIE). Bioinformatics 29, 2757–2764 (2013).
Huang, R. S. et al. Population differences in microRNA expression and biological implications. RNA Biol. 8, 692–701 (2011).
Schubert, M. et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 9, 20 (2018).
Alvarez, M. J. et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 48, 838–847 (2016).
Acknowledgements
M.O., A.T., L.G., and M.P. are supported by the UKRI Biotechnological and Biosciences Research Council (BBSRC) funded Norwich Research Park Biosciences Doctoral Training Partnership (grant numbers BB/M011216/1 and BB/S50743X/1). The work of T.K. and D.M. was supported by the Earlham Institute (Norwich, UK) in partnership with the Quadram Institute (Norwich, UK) and strategically supported by the UKRI BBSRC UK grants (BB/J004529/1, BB/P016774/1, and BB/CSP17270/1). T.K. and D.M. were also funded by a BBSRC ISP grant for Gut Microbes and Health BB/R012490/1 and its constituent projects, BBS/E/F/000PR10353 and BBS/E/F/000PR10355. T.A. and S.T. acknowledge the funding from the Darwin Trust of Edinburgh and from the ERC Consolidator grant METACELL from European Union’s Horizon 2020 programme. T.A. and S.T. acknowledge support from the EMBL Genomics Core Facility and particularly help from Vladimir Benes. B.V. is supported by the Clinical Research Fund (KOOR) University Hospitals Leuven. S.B. was supported by research grants from the Deutsche Forschungsgemeinschaft (DFG): project numbers 415089553 (Heisenberg program), 240245660 (SFB1129), 278001972 (TRR186), and 272983813 (TRR179), the state of Baden Wuerttemberg (AZ: 33.7533.-6-21/5/1) and the Bundesministerium Bildung und Forschung (BMBF) (01KI20198A). M.L.S. was supported by the DFG (416072091) and the BMBF (01KI20239B). D.T. was supported by the Federal Ministry of Education and Research (BMBF, Computational Life Sciences grant no. 031L0181B) to J.S.R.
Author information
Authors and Affiliations
Contributions
M.P and A.T contributed equally to this manuscript. M.P., A.T., L.C., L.G. and D.M. contributed to the study design, data analysis and drafting the manuscript. M.M., M.O. and B.B. contributed to the initial input data processing and bioinformatics pipeline development. A.V., D.T., S.T. contributed with data generation and supervision during data analysis and bioinformatics pipeline development. B.V., T.A., J.S.R., M.L.S., S.B. and T.K. led the initial study design, supervised the work and contributed to writing the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The Authors declare no Competing Non-Financial Interests but the following Competing Financial Interests: B. Verstockt reports financial support for research from Pfizer; lecture fees from Abbvie, Biogen, Chiesi, Falk, Ferring, Galapagos, Janssen, MSD, Pfizer, R-Biopharm, Takeda and Truvion; consultancy fees from Janssen, Guidepont and Sandoz, all outside of the submitted work; JSR received funding from GSK and Sanofi and consultant fees from Travere Therapeutics.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poletti, M., Treveil, A., Csabai, L. et al. Mapping the epithelial–immune cell interactome upon infection in the gut and the upper airways. npj Syst Biol Appl 8, 15 (2022). https://doi.org/10.1038/s41540-022-00224-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-022-00224-x
This article is cited by
-
Mucosal-associated invariant T cells in infectious diseases of respiratory system: recent advancements and applications
Journal of Inflammation (2024)
