
Abstract
Aluminum current collectors are widely used in nonaqueous batteries owing to their cost-effectiveness, lightweightness, and ease of fabrication. However, they are excluded from aqueous batteries due to their severe corrosion in aqueous solutions. Here, we propose hydrolyzation-type anodic additives to form a robust passivation layer to suppress corrosion. These additives dramatically lower the corrosion current density of aluminum by nearly three orders of magnitude to ~10−6 A cm−2. In addition, realizing that electrochemical corrosion accompanies anode prelithiation, we propose a prototype of self-prolonging aqueous Li-ion batteries (Al ||LiMn2O4 ||TiO2), whose capacity retention rises from 49.5% to 70.1% after 200 cycles. A sacrificial aluminum electrode where electrochemical corrosion is utilized is introduced as an electron supplement to prolong the cycling life of aqueous batteries. Our work addresses the short-life issue of aqueous batteries resulting from the corrosion of the current collector and lithium loss from side reactions.
Similar content being viewed by others


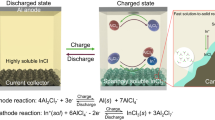
Introduction
Aqueous Li-ion batteries (ALIBs) show promise as large-scale energy storage technology due to their nonflammability and environmental friendliness1,2,3,4,5,6. However, their cycle life is inadequate for the demand in energy storage due to irreversible parasitic side reactions from their cathode and anode sides.
The cathode side expectedly features cycling fading in the form of irreversible oxidation corrosion of metal current collectors in aqueous electrolytes4,6,7,8,9. Thus, cost-effective, manufacture-friendly, and lightweight Al foils are excluded as current collectors in ALIBs because of the harsh aqueous environment attacks on the Al2O3 passivation film and continuous corrosion of Al, which damage the electronic matrix and result in contact loss and high resistance of electrodes10. To address these issues, scholars have used durable metal current collectors, including titanium11, nickel12, stainless steel3,9, and carbon foil13,14,15,16,17, to reduce the effects of corrosion on ALIBs. However, almost all of these candidates fail as substitutes to Al current collectors, which can simultaneously satisfy the lightweight, conductivity, mechanical strength, and cost-effectiveness requirements.
Different from nonaqueous electrolytes that passivate Al through the addition of effective salts, such as LiPF6, LiBF4, LiDFOB, or optimization of solvents with a low dielectric constant10,18,19,20,21,22,23,24,25,26,27,28, the passivation of Al metal foil in aqueous solutions is considerably more challenging because of the incompatibility of the salts and water. On the one hand, solvent water possesses a relatively high dielectric constant (78.4 F m−1 at 25 °C), which promotes the diffusion of corrosion products and accelerates Al corrosion18,19; on the other hand, the formation of solid electrolyte interphase (SEI) in aqueous electrolytes highly depends on organic salts, such as bis(trifluoromethane sulfonyl)imide (LiTFSI), and lithium triflouromethanesulfonate (LiOTF)3, whose anions are aggressive against Al regardless of the use of nonaqueous or aqueous electrolytes18,19,20,26,27,28,29. Therefore, addressing the corrosion of Al by aqueous LiTFSI electrolyte poses an important challenge in the field of aqueous batteries. Although the corrosion of Al current collectors can be suppressed through the increase in salt concentration4,6 since high concentration and high viscosity decrease the dissolving capability and hinder the departure of corrosion products from the of Al surface4,6,20,21,22, high concentration also increases the cost against the original intention of employing low-cost Al current collectors.
From the anode side, another failure mechanism of cycling life results from lithium loss caused by the formation of a SEI and hydrogen evolution reaction (HER), which lead to a rapid capacity degradation. Typically, SEI formation in traditional nonaqueous LIBs irreversibly consumes 6%–15% Li ions, which are extracted from the cathode material, and reduces the specific energy by 5%–20% during the initial cycle30,31. However, as the extra HER that consumes the Li resource does not contribute to SEI formation, irreversible Li consumption is expectedly more severe in ALIBs5,32; this condition results in a high P/N (cathode-to-anode mass ratio)>1.5, which offsets the lithium compensation for the capacity loss from HER and SEI.
Based on the above information, the critical challenge in ALIB application is the stabilization of the Al current collector in low-concentration aqueous electrolytes. Here, we proposed a prolonged life strategy for aqueous batteries that involves the electrochemical regulation of the corrosion and passivation of Al to make it an electron supplement and a stable current collector. We used the hydrolyzation type-anodic additive (HTA) to achieve electrochemical–chemical passivation of the Al current collector and successfully precipitated Al(OH)3 as a passivating layer on the Al surface. In addition, the passivation process, which involves Al oxidation at the cathode, can supplement electrons to anode, which promotes the compensation of the capacity loss resulting from hydrogen evolution at the anode side. The oxidation of Al is an electron supplement process, with an additional lithium source originating from the electrolyte as supplement. Therefore, in this context, an “electron supplement” was used instead of “lithium supplement.” Furthermore, considering practical industrial applications, we designed a prototype of self-prolonging ALIBs (SP-ALIBs) (Al ||LiMn2O4||TiO2) with an Al-sacrificing electrode, which enabled self-lithium supplementation via periodic switching between either full LiMn2O4||TiO2 or Al||TiO2 in combination with electrochemical regulation.
Results
Design of Al-passivating additives in an aqueous electrolyte
Figure 1 illustrates the schematic of our proposed HTA-passivating mechanism for Al current collectors in aqueous LiTFSI electrolytes. As reported previously18,26, TFSI− aggressively destroys the original Al2O3 layer to form dissolved [Al(TFSI)x3−x] at the initial active spots of the Al surface film (TFSI−(aq) + Al2O3 \(\to\) Al(TFSI)x3−x(aq) + O2 + e−). Then, more active spots emerged, which caused pitting corrosion (Al(s) – e− + TFSI−(aq) \(\to\) Al(TFSI)x3−x(aq)). Therefore, given the inevitability of attacks from TFSI− to the original passivation Al2O3 layer, our design principle was aimed at the chemical conversion of the initial electrochemical corrosion product to insoluble solid-phase aluminum compounds and coverage of the Al surface as a passivation layer. Previous literature proposed the protection of Al from chloride corrosion through the construction of inorganic passivation layers33,34. Given this information, some insoluble aluminum compounds (AlaMb), such as Al(OH)3, can serve as target passivation products due to their relatively low solubility product constant; studies have reported the corrosion resistance of these compounds against chloride ions34,35. We applied the universal corrosion principles in aqueous batteries to prevent the Al current collector from being corroded by TFSI−. To form a robust and dense AlaMb layer and restore the passivation layer on the Al surface, we contemplated on the use of Al ions generated by electrochemical corrosion during battery charging (Al(s) – e− \(\to\) Al3+) to chemically transform Al into solid-phase depositions and form a passivation layer (reaction path: Al(s) – e− + HxMn−(aq) + H2O → Al(OH)3(s) + Hx+1M1−n(aq)) instead of dissolution in electrolytes as [Al(TFSI)x3−x]. First, electrochemical corrosion occurs, followed by chemical passivation that is, electrochemical–chemical passivation. Based on preceding findings, slightly soluble lithium weak acid salts, such as Li3PO4, Li2CO3, Li2SiO3, and LiAlO2, were proposed as passivation additives for Al in aqueous electrolytes, whose anions after hydrolysis can combine with proton. These salts also perform a function similar to pH buffers in proximity to the Al foil and facilitate the formation of Al(OH)3. Supplementary Information Note 2 provides a detailed discussion on the role of HTA.

Schematic of HTA-passivating Al mechanism in an aqueous LiTFSI electrolyte.
To verify the anticorrosion effectiveness of HTA, we performed CA experiments using LiTFSI solution on three-electrode devices (Al-WE||Ag/AgCl-RE||Al-CE), where the potential of Al-WE was set at 4.5 V vs Li/Li+ for 10 minutes and then relaxed to the open-circuit potential (OCV) for 1 minute, for a total of 12 cycles (Fig. 2A). This periodic switching between 4.5 V and OCV eliminated the effect on transport limitations at the interface, which ensured that the experiment proceeded under harsh conditions. HTAs (Li3PO4, Li2CO3, LiAlO2, and Li2SiO3) were added to 1 and 10 m LiTFSI solutions, whose content was saturated (<0.05 m). Al in the blank electrolyte (1 m LiTFSI) had a corrosion current density extremely higher than 10−3 A cm-2, which indicates its severe corrosion in the diluted solution (Fig. 2A). Notably, after the introduction of saturated LiAlO2, Li2CO3, and Li3PO4, the corrosion current density dropped dramatically by nearly three orders of magnitude (10−6 A cm−2), which indicates a satisfying anticorrosion effectiveness. The CA experiments (Fig. 2A, B) were repeated twice to verify our claims (Figs. S7 and S8, respectively). Moreover, Supplementary Information Note 2 discusses the influence of the HTA amount on corrosion resistance.

A, B Chronoamperometry (CA) experiments in 1 and 10 m LiTFSI solutions with saturated HTA (Li3PO4, Li2CO3, LiAlO2, and Li2SiO3). C, D Al contents in electrolytes obtained via inductive coupled plasma (ICP) measurement and SEM images of Al foils after CA experiments using 1 and 10 m LiTFSI solutions with HTA. E Al 2p X-ray photoelectron spectroscopy (XPS) spectra of Al foil after CA experiments using 10 m LiTFSI with HTA before and after 60 s Ar+ sputtering. The blue vertical line is based on the Al2O3 peak of pristine Al.
To further support our claim, we used inductive coupled plasma (ICP) to detect the dissolved Al ion content in the electrolyte and scanning electron microscopy (SEM) to detect corrosion damage on the morphologies of Al foil after CA experiments (Fig. 2C, D). The determined Al contents of the electrolyte with Li3PO4 or Li2CO3 reached 0.67 and 1.8 ppm, respectively, which are considerably lower than that of in blank 1 m LiTFSI (46.29 ppm). In addition, the corrosive pits were scarce and tiny compared with those of Al foil in 1 m LiTFSI. Furthermore, the cycled Al foils in 1 m LiTFSI with additives were washed in ethanol to prevent residual electrolyte from occupying their surface and immersed in 1 m LiTFSI to repeat the CA experiment under the same condition. The foils still exhibited an excellent anticorrosion effect with a low corrosive current density (Figure S3) and low content of dissolved Al (Li3PO4: 0.46 ppm, LiAlO2: 0.73 ppm, Li2CO3: 1.19 ppm, and Li2SiO3: 2.26 ppm; Fig. S4). Thus, a passivation layer formed in the first round of the CA experiment by HTA. Moreover, the passivation layer provided a robust and continuous protection to the Al foil in pure 1 m LiTFSI.
Additional corrosion experiments were conducted in 10 m LiTFSI to align with the concentrations used in actual battery systems. Ti serves as a highly corrosion-resistant current collector in aqueous batteries36. Compared with Ti, Al foils with Li3PO4 (2.35 × 10−6 A cm-2) and Li2CO3 (2.71 × 10−6 A cm-2) exhibit the same order of magnitude of corrosion current density as Ti (5.34 × 10−6 A cm−2; Figs. 2B and S8). Li2CO3 and Li3PO4 are considered much better anti-corrosion additives than Li2SiO3 (3.56 × 10−5 A cm-2), whose corrosion current density is lower by one order of magnitude than Li2SiO3. Evidently, the ICP revealed that the 10 m LiTFSI with Li2CO3 failed to detect the Al signal due to lower value than the detection minimum. The value for Li3PO4 was 0.05 ppm, which is considerably lower than that of Li2SiO3 (7.56 ppm). Such result was probably due to the substantial change in pH from 6.82 (10 m LiTFSI), 6.87 (10 m LiTFSI + Li2CO3), 6.93 (10 m LiTFSI + Li3PO4) to 9.20 (10 m LiTFSI + Li2SiO3; Fig. S2). Thus, in the following section, we focused on the HTAs (Li2CO3 and Li3PO4) in 10 m LiTFSI and evaluated their electrochemical performance.
XPS was conducted to detect the passivation layer that formed on the Al foils after the introduction of HTA (Fig. 2E). Considering the insulating Al2O3 on pristine Al foils, we used its XPS spectrum as a reference (Al2O3, Al 2p: 74.81 eV). Al(OH)3 was detected at 74.37 eV for Li3PO4, 74.27 eV for Li2CO3, and 74.19 eV for Li2SiO3 (Fig. 2E), consistent with the reported value in literature (74.4 eV)37. This finding indicates that the Al3+ originating from the oxidization of Al interacted with the anions of HTAs (HPO42−, H2PO4−, HCO3−, and HSiO3−) and was hydrolyzed to insoluble solid Al(OH)3 to form the passivation film that covered the active corrosion spots on the Al surface film and blocked further corrosive reaction. To further clarify our claims, we compared the pristine samples with their corresponding Ar+ sputtering samples. The blank 10 m LiTFSI showed asymmetric double Al 2p peaks belonging to the metallic Al foil, which indicates the destruction of pristine Al2O3 film and its failure to exert anticorrosion action against the aqueous LiTFSI electrolyte. By contrast, for the HTAs observed after 60 s Ar ion sputtering, signals from the passivation layers Al(OH)3 weakened, but remained with a relatively high intensity. Specifically, during etching, the signals of pristine Al2O3 intensified, which signifies the presence of double passivation layers consisting of an Al(OH)3 top layer and a pristine Al2O3 bottom layer. Thus, ceaseless destruction of the Al2O3 layer by TFSI− was impossible due to the passivation of Al foil because once the corrosion pitting sites appeared, the dissolved Al3+ rapidly reacted with the HTA, which resulted in Al(OH)3 layer covering the undamaged Al2O3 layer around the pitting site. Based on the information mentioned above, a compact and stable passivation layer Al(OH)3 formed on the surface of Al foil. Moreover, we observed our proposed mechanism in aqueous sodium-ion electrolytes, in which Na2CO3 exhibited an effective anticorrosion effect on the Al foil (Figs. S8–S10). Thus, our concept is universal and applies to the stabilization of Al foils in aqueous batteries.
Identification and quantification of Al oxidation corrosion for electron supplement
During corrosion, the current collector provided additional irreversible overcharging capacity as a result of Al oxidation. In consideration of the charge balance of batteries, Al oxidation at the cathode side corresponded to the reduction on the anode side, which balanced the redox reaction. Thereby, in such a redox reaction, the prelithiation of the anode compensates for the extra electron loss on the cathode side. To verify the life extension mechanism via Al oxidation, we evaluated individual Al and Ti current collectors in a full cell (LMO | | 10 m LiTFSI || TiO, P/N = 2) (Fig. 3). An extra 2 cm2 bare Al foil was attached behind the 0.79 cm2 LiMn2O4 electrode to amplify electron supplementation via the oxidation of Al as a prelithiation additive. Figure 3 reveals that compared with the Ti current collector, the battery with an Al current collector exhibited an extra plateau of around 1.2–1.3 V at the first charge process, and such finding was attributed to the corrosion of the Al current collector. Moreover, the extra plateau of approximately 1 V at the following discharge process belongs to the overlithiation of LiMn2O4. This result indicates the corrosive oxidation of the Al current collector at the earlier stage of the first charge, accompanied with the intercalation of Li ions in TiO2. This condition resulted in the partial prelithiation of the anode (LixTiO2) and overlithiation of Li1+xMn2O4 in the following discharge (Fig. 3B). Overall, Al was oxidized into Al ions that were dissolved in the electrolyte. In addition, Li ions from the electrolyte were intercalated in the active material. The electrolyte served as an additional source of lithium, and oxidation of the Al foil was the extra electron supplement. Thus, oxidation of Al current collectors at the cathode side can function as a prelithiation additive to prolong the life of batteries.

A First charge–discharge profiles of LiMn2O4–TiO2 cells with Al or Ti current collector in 10 m LiTFSI. B Schematic of the prelithiation of TiO2 via Al current collector oxidation in the charge process and overlithiation of LiMn2O4 in the discharge process.
Despite the prelithiation via the oxidation of the current collector, the feasibility of electrochemical regulation of corrosion and passivation requires further verification before a proof of concept can be proposed. Suppose the corrosion process cannot be controlled through electrochemical regulation. In such a case, the electrode will be damaged, and continuous irreversible overcharging will overcompensate for the fully lithiated Li0.5TiO2, which will result in the low utilization of capacity and further acceleration of other parasitic reactions, such as HER. Such condition will lead to the degradation of the battery’s lifespan. Fortunately, this problem can be solved with the use of HTA. A small amount of Al has to be corroded first to offer some dissolving Al ion as the reaction source for the following passivation by HTA. Such a mechanism ensures the electrochemical regulation of the corrosion–passivation of Al for the long-term cycle life of battery via the stabilization of the Al current collector and appropriate supplementation of lithium loss.
To further confirm that the oxidation of Al only prelithiates the anode in the initial charge process and the passivated Al by HTA serves as a robust current collector in the following cycle, we designed three-electrode cells with two fresh TiO2 anodes to distinguish Al oxidation in the first and second cycles and an excess LiMn2O4 cathode with a high P/N ≈ 3 to provide sufficient lithium resource to sustain the double TiO2 anodes (Fig. 4A). Accordingly, the charging program of batteries was set up in the following order: (1) cell configuration: ①LMO electrode /②TiO2 electrode, first full charging at 1 C; (2) cell configuration: ①LMO electrode /③TiO2 electrode, second full charging at 1 C rate. As the entire process occurred without discharging, interference was eliminated from overlithiation of LiMn2O4. As shown in Fig. 4B, regardless of whether Li3PO4 and Li2CO3 was used, the Al oxidation occurred at approximately 1.2 V on the first charge and almost disappeared on the second charge (Li2CO3: 1st 59.34 mAh g-1 and 2nd 2.73 mAh g-1; Li3PO4: 1st 77.22 mAh g-1 and 2nd 9.68 mAh g-1). Thus, the corrosion and passivation with the aid of additives mainly transpired during the first charge, accompanied by the electron supplement from the Al oxidation. In the following cycles, the Al current collector was stabilized via passivation to support the long-term calendar life of aqueous batteries.

A Process diagram for continuous charging of dual-anode battery. Charge the battery with ① LiMn2O4 cathode and ② TiO2 anode, then charge it with ① LiMn2O4 cathode and ③ TiO2 anode. B Consecutive charging profiles of batteries with one LiMn2O4 cathode and two TiO2 anodes in 10 m LiTFSI + Li2CO3 or Li3PO4.
Effectiveness of HTA in prolonging the cycle life of batteries
We assembled LiMn2O4 | |TiO2 full cells (P/N ≈ 2) to evaluate the effectiveness of Li3PO4 and Li2CO3 additives (Fig. 5). The Al and Ti current collectors with a blank electrolyte consisting of 10 m LiTFSI served as control samples. To magnify the passivation process and the effects of electron supplement from Al oxidation, we attached an extra Al foil (2 cm2) behind the LiMn2O4 electrode (0.79 cm2), excluding the Ti current collector. As shown in Fig. 5A, Al corrosion commenced at approximately 1.2 V, along with Al passivation by HTA, which reflects an increase in the plateau from 1.2 V to 1.4 V, consistent with the peak of ~1.3 V in the dQ/dV plot (Fig. 5B). Furthermore, full cells can survive in the relatively low-concentration electrolyte (10 m LiTFSI) with HTAs, whose cycle life improved substantially from 12.6% to above 84.7% (Li2CO3) and 89.1% (Li3PO4) after 1500 times. Such result was due to the suppressed continuously irreversible Al corrosion via passivation, which maintained the good electronic conducting contact between the electrode and current collector. In addition, the cycling stability of anticorrosion Ti (65.2%, 1500 cycles) cannot surpass that of Al with HTA, which confirms the effect of the prelithiation of TiO2 during electrochemical–chemical Al passivation. Figure 5D indicates a clear distinction between the efficiency of cells with and without additives. Batteries with HTA showed a rapid increase in efficiency at the initial cycles, with 1500 cycles average values of 99.36% (Li2CO3) and 99.37% (Li3PO4), which are considerably higher than that of 10 m LiTFSI (98.03%). Thus, the electrochemical–chemical Al passivation derived by HTA can stabilize the Al current collector in relatively low-concentration electrolytes and offer an electron supplement for the prolonged cycle life of ALIBs.

A, B First charge–discharge profiles and dQ/dV curves of LiMn2O4 ||TiO2 cells with an Al current collector in 10 m LiTFSI with additives. C Cycling stability of LiMn2O4 ||TiO2 cells with Al and Ti current collectors in 10 m LiTFSI with/without additives. D Comparison of coulombic efficiency with a magnified view inside.
Prototype of SP-ALIBs
From a practical point of view, as the mass loading of commercial Li-ion batteries reaches above 20 mg cm-2. Given the limited reactive mass of the Al current collector, the effectiveness of electron supplement from the Al current collector alone may be extremely low in actual conditions. To solve this problem, we designed a new battery type through introduction of an Al sacrificial prelithiation electrode dedicated to being oxidized as the electron supplement (Fig. 6B). The detailed program is as follows. Prior to cycling, the cell was pre-charged with an Al sacrificial prelithiation electrode to obtain TiO2 anode partially prelithiated (Al ||TiO2). The prelithiation capacity of TiO2 is depicted with the red short bar in Fig. 6A. Subsequently, the LiMn2O4 electrode, instead of the sacrificial Al electrode, initiated the cycling process (LiMn2O4 ||TiO2). As shown in Fig. 6A, when TiO2 was fully lithiated by LiMn2O4, the quantity of Li extracted from LiMn2O4 during the charging process was less than the Li extracted from TiO2 during the discharging process, which caused the overlithiation of LiMn2O4. Overall, Al is a controllable electron supplement. In addition to the initial stage, when capacity faded after long cycles, the cathode can be switched to an Al electrode for another electron supplementation. Preliminarily, we constructed a pouch cell (Al||LiMn2O4-Ti||TiO2) employing a Ti current collector to clarify the extra electron supplement only from the sacrificial prelithiation Al electrode (Figure S15). The anode was initially prelithiated using a sacrificial prelithiation Al electrode (2.4 V constant potential between Al and TiO2 for 20 minutes at 25 °C), which corresponded to the new discharge plateau of 0.98 V with an overlithiation capacity of 26.7 mAh g-1) and subsequently resulted in notably better cycling performance. Finally, we constructed a 0.5 Ah multilayer stacked SP-ALIBs with Al foil as the current collector (Al ||LiMn2O4-Al ||TiO2, 0.5 Ah, and 55.2 Wh kg-1), whose loading mass was 20 mg cm-2 for LiMn2O4 and 12 mg cm-2 for TiO2, which are close to the level of commercial Li-ion batteries. The introduction of the sacrificial Al electrode lowered the energy density. However, as the sacrificial Al electrode had a high theoretical capacity of 2980 mAh g-1, the overall estimated energy density decreased by less than 5%. After the prelithiation of TiO2 anode by the Al electrode (2.8 V constant potential between Al and TiO2 for 22 h at 35 °C), the discharge profile of the initial cycle showed the overlithiation plateau of LiMn2O4 (Fig. 6C). This finding reveals that high voltage and temperature cause the partial destruction of the passivation layer on sacrificial prelithiation Al electrodes generated by HTA and the feasibility of using applied potential and temperature to regulate Al corrosion with time. For the simultaneous protection of the Al current collector and utilization of Al corrosion for electron supplement, an additional sacrificial Al electrode was used, and the operating conditions of the Al current collector and sacrificial electrode were differentiated. In this case, the Al current collector was protected from HTA and functioned well throughout the battery lifespan. Meanwhile, the sacrificial Al electrode was oxidized in harsh conditions to prelithiate TiO2. The capacity retention of SP-ALIBs with HTA (Li2CO3) rose to 70.1% after 200 cycles, which is considerably higher than that of the traditional LiMn2O4 ||TiO2 cell without HTA (49.5%) (Fig. 6D).

A Program for SP-ALIBs. B Schematic of SP-ALIBs. C First charge–discharge profiles of the formation process of the 0.5 Ah SP-ALIB at 0.8 C after the prelithiation of TiO2 anode at 2.8 V for 22 h via the sacrificial prelithiation Al electrode and traditional pouch cell. D Comparison of cycle stabilities of SP-ALIBs and traditional pouch cell at 1 C rate. Inset: photo of a practical SP-ALIB.
Furthermore, a distinct coulombic efficiency difference was observed between SP-ALIBs (average 99.32% for 200 cycles) and normal cells (93.14%). The low efficiency was attributed to the failure of the Al current collector at the cathode and electrolyte pollution by Al3+. Thus, the HTA must convert the corrosion product Al3+ to precipitate it as a passivation layer. We validated the reliability of our design through the assembly and evaluation of three SP-ALIBs (0.5 Ah) to (Fig. S17), which showed an excellent consistency. The prototype of SP-ALIBs achieved the stability of Al current collectors in harsh aqueous environments and supplemented extra electrons via the sacrificial prelithiation Al electrode, which has great potential for industrial application. An open design for electrolyte replenishment should be considered for practical purposes due to electrolyte loss during long-term cycling.
Discussion
In this work, the anticorrosion effectiveness of HTAs was discovered, and their mechanism was investigated. With the assistance of HTA, a robust Al(OH)3 passivation layer formed on the Al surface, which dramatically lowered the corrosion current density by nearly three orders of magnitude (10−6 A cm-2) in 1 m LiTFSI solution. Such a condition made the Al foil corrosion-resistant and comparable to Ti. Moreover, through regulation by HTA, the controllable electrochemical corrosion process on the cathode side can be used to prelithiate the anode. With the robust Al current collector and extra electron supplemented by Al passivation, full cells can survive in relatively low-concentration electrolytes (10 m LiTFSI) with the employment of HTA, whose cycle life improved substantially from 12.6% to above 84.7% (Li2CO3) and 89.1% (Li3PO4) after 1500 times. Such finding implies the feasibility of using Al corrosion–passivation regulation to prolong the life of aqueous batteries. Al passivation ensured that the current collector’s long-term chemical/electrochemical stability and Al oxidation corrosion compensated for the irreversible capacity loss caused by HER and SEI formation. Based on these results, we designed a prototype of SP-ALIBs with an extra Al sacrificial prelithiation electrode as an electron supplement. The capacity retention of the 0.5 Ah prelithiated SP-ALIBs with HTA (Li2CO3) rose to 70.1% at 1C after 200 cycles, and such value is higher than that of the traditional LiMn2O4 || TiO2 cell (49.5%). The average efficiency increased from 93.14% to 99.32%. The discovery of HTA in a broader context revealed the once-overlooked complicated corrosion and passivation processes that occur in batteries in general. Through exploration of anticorrosion additives, the SP-ALIBs will gain more potential to achieve long-life ALIBs, which benefits the sustainable and lifelong industrial applications of ALIB.
Methods
Preparation of LiMn2O4-TiO2, LiMn2O4-TiO2-TiO2, Al-TiO2, and LiMn2O4-Al-TiO2 pouch cells
All pouch cells other than 0.5 Ah were assembled using a glass fiber as a separator, 10 m LiTFSI as the electrolyte (some with HTA), and Al plastic film as packaging. We fabricated the LiMn2O4 cathode by mixing LiMn2O4, carbon black (CB), and polyvinylidene fluoride (PVDF) at a weight ratio of 8:1:1 in N-methyl pyrrolidinone (NMP) in an SK-300SII CE mixing machine (SHASHIN KAGAKU Co., Ltd.) for 20 minutes at a speed of 2000 rsm to produce a black slurry. The slurry was spread uniformly on a clean Al or Ti foil and then dried. TiO2 anode was prepared using the same process and spread on the Al foil using the same CB, PVDF, and NMP at a weight ratio of 8:1:1. An extra 2 cm2 bare Al foil was attached behind the Al current collectors of LiMn2O4 in Figure 345 aside from LiMn2O4 with Ti current collector. The small pouch cells included unilaterally coated LiMn2O4 cathode (diameter: 10 mm, thickness: ~41.2 µm, and mass loading: ~3.2 mg cm−2), unilaterally coated TiO2 anode (diameter: 10 mm, thickness ~29 µm, and mass loading ~1.6 mg cm−2), and separator (area 2.5 cm × 1.3 cm; thickness: ~55 µm). The 10 m LiTFSI was used as an electrolyte. HTA was added to the electrolyte as an additive until saturation and the supernatant was used in the pouch cells. Li2CO3, Li3PO4, Li2SiO3, or LiAlO2 was hard to dissolve in 10 m LiTFSI, and saturation was reached at a concentration below 0.05 m.
The 0.5 Ah SP-ALIBs were assembled with eight layers of LiMn2O4 cathode, seven layers of TiO2 anode, and a sacrificial Al electrode. LiMn2O4 and TiO2 had mass loadings of ~20 (90 wt%) and ~12 mg cm-2 (80 wt%), respectively. The cathode area was 4.5 cm × 5.8 cm, that of the anode was 4.3 cm × 5.6 cm, and the width of the PP separator was 6 cm. The sacrificial Al electrode (4.5 cm × 5.8 cm) was sanded using a sandpaper to facilitate corrosion and placed on one side of the cell. Prior to standard formation, the sacrificial Al electrode prelithiated the cell at a constant voltage of 2.8 V at 35 °C for 22 h.
The electrolyte in this study was consistently expressed in terms of molality (m).
Electrochemical measurements
CA experiments were conducted in a 25 °C constant-temperature chamber using CHI 660E electrochemical workstation in three-electrode cells with 10 mL electrolyte, and the area of the Al working electrode was 1 cm2. The CA experiments on LiTFSI solution were performed in three-electrode devices (Al-WE ||Ag/AgCl-RE ||Al-CE), where the potential of the Al-WE was set at 4.5 V vs Li/Li+ for 10 minutes, relaxed to the OCV for 1 minutes, and cycled for 12 times as above. The batteries were tested using a LAND battery test system (Wuhan, China). The pouch cells in Figs. 3 and 5 were tested at the 3 C rate, and those in Figs. 4 and 6 were tested at a 1 C rate. The small pouch cells in Figs. 3, 4, and 5 had a cutoff voltage from 0.7 V to 2.5 V. The 0.5 Ah pouch cells in Fig. 6 experienced a cutoff voltage of 0.5 V to 2.5 V. All batteries were tested in a 25 °C constant-temperature chamber. Calculations of the specific energy of 0.5 Ah pouch cells was based on the masses of the cathode, anode, separator, and electrolyte.
Characterizations
SEM images of the morphologies of the Al foil after CA experiments were obtained using a Hitachi S-4800 field-emission scanning electron microscope operated at 10 kV. XPS analysis was performed using an ESCALAB 250 Xi, ThermoFisher with Mg/Al Kα radiation. All the binding energies were referenced to the C 1 s line at 284.8 eV. All electrolyte contents were measured on an ICP-OES Agilent 5800.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided in this paper. Extra data are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Li, W., Dahn, J. R. & Wainwright, D. S. Rechargeable lithium batteries with aqueous electrolytes. Science 264, 1115–1118 (1994).
Wang, Y., Yi, J. & Xia, Y. Recent progress in aqueous lithium-ion batteries. Adv. Energy Mater. 2, 830–840 (2012).
Suo, L. et al. Water-in-salt’ electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 350, 938–943 (2015).
Liu, B. et al. Sandwich structure corrosion-resistant current collector for aqueous batteries. ACS Appl. Energy Mater. 4, 4928–4934 (2021).
Yue, J. & Suo, L. Progress in rechargeable aqueous alkali-ion batteries in China. Energy Fuels 35, 9228–9239 (2021).
Kühnel, R. S. et al. Water-in-salt’ electrolytes enable the use of cost-effective aluminum current collectors for aqueous high-voltage batteries. Chem. Commun. 52, 10435–10438 (2016).
Ratajczak, P., Jurewicz, K., Skowron, P., Abbas, Q. & Béguin, F. Effect of accelerated ageing on the performance of high voltage carbon/carbon electrochemical capacitors in salt aqueous electrolyte. Electrochim. Acta 130, 344–350 (2014).
Zhou, X., Peng, C. & Chen, G. Z. 20 V stack of aqueous supercapacitors with carbon (−), titanium bipolar plates and CNT-polypyrrole composite (+). AIChE J. 58, 974–983 (2012).
Wen, Y. H. et al. Carbon coated stainless steel mesh as a low-cost and corrosion-resistant current collector for aqueous rechargeable batteries. J. Mater. Chem. A 5, 15752–15758 (2017).
Guo, L., Thornton, D. B., Koronfel, M. A., Stephens, I. E. L. & Ryan, M. P. Degradation in lithium ion battery current collectors. J. Phys. Energy https://doi.org/10.1088/2515-7655/ac0c04 (2021).
Liu, S., Ye, S. H., Li, C. Z., Pan, G. L. & Gao, X. P. Rechargeable aqueous lithium-ion battery of TiO2∕LiMn2O4 with a high voltage. J. Electrochem. Soc. https://doi.org/10.1149/2.094112jes (2011).
Zhao, M., Huang, G., Zhang, B., Wang, F. & Song, X. Characteristics and electrochemical performance of LiFe0.5Mn0.5PO4/C used as cathode for aqueous rechargeable lithium battery. J. Power Sources 211, 202–207 (2012).
Dyatkin, B. et al. Development of a green supercapacitor composed entirely of environmentally friendly materials. ChemSusChem 6, 2269–2280 (2013).
Lahan, H. & Das, S. K. Active role of inactive current collector in aqueous aluminum-ion battery. Ionics 24, 2175–2180 (2018).
Luo, W. et al. Highly conductive, light weight, robust, corrosion-resistant, scalable, all-fiber based current collectors for aqueous acidic batteries. Adv. Energy Mater. 8, 6 (2018).
Yao, Y. G. et al. Epitaxial welding of carbon nanotube networks for aqueous battery current collectors. ACS Nano 12, 5266–5273 (2018).
Ziv, B. et al. Investigation of graphite foil as current collector for positive electrodes of Li-ion batteries. J. Electrochem. Soc. 160, A581–A587 (2013).
Wang, X., Yasukawa, E. & Mori, S. J. E. A. Inhibition of anodic corrosion of aluminum cathode current collector on recharging in lithium imide electrolytes. Electrochim. Acta 45, 2677–2684 (2000).
Kawamura, T. et al. Methyl difluoroacetate inhibits corrosion of aluminum cathode current collector for lithium ion cells. Electrochem. Solid-State Lett. 8, A459 (2005).
Piao, N., Wang, L. & He, X. J. Anodic stabilities of various metals as the current collector in high concentration electrolytes for lithium batteries. J. Electrochem. Soc. 168, 030509 (2021).
McOwen, D. W. et al. Concentrated electrolytes: decrypting electrolyte properties and reassessing Al corrosion mechanisms. Energy Environ. Sci. 7, 416–426 (2014).
Matsumoto, K. et al. Suppression of aluminum corrosion by using high concentration LiTFSI electrolyte. J. Power Sources 231, 234–238 (2013).
Nakajima, T., Mori, M., Gupta, V., Ohzawa, Y. & Iwata, H. Effect of fluoride additives on the corrosion of aluminum for lithium ion batteries. Solid State Sci. 4, 1385–1394 (2002).
Song, S.-W., Richardson, T. J., Zhuang, G. V., Devine, T. M. & Evans, J. W. Effect on aluminum corrosion of LiBF4 addition into lithium imide electrolyte; a study using the EQCM. Electrochim. Acta 49, 1483–1490 (2004).
Yan, G. et al. Lithium difluoro(oxalato)borate as an additive to suppress the aluminum corrosion in lithium bis(fluorosulfony)imide-based nonaqueous carbonate electrolyte. J. Solid State Electrochem. 20, 507–516 (2016).
Yang, H., Kwon, K., Devine, T. M. & Evans, J. W. J. Aluminum corrosion in lithium batteries an investigation using the electrochemical quartz crystal microbalance. J. Electrochem. Soc. 147, 4399 (2000).
Krause, L. J. et al. Corrosion of aluminum at high voltages in non-aqueous electrolytes containing perfluoroalkylsulfonyl imides; new lithium salts for lithium-ion cells. J. Power Sources 68, 320–325 (1997).
Morita, M., Shibata, T., Yoshimoto, N. & Ishikawa, M. Anodic behavior of aluminum current collector in LiTFSI solutions with different solvent compositions. J. Power Sources 119-121, 784–788 (2003).
Theivaprakasam, S. et al. Passivation behaviour of aluminium current collector in ionic liquid alkyl carbonate (hybrid) electrolytes. npj Mater. Degrad. https://doi.org/10.1038/s41529-018-0033-6 (2018).
Ding, R. et al. Recent advances in cathode prelithiation additives and their use in lithium–ion batteries. J. Electroanal. Chem. https://doi.org/10.1016/j.jelechem.2021.115325 (2021).
Huang, Z. et al. Progress and challenges of prelithiation technology for lithium‐ion battery. Carbon Energy 4, 1107–1132 (2022).
Lv, T. & Suo, L. Water-in-salt widens the electrochemical stability window: thermodynamic and kinetic factors. Curr. Opin. Electrochem. https://doi.org/10.1016/j.coelec.2021.100818 (2021).
Szklarska-Smialowska, Z. J. C. Pitting corrosion of aluminum. Corros. Sci. 41, 1743–1767 (1999).
Rudd, W. J. & Scully, J. C. The function of the repassivation process in the inhibition of pitting corrosion on aluminium. Corros. Sci. 20, 611–631 (1980).
Buchheit, R. G., Bode, M. D. & Stoner, G. E. Corrosion-resistant, chromate-free talc coatings for aluminum. Corrosion 50, 205–214 (1994).
Giurlani, W. et al. Electrochemical stability of steel, Ti, and Cu current collectors in water-in-salt electrolyte for green batteries and supercapacitors. J. Solid State Electrochem. 26, 85–95 (2022).
Rueda, F. et al. Characterization of Venezuelan laterites by X-ray photoelectron spectroscopy. J. Electron. Spectrosc. Relat. Phenom. 82, 135–143 (1996).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFB2404500), the National Natural Science Foundation of China (U22B20124), CAS Youth Interdisciplinary Team, and the Center for Clean Energy.
Author information
Authors and Affiliations
Contributions
L.S. conducted the project. B.L. and L.S. conceived the concept and designed this work. B.L. and L.S. wrote the paper. B.L. conducted the electrochemical experiments, SEM, and ICP measurements. T.L. (Tianshi Lv) conducted the XPS measurements. T.L. (Tianshi Lv), A.Z. and X.Z. participated in constructing the 0.5Ah batteries. Z.L. participated in drawing the schematic diagram. T.L. (Ting Lin) assisted in analyzing the composition of the passivation layer. All authors participated in the analysis of the results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, B., Lv, T., Zhou, A. et al. Aluminum corrosion–passivation regulation prolongs aqueous batteries life. Nat Commun 15, 2922 (2024). https://doi.org/10.1038/s41467-024-47145-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47145-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
