
Abstract
Aneurysmal subarachnoid haemorrhage (aSAH) presents a challenge to clinicians because of its multisystem effects. Advancements in computed tomography (CT), endovascular treatments, and neurocritical care have contributed to declining mortality rates. The critical care of aSAH prioritises cerebral perfusion, early aneurysm securement, and the prevention of secondary brain injury and systemic complications. Early interventions to mitigate cardiopulmonary complications, dyselectrolytemia and treatment of culprit aneurysm require a multidisciplinary approach. Standardised neurological assessments, transcranial doppler (TCD), and advanced imaging, along with hypertensive and invasive therapies, are vital in reducing delayed cerebral ischemia and poor outcomes. Health care disparities, particularly in the resource allocation for SAH treatment, affect outcomes significantly, with telemedicine and novel technologies proposed to address this health inequalities. This article underscores the necessity for comprehensive multidisciplinary care and the urgent need for large-scale studies to validate standardised treatment protocols for improved SAH outcomes.
Similar content being viewed by others

Background
Subarachnoid haemorrhage (SAH), a neurovascular emergency with an incidence in the UK, is approximately 8 per 100,000 population, peaking at 50–60 years and is 1.6 times more common in women than men1. The spontaneous rupture of an intracranial aneurysm (80–85%) is the most common cause of SAH2. Aneurysmal SAH (aSAH) results in substantial morbidity, mortality and burden on the healthcare system; its downstream effects trigger a cascade of events resulting in organ dysfunction. After initial ictus, these complex ensuing processes cause significant morbidity and mortality. This complex neurovascular syndrome requires an established multidisciplinary team and is best managed in dedicated neurosciences units3.
Anatomy
Most aneurysms are found in the anterior circulation of the Circle of Willis (Fig. 1). Aneurysms in the posterior circulation of the vertebral and basilar systems are less frequent, accounting for only 12% of intracranial aneurysms4. History of familial aneurysms (at least one first-degree relative with an intracranial aneurysm5) and certain genetic diseases such as autosomal dominant polycystic kidney disease, Ehlers-Danlos syndrome type IV, Marfan’s syndrome, and neurofibromatosis type 1 have been identified as predisposing factors to cerebral aneurysms6,7.

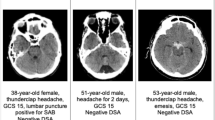
A Computerised tomography scan showing subarachnoid and intraventricular haemorrhage; B Culprit aneurysm shown on 3D rendering from CT angiography, treated with an external ventricular drain; C Closer view of unruptured aneurysm.
Prognosis
Rupture
The incidence of rupture is approximately 0.95% annually. The factors associated with an increased risk of rupture of a cerebral aneurysm are hypertension, smoking, cocaine and alcohol usage. The risk of rupture also varies according to the size (>7 mm) and location, with anterior and posterior aneurysms at a higher risk of rupture than middle cerebral artery (MCA) aneurysms. A daughter sac (irregular protrusion on the sac wall of the aneurysm) and a large size ratio, defined as the aneurysm to vessel size ratio, also increase the risk of aneurysmal rupture8,9.
Outcomes
They vary significantly, from complete recovery to severe disability or death. The most important early predictor of outcome is intact consciousness, with patients having better Glasgow coma score (GCS) at presentation faring better10,11: Survival after aSAH has increased by 17% in the past few decades due to early diagnosis, early minimally invasive interventions of aneurysm securement, nimodipine usage and intensive care support12. Cumulative case fatality rates after SAH are 25–30% on day one, 40–45% within week one, 50–60% after the first month, and 55–60%, 65% and 65–70% at 6, 12 and 60 months respectively. 12% of patients die before receiving medical attention13. Poor outcomes in patients with aSAH are often associated with several key factors. These include older age, worsening neurological conditions, posterior circulation aneurysm rupture, larger aneurysm sizes, and an increased presence of SAH on initial CT scans. Complications such as intracerebral haematoma or intraventricular haemorrhage and elevated admission systolic blood pressure also contribute to unfavourable outcomes. Previous diagnoses of hypertension, myocardial infarction, liver disease, or SAH further compound the risk14,15. Intensive care management of aSAH patients primarily focuses on addressing delayed cerebral ischaemia, which affects 20–45% of patients and is linked with worse neurologic outcomes and mortality. However, aSAH also often leads to medical complications like fever, anaemia, and hyperglycaemia, which further influence the patient’s prognosis and hospital stay duration12,16,17.
Diagnosis
SAH should be considered in all patients presenting with sudden-onset severe headaches. Among other presenting features, seizures are observed in approximately 6% of patients18. Focal neurological deficits are usually associated with an intraparenchymal haematoma or the location of an aneurysm, e.g. an oculomotor nerve palsy with pupillary dysfunction is associated with enlargement or rupture of the ipsilateral posterior communicating artery aneurysm.
Among patients presenting with an acute nontraumatic headache that has reached maximal intensity within one hour and intact neurology, the Ottawa rules are very sensitive for identifying an SAH but with low specificity19. A non-contrast CT scan of the head (CTH), usually modern third-generation CTH, is 100% specific and highly sensitive for aSAH if scanned in the first 6 h of headache onset20, the sensitivity decreases to 97% in the first 72 h and further decreases by 50% in 5 days12. If CTH performed ≥6 h after the ictus is equivocal or negative, a lumbar puncture performed within 6–12 h of symptom onset typically shows xanthochromia. Unlike CT, xanthochromia is present in the cerebrospinal fluid (CSF) in all patients up to 2 weeks post ictus; false positive results can occur from a traumatic lumbar puncture21.
For handling aSAH, computed tomographic and digital subtraction angiography play significant roles22. CTA, as a less invasive and readily accessible option, is the first investigation to consider. Apart from identifying aneurysms, it also aids in planning the repair23. However, in inconclusive CTA results, Digital Subtraction Angiography (DSA), the gold standard in angiography, is recommended24. Furthermore, stereotactic angiography with 3-dimensional rotational capabilities proves valuable for detecting aSAH aneurysms, except when previously diagnosed by a noninvasive angiogram. It contributes to treatment planning and assessing an aneurysm’s suitability for endovascular coiling or surgery25. DSA has the added benefit of offering an endovascular treatment and minimal complication rates of 1%. Importantly, studies comparing CTA and DSA indicate strong agreement, suggesting they both provide high sensitivity and specificity in diagnosing vasospasm26.
Magnetic resonance angiography (MRA) is an alternative to CTA or DSA for aneurysm detection, especially in patients allergic to iodine. MRA does not expose the patients to radiation, and the time-of-flight sequence does not require contrast. MRA is not very sensitive when done early after symptom onset due to high oxygen concentration in CSF27. Magnetic resonance angiogram (MRA) is 95% sensitive for aneurysms >3 mm28; the main disadvantages are availability, cost, and acquisition time.
Pathobiology
Cerebral saccular aneurysms are acquired defects that develop at branch points of major arteries of the circle of Willis. Haemodynamic stress induces degeneration of the internal elastic lamina with thinning and loss of the tunica media, thus forming aneurysms. Aneurysm rupture causes a sudden rise in intracranial pressure (ICP)29,30, reducing cerebral perfusion and leading to a transient or persistent ischaemic state31. Ongoing ischaemia is associated with mortality and morbidity.
The complex interplay of events resulting in global early brain injury (EBI) such as transient global cerebral ischaemia with diffusion hypoxia due to microthrombosis, Blood-Brain Barrier (BBB) dysfunction, ionic disequilibrium within neurons, and neuroinflammation outside the immediately affected vascular territories makes complete understanding of this phenomenon elusive32. The occurrence of cerebral energy utilisation dysfunction, with normal or hyperaemic cerebral blood flow, introduces further difficulties in understanding pathobiology, indicating a possible non-ischaemic mechanism contributing to EBI. This non-ischaemic damage could be linked with widespread cortical spreading depolarisation or mitochondrial dysfunction, leading to aberrant cerebral energy metabolism33. Despite these insights, the exact mechanisms of EBI remain uncertain; detailed studies and investigations continue to unfold the reasons for EBI34.
In the initial hours after aSAH, the above results in cytotoxic oedema (intraneuronal), progressing to vasogenic oedema (perivascular and extracellular oedema). Cerebral oedema is associated with further inflammation of nervous tissue, excitotoxicity, impaired cerebral autoregulation, microthrombosis, and oxidative stress35.
The emerging concept of immune-mediated cerebral damage after aSAH has been clearly defined in experimental and clinical conditions. These immune responses can be assessed using biomarkers like neuron-specific enolase, S100B, high sensitive-CRP, serum procalcitonin, glial fibrillary acid protein and ubiquitin carboxy-terminal hydrolase L1. These biomarkers, used in research and clinical settings, reflect inflammation, neuronal damage, and progression after SAH36,37,38,39. Once blood enters the subarachnoid space, the neutrophil count increases globally, systemic interleukin 1 (IL-1) and IL-6 levels rise rapidly, whereas IL-10 levels decrease40. Neuronal cell death ensues when the neuronal inflammatory processes trigger astrocytes and microglial cells (triggering EBI) in the face of cerebral metabolic distress. This is compounded by sympathetic nervous system activation, cerebral autoregulatory failure, inflammation, and platelet activation, leading to microthrombosis with cortical spreading depolarisation41. These factors imply that aSAH could be a systemic inflammatory condition for which innovative therapies might be more impactful than current vasospasm/DCI interventions. Nonetheless, this notion remains under further investigation.
Global cerebral oedema is EBI’s most prominent imaging manifestation, quantified using the subarachnoid haemorrhage early brain oedema score (SEBES). The risk of poor prognosis is doubled in patients with features of EBI. Plasma catecholamines remain elevated for several days post-SAH, and mortality and morbidity are proportional to serum catecholamine concentrations42.
DCI usually occurs after three days and is seen for up to 21 days43; it is caused by vasospasm, enhanced apoptosis, BBB breakdown, microthrombosis with microcirculatory dysfunction and CSD. DCI was believed to be mainly associated with the narrowing of cerebral arteries beginning days after aSAH, defined as cerebral vasospasm. The CONSCIOUS trial (Clazosentan to Overcome Neurological Ischemia and Infarction Occurring After Subarachnoid Hemorrhage) suggested that vasospasm prevention does not reduce all-cause mortality or DCI. DCI have a multifactorial aetiology related to EBI, arteriolar constriction and thrombosis, cortical spreading ischaemia, and angiographic vasospasm44. Over time, the extravasated blood modulates some core factors, such as EBI, resulting in DCI. Overall, the inability of cerebral perfusion to match metabolic demands leads to DCI.
After haemorrhage, free haemoglobin toxicity with transient global ischaemia drives vasoconstriction and neuronal dysfunction44. Nitric oxide pathway modulation is a crucial feature of DCI due to decreased production and increased scavenging, linking vascular dysfunction to inflammation and cortical spreading ischaemia. Usually, autoregulatory microvascular dilatation is mediated by astrocyte-derived vasoactive molecules. In aSAH, these mechanisms are inverted, causing vasoconstriction, leading to local hypoperfusion, blood rerouting, and hyperperfusion45. These ischaemic and non-ischaemic injuries warrant novel neurotherapies to reduce aSAH-related mortality and morbidity.
Grading scales
Clinical and radiological grading of subarachnoid and intraventricular haemorrhage is traditionally performed on admission. The preferred scale is the WFNS, which is based on the Glasgow Coma Scale. Severe scores (4 and 5) on this scale indicate a poor outcome. Additionally, the mFisher scale (radiological scale) aids in assessing the extent of bleeding. Other scales such as Apache II, SOFA, and SAPS provide comprehensive health evaluations, while the Hunt and Hess and FOUR scales also find use (Table 1). Newer scales, including SEBES, Hidjra, and VASOGRADE, are now being adopted. These grading systems facilitate multidisciplinary communication and prognostication and identify neurological deterioration early. Factors like patient age, existing health conditions, hyperglycaemia, sepsis, fever, delayed cerebral ischaemia, and rebleeding are also linked with poorer outcomes10,11,46,47.
Management of aSAH
Critical care management of patients with SAH is challenging and requires awareness of all potential medical and neurological complications, with timely intervention and treatment. Care of patients with aSAH requires a multidisciplinary team approach. Outcomes are improved at high-volume centres (institutions that care for at least 35 patients with SAH annually) with dedicated neurocritical care units and specialised team members3,48. Aims of critical care management summarised in (Fig. 2) include initial stabilisation of the patient to prevent rebleeding and allow for definite early treatment, limiting secondary neurological injury and early recognition and treatment of complications49,50.

NCCT non-contrast CT, CTA CT angiography, DSA digital subtraction angiography, MRI magnetic resonance imaging, MRA magnetic resonance angiography, ABCDE airway breathing circulation disability exposure, GCS Glasgow coma scale, SBP systolic blood pressure, MAP mean arterial blood pressure, ICP intracranial pressure, EVD external ventricular drain, Hb haemoglobin, ECG electrocardiogram, DCI delayed cerebral ischaemia, CPP cerebral perfusion pressure, TCD transcranial doppler, TCCD transcranial colour doppler, EEG elecroencephalogramPtiO2-brain tissue oxygenation, CMD cerebral microdialysis, CBF cerebral blood flow, ARDS acute respiratory distress syndrome, IAP intraabdominal pressure, DVT deep vein thrombosis, HAI hospital-acquired infection, LOC loss of consciousness. Created with Biorender.com.
Management before securing the aneurysm
The initial treatment is the same as any critically ill patient: concurrent resuscitation and management of acute neurological decline, avoiding hypoxia and hyperoxia, if airway compromise, GCS < 8 or drop in GCS > 2 points, secure airway. Avoid hypotension, monitor for arrhythmia, avoid hypotonic fluids, maintain normovolaemia, and treat seizures and fever. Initiate neuromonitoring, including pupillary size, shape and reactivity to light every 20 min once the patient is sedated and paralysed.
In patients with aSAH, rebleeding carries very high morbidity and mortality51. Rates of aneurysmal rebleeding have drastically improved since the 1980s, when >35% of patients rebled before securing the aneurysm52. Currently, 10–15 % of aSAH rebleed, with 50% occurring in the first 6 h53,54.The risk factors associated with rebleeding are higher grade aneurysms, hypertension, lower GCS, larger or irregular aneurysms, and delayed securing53.
Guidelines recommend reducing blood pressure; mean arterial pressure (MAP) > 95 mmHg may be detrimental55,56,57. The European Stroke Organisation guidelines recommend treatment for BP if systolic BP > 180 mmHg, adjusting the MAP with analgesics, nimodipine and titratable antihypertensives as necessary. The American Heart Association/American Stroke Association (AHA/ASA) guidelines recommend treatment with a titratable agent50. However, they suggest a lower systolic BP threshold of <160 mmHg. After decades of research, we still need better evidence on BP targets. Give nimodipine as early as possible to all patients with aSAH; it is a Class I recommendation with level A evidence as per AHA/ASA. Avoid aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) before securing the aneurysm.
Management of aneurysm
Definitive treatment hinges on early securement of the aneurysm, within 72 h or ideally within 48 h of diagnosis58,59. Most rebleeding happens within the first 24 h60, so early securement (within 24 h) may be safe61,62. The securement method depends on many factors, particularly the patient’s age, morphology and location of the aneurysm, and presence of intraparenchymal haemorrhage. Following the international subarachnoid aneurysm trial (ISAT), coiling is preferred as it is associated with improved mortality and functional outcomes, with a marginally higher rate of aneurysm recurrence63,64,65. The choice of securement method should be made after a multidisciplinary team discussion between the interventional radiologists, neurosurgeons, and neurocritical specialists. Tranexamic acid may reduce rebleeding and improve overall mortality66,67,68. Current guidelines recommend a short course of antifibrinolytic agent and discontinuing after securing the aneurysm, a maximum of 72 h of tranexamic acid. Prolonged use of antifibrinolytics increases the risk of thrombotic events and DCI67,69.
Management after securing the aneurysm
Early brain injury
EBI (Fig. 3) occurs within the first 72 h, has a long-lasting impact, and is associated with the development of DCI70,71. It increases mortality and morbidity. Those at the highest risk include patients with high-grade haemorrhage, large intracranial blood volume, and prolonged loss of consciousness. Early CT features of brain oedema and ischaemia evidence EBI. Early brain hypoperfusion plays a crucial role in the genesis of EBI71,72. There are no current therapeutic modalities to treat established EBI.

The time ranges at bottom depict approximate/presumed periods when the various processes occur. CSF cerebrospinal fluid, CBF cerebral blood flow, CBV cerebral blood volume, ICP intracranial pressure, Oxy-Hb oxyhaemoglobin, EDHF endothelial derived hyperpolarising factor, ET-1 endothelin 1, FasL Fas ligand, AIF apoptosis inducing factor, IL-6 interleukin 6, NO nitric oxide, PAF platelet-activating factor, PGI2 prostacyclin, TNFR tumour necrosis factor receptor, vWF von Willebrand factor, DCI delayed cerebral ischaemia, BBB blood brain barrier.
Early management of aSAH should focus on preventing the development of EBI by providing adequate brain oxygen delivery and meeting the brain’s metabolic demands. Measures include early securement of aneurysm, avoiding considerable BP changes, treating hydrocephalus, seizures, and cardiopulmonary complications. Optimise cerebral perfusion pressure (CPP) to target >70 mmHg but <95 mmHg early in the treatment, as recent data suggest improved outcomes by improving perfusion and reducing brain metabolic stress. Aggressively treat anaemia to maintain adequate oxygen delivery and control fever to reduce metabolic demand on the brain34,58. Advanced multimodality monitoring may assist in individualising treatment strategies. Experimental studies involving agents targeting neuroinflammation like NSAIDs, thromboxane synthase inhibitors, steroids, nitric oxide, and various immunosuppressants have failed to demonstrate benefit. Agents like ketamine targeting cortical spreading depolarisations are under evaluation72,73.
Hydrocephalus
This complication develops within 72 h of aSAH, characterised by ventricular enlargement. Incidence is 20-30% of patients with aSAH. Hydrocephalus can dramatically affect the level of consciousness with improved drainage via CSF diversion (e.g. with an external ventricular drain or lumbar drain).
Seizures
These are also early complications after an aSAH. Most seizures occur within the first 24 h and may indicate a rebleed74. Seizures are common in young patients with MCA aneurysms and the presence of intraparenchymal lesions (e.g. arteriovenous malformations, bleeds or infarcts). Nonconvulsive status epilepticus may be present in up to 20% of patients, especially those with a high-grade haemorrhage or comatose75. Antiepileptics are associated with poor cognitive outcomes and increased hospital complications, so routine prophylaxis is not recommended76,77,78,79. Patients with poor-grade aneurysms and depressed conscious states without clear explanation should have continuous electroencephalograms to look for subclinical seizures and nonconvulsive status epilepticus80,81.
DCI and vasospasm
Development of vasospasm and DCI (Fig. 3) commonly occurs 3–14 days post-aSAH. DCI with cerebral infarction is the leading cause of morbidity in survivors. DCI was purely attributed to vasospasm in the past, but there is radiological evidence of vasospasm in 70% of patients with aSAH without any focal neurology. Recent evidence suggests a complex interplay of pathologies resulting in the development of EBI followed by a DCI82. DCI is characterised by any neurological deterioration (focal deficit or decline in GCS by ≥2 points), lasting more than one hour and no other cause. It is reported to occur in about 30% of aSAH patients and is a significant cause of mortality and disability83.
Effective treatment of vasospasm does not correlate with decreased incidence of DCI or improved outcomes, as seen in the Clazosentan trials84. Also, nimodipine does not affect vasospasm yet improves DCI and patient outcomes85,86. Other clinical trials aimed at vasospasm, including magnesium (MASH trial and subsequent meta-analyses)87,88, statins89 and methylprednisolone90, have failed to show outcome benefits. These negative trials reinforce our understanding of DCI’s complex and multifactorial pathophysiology.
Vasospasm development gives an insight into those at high risk for developing DCI. Transcranial Doppler (TCD) and Transcranial Colour Doppler (TCCD) have revolutionised the detection of vasospasm and are widely used at the bedside. It is a noninvasive tool for identifying vasospasm91. It has a Class IIa Level of Evidence B recommendation to monitor the development of vasospasm58. TCD evidence of vasospasm has a high prediction for the development of DCI. It has a sensitivity of 90% (95% CI 77–96%), specificity of 71% (95% CI 51–84%), Positive predictive value (PPV) of 57% (95% CI 38–71%), and negative predictive value (NPV) of 92% (95%, CI 83–96%)92. Pitfalls include availability and operator-dependent variability. TCD is most sensitive to MCA vasospasm. Velocities >120 cm/s in the MCA have a high negative predictive value, and velocities >180 cm/s have a high positive predictive value for the presence of vasospasm. Lindegaard ratio (LR) is the MCA’s mean velocity divided by the ipsilateral extracranial internal carotid artery (ICA). This ratio is usually <3. LR is used to distinguish whether the MCA velocity is secondary to hyperaemia (can also result in flow velocities >120 cm/s) or due to vasospasm. LR < 3 suggests hyperaemia, 3–6 indicates mild vasospasm, >6 severe vasospasm. Serial examinations and trends in velocity are more critical than a single reading. TCD still lacks good evidence for its routine usage outside research49.
If symptomatic vasospasm is suspected, immediate CT angiogram (CTA) and CT perfusion (CTP) may be indicated. CTA has become more widely used as it is highly sensitive and specific93,94. CTP is emerging as a potential test for hypoperfusion in the presence or absence of large vessel vasospasm. CTP may be able to identify DCI when it is still reversible95. Digital subtraction angiogram (DSA) remains the gold standard for vasospasm identification and should be performed if clinical large vessel vasospasm is suspected. This has the advantage of being a diagnostic tool, and treatment can be provided in the same session.
Multimodality monitoring
Multimodality monitoring in aSAH patients offers a comprehensive approach to assessing the risk of delayed cerebral ischemia. Brain tissue oxygen (PbtO2) monitoring measures invasive partial pressure of oxygen, with normal levels below 20 mmHg indicating risk of ischemia96,97. Decreasing levels below 15 mmHg warrants immediate measures to enhance cerebral tissue oxygenation. Interstitial glucose, glycerol, lactate, pyruvate, glutamate, and numerous inflammatory biomarkers constitute markers for cerebral microdialysis (CMD), another monitoring tool. A heightened lactate-pyruvate ratio (LPR) signals anaerobic metabolism, suggesting an essential role in initiating DCI. Information on systemic haemodynamics, vital for managing critically unstable aSAH patients in the ICU, is obtainable from the cardiac output and index. Noninvasive brain oxygenation monitoring, crucial for the early identification of hypoperfusion linked to DCI, is delivered through near-infrared spectroscopy. The transcranial doppler ultrasonography monitors cerebral blood flow velocity and anticipates vasospasm, a common DCI complication. Employing dual or multichannel systems enables simultaneous extensive monitoring, supporting timely intervention and management to impede DCI progression.
Nimodipine and other therapeutic interventions for DCI in aSAH patients
Currently, nimodipine is the only proven standard neurotherapeutic regimen to prevent and treat cerebral vasospasm and DCI. Mechanisms of action are likely more complex than simple inhibition of vasoconstriction and include reduction of vasospasm, neuroprotection, enhancement of fibrinolytic activity and thus reduction of microthrombosis, and diminishment of cortical spreading depolarisations98,99. Systemic arterial hypotension remains the most significant adverse effect of nimodipine treatment100. Nimodipine in hemodynamically unstable patients will increase the therapeutic intensity levels, such as using vasopressors, fluids, or frequent reduction and adjustment of its dosage. Nimodipine should be prescribed to all aSAH patients for up to 21 days101. The available therapies for DCI include induced hypertension and endovascular treatment. Hypertension is the only “Triple H” therapy component that effectively increases perfusion and brain oxygenation102,103. Both hypervolaemia and haemodilution lead to more deleterious effects104,105,106. Blood pressure should be titrated, weighing the risks and benefits stepwise until clinical symptoms improve. If the neurologic deficit persists despite hypertensive therapy, consider endovascular therapy to improve long-term outcomes107. Intravascular vasodilators (milrinone, verapamil, nicardipine) are associated with significant improvement in angiographic spasm and neurological signs, but phase three studies are required before their routine use. Angioplasty can be considered if the spasm is refractory to hypertensive therapy. However, prophylactic use of angioplasty is not associated with improved clinical outcomes and may be related to increased risk of arterial rupture; it is not currently recommended108.
Medical complications
Medical complications are common in severe aSAH, affecting the outcomes. Among the prevalent complications are infections such as central line infections, ventilator-associated pneumonia, and deep venous thrombosis. Both prevention and keen surveillance are crucial, along with precise treatment plans to manage these complications. Hospital-acquired infections have been linked to nutritional deficiencies, specifically low glutamine levels, and adverse outcomes109,110. Fever increases brain metabolism and exacerbates brain hypoxia in aSAH patients. Therefore, fever prevention/treatment is necessary to reduce secondary brain injury and improve outcomes. Pharmacological antipyretics may not suffice, necessitating additional strategies for maintaining normothermia. It’s essential in critical care to maintain normoglycemia, avoiding aggressive insulin strategies that could cause intracerebral hypoglycaemia in SAH111.
Anaemia is also associated with poor outcomes in aSAH112. Optimal haemoglobin targets remain controversial and warrant further research. Optimal fluid resuscitation strategies are unclear; hyper and hypovolaemia are deleterious, so euvolaemia is advocated113. In the context of resuscitation strategies, maintaining optimal oxygenation is key; hypoxia and hyperoxia should be prevented114. Initiation of pharmacological DVT prophylaxis within 24 h of securing the aneurysm is recommended, though controversy persists115,116.
Early mobilisation strategies benefit all patients with aSAH, including those with EVDs117. The timing for procedures like tracheostomy and percutaneous endoscopic gastrostomy is debatable. In a small retrospective case series, there was no difference in 6-month functional outcomes for early versus delayed tracheostomy (≥10 days) following ischaemic stroke, intracerebral haemorrhage, or aSAH118.
Fever is a common association in patients with aSAH, linked to worse outcomes. Central fevers can occur with or without infections, leading to potentially unnecessary antibiotic treatment. Fever prevention and treatment reduces secondary brain injury and improves outcomes119 as fever increases brain metabolism and causes brain hypoxia, exacerbating DCI in patients with aSAH. Pharmacological antipyretics might not be sufficient, so adjunctive strategies for normothermia might be necessary.
Cardiopulmonary
Myocardial injury is a common occurrence following subarachnoid haemorrhage, with initial symptoms including troponin elevations in 28% of patients within the first 24 h120, arrhythmias in 35%121, and wall motion abnormalities in 28%122. Deep septal T-wave inversions, cerebral T waves, and QTC prolongation associated with insular injury are observed on ECG. Nearly all patients experience some ECG abnormality, with severity proportional to the cerebral insult123, although findings can vary significantly. Life-threatening arrhythmias, however, occur in only 5% of cases121. The degree of troponin increase correlates with the severity of intracranial injury124, left ventricular dysfunction, and mortality125,126.
Neurogenic stress cardiomyopathy typically requires both transient and reversible left ventricular dysfunction, and a negative coronary angiogram for diagnosis. Retrospective studies estimate incidence among aSAH patients at 1–5%127,128.This is due to acute brain-heart interactions following severe brain injury129. The pathophysiology of neurogenic cardiac injury is complicated130,131, encompassing autonomic dysregulation, excessive catecholamine release, myocyte injury, mitochondrial dysfunction, and ongoing inflammation132.
Catecholamine release and sympathetic overstimulation lead to cardiopulmonary dysfunction, which can manifest as subendocardial ischaemia, stunned myocardium, atrial and ventricular arrhythmias and ECG changes (T-wave inversion, ST depressions, or even ST elevations) in the absence of structural coronary artery disease. Echocardiography may show regional wall motion abnormalities, including non-ischaemic cardiomyopathies, such as takotsubo cardiomyopathy with apical ballooning133. Cardiac monitoring with electrocardiogram, cardiac enzymes, and echocardiogram is essential in aSAH when there is an insufficient response to ionotropic support during induced hypertension. The development of acute heart failure and hypotension are associated with poor outcomes. SAH patients with symptomatic vasospasm with high initial troponin I level, indicative of neurogenic cardiac injury, are at twice the risk of medical treatment failure107. Acute respiratory distress syndrome, and neurogenic and cardiogenic pulmonary oedema have also been described in patients with aSAH. Hypoxia and hypotension harm the brain, contributing to secondary brain injuries. We aim to maintain brain perfusion and oxygenation while treating cardiopulmonary complications.
Hyponatraemia
Hyponatraemia is commonly seen in association with aSAH. Hyponatraemia (serum sodium of <135 mmol/L) affects 27–44% of patients after aSAH, most commonly from the syndrome of inappropriate antidiuretic hormone (SIADH) or cerebral salt wasting (CSW). CSW is a debated entity amongst clinicians, with some studies suggesting it doesn’t exist as an aetiology for hyponatraemia134. Hypovolaemia, hyponatraemia or subsequent fluid restriction increase the risk of DCI with poor outcomes. Therefore, in clinical practice, hyponatraemia associated with SAH management is based on maintaining euvolaemia, avoiding hypo- and hypervolaemia and repletion of volume and sodium losses135.
Managing non-culprit (unruptured) aneurysms
A multidisciplinary team comprising an interventional neuroradiologist and a neurosurgeon should discuss the options, including endovascular coiling, neurosurgical clipping, or conservative management. Follow-up monitoring considers factors including aneurysm size, lifetime risk of rupture, risk of treatment options, and comorbidities. These should be discussed with the patient and their family where appropriate49.
Management of SAH in young people
It is an uncommon disease in children and young adults, with a male-to-female ratio of 1:8 ratio136. Anatomically, most of the aneurysms are located at the tip of the internal carotid artery, followed by the posterior circulation, with 60% presenting as an SAH. 30% of SAH in the paediatric population is due to a structural abnormality of the blood vessel wall abnormalities, such as Marfan syndrome, Ehler-Danlos syndrome type IV, fibromuscular dysplasia or arterio-venous malformations. These patients should all be investigated for connective tissue disorders as a part of their work to manage the SAH.
SAH and inequality of healthcare accessibility
Inequalities within the health care systems, limited access to experienced neurosciences units and lack of skilled staff to manage the SAH within and between countries significantly impact the SAH outcomes. The first step to reducing these inequalities requires access to good-quality data and acknowledging these disparities137. An example of such discrepancy is the differences in outcomes of high-volume versus low-volume centres for SAH management. Telemedicine can be a valuable tool that can be leveraged in resource-limited or low-volume settings, providing equitable access to care and expert intervention138. Greater attention to community-based educational initiatives to improve stroke awareness and timely intervention enhances patient outcomes139. Management of SAH requires urgent large-scale phase three studies to establish standardised protocols and multidisciplinary neurovascular training programmes. The evolution of technology use in resource-poor settings, such as machine learning and artificial intelligence, can advance health equity in aSAH management by predicting disparities and aiding diagnosis and treatment decisions140. Techniques, such as novel robotic transcranial Doppler monitoring, may mitigate the requirement for trained staff to manage these patients141. Once established, adherence to evidence-based guidelines should be encouraged by data-driven performance analysis142,143,144. Reduction of long-term morbidity after SAH, such as neurocognitive and social decline, requires a close integration of health and welfare services145,146,147.
Discussion
While advancements in medical technology and treatment protocols have significantly improved outcomes, SAH remains a challenging condition with profound implications for patient care. The highly individualised nature of SAH, due to factors such as aneurysm location patient comorbidities, and pathophysiology underscores the importance of personalised treatment strategies. The incidence of multi-system complications further exemplifies the need for an interdisciplinary approach, which is critical for optimising patient outcomes.
As we continue to face these multifaceted challenges, there is a pressing need for ongoing research to refine existing treatment protocols and to develop new, innovative approaches to manage SAH. Future studies should aim to address the gaps in our understanding of SAH pathophysiology and to evaluate strategies that could mitigate the systemic inequities that currently limit the accessibility of optimal care.
The collaboration between neuroscientists, clinicians, and health policy experts will be crucial in this endeavour. The development of novel therapeutics and improved management techniques, backed by robust data collection and analysis, holds the promise of enhancing the quality of life for individuals affected by SAH.
References
Linn, F. H., Rinkel, G. J., Algra, A. & van Gijn, J. Incidence of subarachnoid hemorrhage: role of region, year, and rate of computed tomography: a meta-analysis. Stroke 27, 625–629 (1996).
Maher, M., Schweizer, T. A. & Macdonald, R. L. Treatment of spontaneous subarachnoid hemorrhage: guidelines and gaps. Stroke 51, 1326–1332 (2020).
Boogaarts, H. D. et al. Caseload as a factor for outcome in aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J. Neurosurg. 120, 605–611 (2014).
Storrow, A. B. & Wrenn, K. Aneurysmal subarachnoid hemorrhage. N. Engl. J. Med 354, 1755–1757 (2006).
Broderick, J. P. et al. Greater rupture risk for familial as compared to sporadic unruptured intracranial aneurysms. Stroke 40, 1952–1957 (2009).
Leblanc, R. Familial cerebral aneurysms. Stroke 27, 1050–1054 (1996).
Ronkainen, A. et al. Familial intracranial aneurysms. Lancet 349, 380–384 (1997).
Tominari, S. et al. Prediction model for 3-year rupture risk of unruptured cerebral aneurysms in Japanese patients. Ann. Neurol. 77, 1050–1059 (2015).
Lindgren, A. E. et al. Irregular shape of intracranial aneurysm indicates rupture risk irrespective of size in a population-based Cohort. Stroke 47, 1219–1226 (2016).
Hunt, W. E. & Hess, R. M. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J. Neurosurg. 28, 14–20 (1968).
Teasdale, G. M. et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J. Neurol. Neurosurg. Psychiatry 51, 1457 (1988).
Macdonald, R. L. & Schweizer, T. A. Spontaneous subarachnoid haemorrhage. Lancet 389, 655–666 (2017).
Huang, J. & van Gelder, J. M. The probability of sudden death from rupture of intracranial aneurysms: a meta-analysis. Neurosurgery 51, 1101–1105 (2002).
Tidswell, P., Dias, P. S., Sagar, H. J., Mayes, A. R. & Battersby, R. D. Cognitive outcome after aneurysm rupture: relationship to aneurysm site and perioperative complications. Neurology 45, 875–882 (1995).
Rosengart, A. J., Schultheiss, K. E., Tolentino, J. & Macdonald, R. L. Prognostic factors for outcome in patients with aneurysmal subarachnoid hemorrhage. Stroke 38, 2315–2321 (2007).
Claassen, J. et al. Effect of cisternal and ventricular blood on risk of delayed cerebral ischemia after subarachnoid hemorrhage: the Fisher scale revisited. Stroke 32, 2012–2020 (2001).
Wartenberg, K. E. et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit. Care Med. 34, 617–623 (2006).
Behrouz, R. et al. Focal neurological deficit at onset of aneurysmal subarachnoid hemorrhage: frequency and causes. J. Stroke Cerebrovasc. Dis. 25, 2644–2647 (2016).
Perry, J. J. et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 310, 1248–1255 (2013).
Walton, M. et al. Management of patients presenting to the emergency department with sudden onset severe headache: systematic review of diagnostic accuracy studies. Emerg. Med. J. 39, 818–825 (2022).
Perry, J. J. et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 343, d4277 (2011).
Kidwell, C. S. & Wintermark, M. Imaging of intracranial haemorrhage. Lancet Neurol. 7, 256–267 (2008).
Dawkins, A. A. et al. Complications of cerebral angiography: a prospective analysis of 2,924 consecutive procedures. Neuroradiology 49, 753–759 (2007).
Agid, R. et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: When is digital subtraction angiography still needed? AJNR Am. J. Neuroradiol. 31, 696–705 (2010).
Zhang, L. J. et al. Dual-energy CT angiography in the evaluation of intracranial aneurysms: image quality, radiation dose, and comparison with 3D rotational digital subtraction angiography. AJR Am. J. Roentgenol. 194, 23–30 (2010).
Wilson, C. D. & Shankar, J. J. Diagnosing vasospasm after subarachnoid hemorrhage: CTA and CTP. Can. J. Neurol. Sci. 41, 314–319 (2014).
Verma, R. K. et al. Detecting subarachnoid hemorrhage: comparison of combined FLAIR/SWI versus CT. Eur. J. Radio. 82, 1539–1545 (2013).
Sailer, A. M., Wagemans, B. A., Nelemans, P. J., de Graaf, R. & van Zwam, W. H. Diagnosing intracranial aneurysms with MR angiography: systematic review and meta-analysis. Stroke 45, 119–126 (2014).
Magni, F., Pozzi, M., Rota, M., Vargiolu, A. & Citerio, G. High-resolution intracranial pressure burden and outcome in subarachnoid hemorrhage. Stroke 46, 2464–2469 (2015).
Lauzier, D. C. et al. Early brain injury after subarachnoid hemorrhage: incidence and mechanisms. Stroke 54, 1426–1440 (2023).
Carteron, L. et al. Non-ischemic cerebral energy dysfunction at the early brain injury phase following aneurysmal subarachnoid hemorrhage. Front. Neurol. 8, 325 (2017).
Reis, C. et al. Pathophysiology of Subarachnoid Hemorrhage, Early Brain Injury, and Delayed Cerebral Ischemia, Primer on Cerebrovascular Diseases. 2nd edn, 125–130 Ch. 25 (Academic Press, 2017).
Kramer, D. R., Fujii, T., Ohiorhenuan, I. & Liu, C. Y. Cortical spreading depolarization: Pathophysiology, implications, and future directions. J. Clin. Neurosci. 24, 22–27 (2016).
Rass, V. & Helbok, R. Early brain injury after poor-grade subarachnoid hemorrhage. Curr. Neurol. Neurosci. Rep. 19, 78 (2019).
Hayman, E. G., Wessell, A., Gerzanich, V., Sheth, K. N. & Simard, J. M. Mechanisms of global cerebral edema formation in aneurysmal subarachnoid hemorrhage. Neurocrit. Care 26, 301–310 (2017).
Lubieniecka, J. M. et al. Biomarkers for severity of spinal cord injury in the cerebrospinal fluid of rats. PLoS One 6, e19247 (2011).
Thelin, E. P. et al. Serial sampling of serum protein biomarkers for monitoring human traumatic brain injury dynamics: a systematic review. Front Neurol. 8, 300 (2017).
Missler, U., Wiesmann, M., Friedrich, C. & Kaps, M. S-100 protein and neuron-specific enolase concentrations in blood as indicators of infarction volume and prognosis in acute ischemic stroke. Stroke 28, 1956–1960 (1997).
Delgado-Alvarado, M., Gago, B., Navalpotro-Gomez, I., Jiménez-Urbieta, H. & Rodriguez-Oroz, M. C. Biomarkers for dementia and mild cognitive impairment in Parkinson’s disease. Mov. Disord. 31, 861–881 (2016).
Gris, T. et al. Innate immunity activation in the early brain injury period following subarachnoid hemorrhage. J. Neuroinflam. 16, 253 (2019).
Atangana, E. et al. Intravascular inflammation triggers intracerebral activated microglia and contributes to secondary brain injury after experimental subarachnoid hemorrhage (eSAH). Transl. Stroke Res. 8, 144–156 (2017).
Dilraj, A., Botha, J. H., Rambiritch, V., Miller, R. & van Dellen, J. R. Levels of catecholamine in plasma and cerebrospinal fluid in aneurysmal subarachnoid hemorrhage. Neurosurgery 31, 42–50 (1992).
Westermaier, T., Jauss, A., Eriskat, J., Kunze, E. & Roosen, K. Time-course of cerebral perfusion and tissue oxygenation in the first 6 h after experimental subarachnoid hemorrhage in rats. J. Cereb. Blood Flow. Metab. 29, 771–779 (2009).
Reis, C. et al. In Primer on Cerebrovascular Diseases (Second Edition) (eds Louis R. Caplan et al.) 125–130 (Academic Press, 2017).
Dreier, J. P. et al. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J. Cereb. Blood Flow. Metab. 18, 978–990 (1998).
van Heuven, A. W., Dorhout Mees, S. M., Algra, A. & Rinkel, G. J. Validation of a prognostic subarachnoid hemorrhage grading scale derived directly from the Glasgow Coma Scale. Stroke 39, 1347–1348 (2008).
Klimo, P. Jr. & Schmidt, R. H. Computed tomography grading schemes used to predict cerebral vasospasm after aneurysmal subarachnoid hemorrhage: a historical review. Neurosurg. Focus 21, E5 (2006).
Cross, D. T. 3rd et al. Mortality rates after subarachnoid hemorrhage: variations according to hospital case volume in 18 states. J. Neurosurg. 99, 810–817 (2003).
Subarachnoid haemorrhage caused by a ruptured aneurysm: diagnosis and management. NICE Guideline. https://www.nice.org.uk/guidance/ng228, Accessed on January 2024.
Greenberg, S. M. et al. 2022 Guideline for the management of patients with spontaneous intracerebral hemorrhage: a guideline from the American Heart Association/American Stroke Association. Stroke 53, e282–e361 (2022).
Hijdra, A., Braakman, R., van Gijn, J., Vermeulen, M. & van Crevel, H. Aneurysmal subarachnoid hemorrhage. Complications and outcome in a hospital population. Stroke 18, 1061–1067 (1987).
Ohkuma, H., Tsurutani, H. & Suzuki, S. Incidence and significance of early aneurysmal rebleeding before neurosurgical or neurological management. Stroke 32, 1176–1180 (2001).
Naidech, A. M. et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch. Neurol. 62, 410–416 (2005).
Steiner, T. et al. European stroke organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc. Dis. 35, 93–112 (2013).
Darkwah Oppong, M. et al. Blood pressure and outcome after aneurysmal subarachnoid hemorrhage. Sci. Rep. 12, 8006 (2022).
Hoh, B. L. et al. 2023 Guideline for the management of patients with aneurysmal subarachnoid hemorrhage: a guideline from the American Heart Association/American Stroke Association. Stroke 54, e314–e370 (2023).
Connolly, E. S. et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage. Stroke 43, 1711–1737 (2012).
Connolly, E. S. Jr. et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke 43, 1711–1737 (2012).
Phillips, T. J., Dowling, R. J., Yan, B., Laidlaw, J. D. & Mitchell, P. J. Does treatment of ruptured intracranial aneurysms within 24 hours improve clinical outcome? Stroke 42, 1936–1945 (2011).
van Donkelaar, C. E. et al. Predictive factors for rebleeding after aneurysmal subarachnoid hemorrhage: rebleeding aneurysmal subarachnoid hemorrhage study. Stroke 46, 2100–2106 (2015).
Laidlaw, J. D. & Siu, K. H. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J. Neurosurg. 97, 250–258 (2002).
Chua, M. H. et al. Documentation of improved outcomes for intracranial aneurysm management Over a 15-Year interval. Stroke 47, 708–712 (2016).
Molyneux, A. J. et al. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 366, 809–817 (2005).
Lindgren, A. et al. Endovascular coiling versus neurosurgical clipping for people with aneurysmal subarachnoid haemorrhage. Cochrane Database Syst. Rev. 8, CD003085 (2018).
Molyneux, A. J., Birks, J., Clarke, A., Sneade, M. & Kerr, R. S. The durability of endovascular coiling versus neurosurgical clipping of ruptured cerebral aneurysms: 18 year follow-up of the UK cohort of the International Subarachnoid Aneurysm Trial (ISAT). Lancet 385, 691–697 (2015).
Hillman, J. et al. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J. Neurosurg. 97, 771–778 (2002).
Post, R. et al. Short-term tranexamic acid treatment reduces in-hospital mortality in aneurysmal sub-arachnoid hemorrhage: a multicenter comparison study. PLoS One 14, e0211868 (2019).
Diringer, M. N. et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit. Care 15, 211–240 (2011).
Starke, R. M. et al. Impact of a protocol for acute antifibrinolytic therapy on aneurysm rebleeding after subarachnoid hemorrhage. Stroke 39, 2617–2621 (2008).
Klass, A., Sanchez-Porras, R. & Santos, E. Systematic review of the pharmacological agents that have been tested against spreading depolarizations. J. Cereb. Blood Flow. Metab. 38, 1149–1179 (2018).
Cahill, J., Calvert, J. W. & Zhang, J. H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow. Metab. 26, 1341–1353 (2006).
Santos, E. et al. Lasting s-ketamine block of spreading depolarizations in subarachnoid hemorrhage: a retrospective cohort study. Crit. Care 23, 427 (2019).
Helbok, R. et al. What should a clinician do when spreading depolarizations are observed in a patient? Neurocrit Care 32, 306–310 (2020).
Sundaram, M. B. & Chow, F. Seizures associated with spontaneous subarachnoid hemorrhage. Can. J. Neurol. Sci. 13, 229–231 (1986).
Claassen, J. et al. Nonconvulsive seizures in subarachnoid hemorrhage link inflammation and outcome. Ann. Neurol. 75, 771–781 (2014).
Naidech, A. M. et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 36, 583–587 (2005).
Lanzino, G., D’Urso, P. I. & Suarez, J., Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid, H. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit. Care 15, 247–256 (2011).
Claassen, J. et al. Predictors and clinical impact of epilepsy after subarachnoid hemorrhage. Neurology 60, 208–214 (2003).
Rosengart, A. J. et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J. Neurosurg. 107, 253–260 (2007).
Claassen, J. et al. Recommendations on the use of EEG monitoring in critically ill patients: consensus statement from the neurointensive care section of the ESICM. Intensive Care Med 39, 1337–1351 (2013).
Claassen, J. et al. Prognostic significance of continuous EEG monitoring in patients with poor-grade subarachnoid hemorrhage. Neurocrit Care 4, 103–112 (2006).
Geraghty, J. R. & Testai, F. D. Delayed cerebral ischemia after subarachnoid hemorrhage: beyond vasospasm and towards a multifactorial pathophysiology. Curr. Atheroscler. Rep. 19, 50 (2017).
Rowland, M. J., Hadjipavlou, G., Kelly, M., Westbrook, J. & Pattinson, K. T. S. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br. J. Anaesth. 109, 315–329 (2012).
Macdonald, R. L. et al. Randomized trial of clazosentan in patients with aneurysmal subarachnoid hemorrhage undergoing endovascular coiling. Stroke 43, 1463–1469 (2012).
Pickard, J. D. et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 298, 636–642 (1989).
Allen, G. S. et al. Cerebral arterial spasm–a controlled trial of nimodipine in patients with subarachnoid hemorrhage. N. Engl. J. Med 308, 619–624 (1983).
van den Bergh, W. M. et al. Magnesium sulfate in aneurysmal subarachnoid hemorrhage: a randomized controlled trial. Stroke 36, 1011–1015 (2005).
Wong, G. K. et al. Intravenous magnesium sulphate for aneurysmal subarachnoid hemorrhage (IMASH): a randomized, double-blinded, placebo-controlled, multicenter phase III trial. Stroke 41, 921–926 (2010).
Kirkpatrick, P. J. & Turner, C. L. Simvastatin in subarachnoid haemorrhage: beyond the short-term–authors’ reply. Lancet Neurol. 13, 1073–1074 (2014).
Gomis, P. et al. Randomized, double-blind, placebo-controlled, pilot trial of high-dose methylprednisolone in aneurysmal subarachnoid hemorrhage. J. Neurosurg. 112, 681–688 (2010).
Kumar, G., Shahripour, R. B. & Harrigan, M. R. Vasospasm on transcranial Doppler is predictive of delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J. Neurosurg. 124, 1257–1264 (2016).
Chaudhary, S. R. et al. Prospective evaluation of multidetector-row CT angiography for the diagnosis of vasospasm following subarachnoid hemorrhage: a comparison with digital subtraction angiography. Cerebrovasc. Dis. 25, 144–150 (2008).
Yoon, D. Y., Choi, C. S., Kim, K. H., Cho, B. M. & Multidetector-row, C. T. angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am. J. Neuroradiol. 27, 370–377 (2006).
Cremers, C. H. et al. CT perfusion and delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J. Cereb. Blood Flow. Metab. 34, 200–207 (2014).
Rouanet, C. & Silva, G. S. Aneurysmal subarachnoid hemorrhage: current concepts and updates. Arq. Neuropsiquiatr. 77, 806–814 (2019).
Rass, V. et al. Protocolized brain oxygen optimization in subarachnoid hemorrhage. Neurocrit Care 31, 263–272 (2019).
Chen, H. I. et al. Detection of cerebral compromise with multimodality monitoring in patients with subarachnoid hemorrhage. Neurosurgery 69, 53–63 (2011).
Vergouwen, M. D., Vermeulen, M., de Haan, R. J., Levi, M. & Roos, Y. B. Dihydropyridine calcium antagonists increase fibrinolytic activity: a systematic review. J. Cereb. Blood Flow. Metab. 27, 1293–1308 (2007).
Carlson, A. P., Hänggi, D., Macdonald, R. L. & Shuttleworth, C. W. Nimodipine reappraised: an old drug with a future. Curr. Neuropharmacol. 18, 65–82 (2020).
Kieninger, M. et al. Incidence of arterial hypotension in patients receiving peroral or continuous intra-arterial nimodipine after aneurysmal or perimesencephalic subarachnoid hemorrhage. Neurocrit. Care 31, 32–39 (2019).
Hajizadeh Barfejani, A., Rabinstein, A. A., Wijdicks, E. F. M. & Clark, S. L. Poor utilization of nimodipine in aneurysmal subarachnoid hemorrhage. J. Stroke Cerebrovasc. Dis. 28, 2155–2158 (2019).
Haegens, N. M. et al. Induced hypertension in preventing cerebral infarction in delayed cerebral ischemia after subarachnoid hemorrhage. Stroke 49, 2630–2636 (2018).
Raabe, A. et al. Relative importance of hypertension compared with hypervolemia for increasing cerebral oxygenation in patients with cerebral vasospasm after subarachnoid hemorrhage. J. Neurosurg. 103, 974–981 (2005).
Lennihan, L. et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 31, 383–391 (2000).
Egge, A. et al. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 49, 593–605 (2001).
Ekelund, A. et al. Effects of iso- and hypervolemic hemodilution on regional cerebral blood flow and oxygen delivery for patients with vasospasm after aneurysmal subarachnoid hemorrhage. Acta Neurochir. (Wien.) 144, 703–712 (2002).
Suwatcharangkoon, S. et al. Medical treatment failure for symptomatic vasospasm after subarachnoid hemorrhage threatens long-term outcome. Stroke 50, 1696–1702 (2019).
Zwienenberg-Lee, M. et al. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 39, 1759–1765 (2008).
Dasenbrock, H. H. et al. Hospital-Acquired Infections after Aneurysmal Subarachnoid Hemorrhage: A Nationwide Analysis. World Neurosurg. 88, 459–474 (2016).
Badjatia, N. et al. Serum glutamine and hospital-acquired infections after aneurysmal subarachnoid hemorrhage. Neurology 91, e421–e426 (2018).
Oddo, M. et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit. Care Med 36, 3233–3238 (2008).
Ayling, O. G. S., Ibrahim, G. M., Alotaibi, N. M., Gooderham, P. A. & Macdonald, R. L. Anemia After Aneurysmal Subarachnoid Hemorrhage Is Associated With Poor Outcome and Death. Stroke 49, 1859–1865 (2018).
Rass, V. et al. Fluid Intake But Not Fluid Balance Is Associated With Poor Outcome in Nontraumatic Subarachnoid Hemorrhage Patients. Crit. Care Med 47, e555–e562 (2019).
Jeon, S. B. et al. Hyperoxia may be related to delayed cerebral ischemia and poor outcome after subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatry 85, 1301–1307 (2014).
Manoel, A. L. et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can. J. Neurol. Sci. 41, 554–561 (2014).
Haines, S. J. Commentary: What We Might or Might Not Know About Venous Thromboembolism Prophylaxis in Neurosurgery. Neurosurgery 86, E455–E468 (2020).
Yataco, R. A. et al. Early Progressive Mobilization of Patients with External Ventricular Drains: Safety and Feasibility. Neurocrit Care 30, 414–420 (2019).
Deng, H., Fang, Q., Chen, K. & Zhang, X. Early versus late tracheotomy in ICU patients: A meta-analysis of randomized controlled trials. Med. (Baltim.) 100, e24329 (2021).
Badjatia, N. et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 66, 696–700 (2010).
Deibert, E. et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J. Neurosurg. 98, 741–746 (2003).
Solenski, N. J. et al. Medical complications of aneurysmal subarachnoid hemorrhage: a report of the multicenter, cooperative aneurysm study. Participants of the Multicenter Cooperative Aneurysm Study. Crit. Care Med 23, 1007–1017 (1995).
Banki, N. et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J. Neurosurg. JNS 105, 15–20 (2006).
Brouwers, P. J. et al. Serial electrocardiographic recording in aneurysmal subarachnoid hemorrhage. Stroke 20, 1162–1167 (1989).
Banki, N. M. et al. Acute Neurocardiogenic Injury After Subarachnoid Hemorrhage. Circulation 112, 3314–3319 (2005).
Naidech, A. M. et al. Cardiac Troponin Elevation, Cardiovascular Morbidity, and Outcome After Subarachnoid Hemorrhage. Circulation 112, 2851–2856 (2005).
Vannemreddy, P., Venkatesh, P., Dinesh, K., Reddy, P. & Nanda, A. 151-154 (Springer Vienna).
Abd, T. T. et al. Incidence and clinical characteristics of takotsubo cardiomyopathy post-aneurysmal subarachnoid hemorrhage. Int J. Cardiol. 176, 1362–1364 (2014).
Lee, V. H. et al. Tako-tsubo cardiomyopathy in aneurysmal subarachnoid hemorrhage: an underappreciated ventricular dysfunction. J. Neurosurg. JNS 105, 264–270 (2006).
Mrozek, S., Gobin, J., Constantin, J. M., Fourcade, O. & Geeraerts, T. Crosstalk between brain, lung and heart in critical care. Anaesth. Crit. Care Pain. Med 39, 519–530 (2020).
Sposato, L. A., Fridman, S., Whitehead, S. N. & Lopes, R. D. Linking stroke-induced heart injury and neurogenic atrial fibrillation: a hypothesis to be proven. J. Electrocardiol. https://doi.org/10.1016/j.jelectrocard.2018.02.006 (2018).
Sposato, L. A. et al. Post-Stroke Cardiovascular Complications and Neurogenic Cardiac Injury: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 76, 2768–2785 (2020).
Krishnamoorthy, V., Mackensen, G. B., Gibbons, E. F. & Vavilala, M. S. Cardiac Dysfunction After Neurologic Injury: What Do We Know and Where Are We Going? Chest 149, 1325–1331 (2016).
Macmillan, C. S., Grant, I. S. & Andrews, P. J. Pulmonary and cardiac sequelae of subarachnoid haemorrhage: time for active management? Intensive Care Med 28, 1012–1023 (2002).
Hannon, M. J. et al. Hyponatremia following mild/moderate subarachnoid hemorrhage is due to SIAD and glucocorticoid deficiency and not cerebral salt wasting. J. Clin. Endocrinol. Metab. 99, 291–298 (2014).
Rabinstein, A. A. & Bruder, N. Management of hyponatremia and volume contraction. Neurocrit Care 15, 354–360 (2011).
Sorteberg, A. & Dahlberg, D. Intracranial Non-traumatic Aneurysms in Children and Adolescents. Curr. Pediatr. Rev. 9, 343–352 (2013).
Leifer, D. et al. Association Between Hospital Volumes and Clinical Outcomes for Patients With Nontraumatic Subarachnoid Hemorrhage. J. Am. Heart Assoc. 10, e018373 (2021).
Almathami, H. K. Y., Win, K. T. & Vlahu-Gjorgievska, E. Barriers and Facilitators That Influence Telemedicine-Based, Real-Time, Online Consultation at Patients’ Homes: Systematic Literature Review. J. Med Internet Res 22, e16407 (2020).
Forster, A. et al. In Development and evaluation of tools and an intervention to improve patient- and carer-centred outcomes in Longer-Term Stroke care and exploration of adjustment post stroke: the LoTS care research programme (NIHR Journals Library Copyright © Queen’s Printer and Controller of HMSO 2014. This work was produced by Forster et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK., 2014).
Bhatt, P., Liu, J., Gong, Y., Wang, J. & Guo, Y. Emerging Artificial Intelligence-Empowered mHealth: Scoping Review. JMIR Mhealth Uhealth 10, e35053 (2022).
Clare, K. et al. Safety and efficacy of a novel robotic transcranial doppler system in subarachnoid hemorrhage. Sci. Rep. 12, 2266 (2022).
Yaria, J. et al. Quality of stroke guidelines in low- and middle-income countries: a systematic review. Bull. World Health Organ 99, 640–652e (2021).
Ohkuma, H., Shimamura, N., Naraoka, M. & Katagai, T. Aneurysmal Subarachnoid Hemorrhage in the Elderly over Age 75: A Systematic Review. Neurol. Med Chir. (Tokyo) 57, 575–583 (2017).
Lens, C. et al. Variation in stroke care at the hospital level: A cross-sectional multicenter study. Front Neurol. 13, 1004901 (2022).
Gerner, S. T. et al. Long-Term Complications and Influence on Outcome in Patients Surviving Spontaneous Subarachnoid Hemorrhage. Cerebrovasc. Dis. 49, 307–315 (2020).
Vetkas, A., Prans, E., Kõks, S., Rätsep, T. & Asser, T. Aneurysmal subarachnoid haemorrhage: Effect of CRHR1 genotype on mental health-related quality of life. Sci. Rep. 10, 724 (2020).
Zhou, X. et al. Racial differences in time to blood pressure control of aneurysmal subarachnoid hemorrhage patients: A single-institution study. PLoS One 18, e0279769 (2023).
Author information
Authors and Affiliations
Contributions
S.T., T.V., P.B. contributed to manuscript conception, design, preparation and review. E.L., T.W., Z.A., N.G. contributed to manuscript preparation and review. All agreed on the final content of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bryan Boling and Agnieszka Uryga for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thilak, S., Brown, P., Whitehouse, T. et al. Diagnosis and management of subarachnoid haemorrhage. Nat Commun 15, 1850 (2024). https://doi.org/10.1038/s41467-024-46015-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46015-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
