
Abstract
Rechargeable calcium (Ca) metal batteries are promising candidates for sustainable energy storage due to the abundance of Ca in Earth’s crust and the advantageous theoretical capacity and voltage of these batteries. However, the development of practical Ca metal batteries has been severely hampered by the current cathode chemistries, which limit the available energy and power densities, as well as their insufficient capacity retention and low-temperature capability. Here, we describe the rechargeable Ca/Cl2 battery based on a reversible cathode redox reaction between CaCl2 and Cl2, which is enabled by the use of lithium difluoro(oxalate)borate as a key electrolyte mediator to facilitate the dissociation and distribution of Cl-based species and Ca2+. Our rechargeable Ca/Cl2 battery can deliver discharge voltages of 3 V and exhibits remarkable specific capacity (1000 mAh g−1) and rate capability (500 mA g−1). In addition, the excellent capacity retention (96.5% after 30 days) and low-temperature capability (down to 0 °C) allow us to overcome the long-standing bottleneck of rechargeable Ca metal batteries.
Similar content being viewed by others
Introduction
Modern electrification has witnessed the ever-growing demand for rechargeable batteries with high sustainability and energy storage capabilities1,2,3,4. Rechargeable calcium (Ca) metal batteries are among the most promising candidates because of their advantageous features, such as high crustal abundance, high theoretical capacity, and ideal redox potential5,6,7. However, compared with the conventional Li counterparts, current Ca metal batteries based on intercalation/deintercalation cathodes, such as metal oxides, suffer from relatively limited electrochemical performance, e.g., low specific capacities (<200 mAh g−1), discharge voltages (<2.6 V), and rate capability (<100 mA g−1)8,9,10,11, which requires the development of new cathode reactions to overcome these limitations on electrochemical performance. In addition, the capacity retention and low-temperature performance of Ca metal batteries remain inaccessible to date, due to the inferior electrochemical stability and sluggish cathode/anode reaction kinetics. Therefore, it is crucial yet challenging to develop new cathode chemistry to realize practical applications of rechargeable Ca metal batteries12,13,14.
Reversible CaCl2/Cl2 redox reaction is among the most attractive cathode reactions for Ca metal batteries, enabling the delivery of high specific capacities (483 mAh g−1 based on the mass of CaCl2) at discharge voltages of 3 V. However, it is currently unavailable so far, owing to the challenging control of Ca-based electrolytes and the generated electrolyte/electrode interfaces3. On the cathode side, for instance, the strong electrostatic force of Ca2+ is prone to inhibit its efficient dissociation and distribution in the electrolyte15,16, making the desired CaCl2/Cl2 cathode conversion thermodynamically and kinetically unfavorable. On the anode side, the high reactivity of Ca metal requires rational regulation of electrolyte solvation to ensure high electrochemical reversibility17,18. Therefore, it is desired but challenging for electrolyte designing and screening to unlock reversible CaCl2/Cl2 redox reactions for practical Ca metal batteries.
Here, we show that reversible CaCl2/Cl2 redox reaction can be fully unlocked in Ca metal batteries using a CaCl2–AlCl3–SOCl2 electrolyte regulated by a lithium difluoro(oxalate)borate (LiDFOB) mediator. LiDFOB has been demonstrated to facilitate the dissociation and distribution of Cl-based species and Ca2+, thus allowing the CaCl2/Cl2 redox reaction. Our rechargeable Ca/Cl2 battery demonstrates discharge voltages of 3 V and remarkable specific capacity (up to 1000 mAh g−1) and rate capability (up to 500 mA g−1) because of the fast kinetics of the CaCl2/Cl2 redox reaction. In addition, the shelf life (>30 days) and low-temperature performance (down to 0 °C) achieved by this battery overcome the long-standing bottlenecks that have plagued the practical application of rechargeable Ca metal batteries. Our results provide a new paradigm for bridging Cl-based cathode and multivalent metal anode chemistry to achieve sustainable and high-performance energy storage.
Results
To develop a rechargeable Ca/Cl2 battery, we used a graphite cathode and a Ca metal anode coupled with a Cl-based electrolyte composed of CaCl2, AlCl3, and LiDFOB salts in SOCl2 (named CALS electrolyte) (Fig. 1a and Supplementary Fig. 1; see preparation details in “Methods”). This rechargeable Ca/Cl2 battery delivered a high reversible specific capacity of 1000 mAh g−1 and a high discharge voltage of more than 3 V (Fig. 1b), making it highly competitive compared with state-of-the-art cathodes used in Ca metal batteries, e.g., metal oxides19, sulfur20, and oxygen21 (Fig. 1c and Supplementary Fig. 2).

a Schematic illustration of a rechargeable Ca/Cl2 battery based on a graphite cathode, a Ca metal anode, and a CALS electrolyte composed of a mixture of CaCl2, AlCl3, and LiDFOB dissolved in SOCl2. b Galvanostatic charge–discharge curve of the rechargeable Ca/Cl2 battery. The specific charge capacity and current density are 1000 mAh g−1 and 100 mA g−1, respectively. c Comparison of the reversible specific capacity and discharge voltage of the rechargeable Ca/Cl2 battery with those of other representative batteries with Ca cathodes, e.g., manganese dioxide19, sulfur20, and oxygen21. All specific capacities were based on the mass of the active materials on the cathode. A comparison of the areal capacities was also provided in Supplementary Fig. 2.
Notably, CaCl2 was in-situ formed on the graphite cathode during the first discharge of the battery, as shown in Eq. (1), and could stably exist at the cathode because the dissolution of CaCl2 in the electrolyte has reached saturation. At the anode, the stripping of Ca metal occurred as described in Eq. (2). The overall reaction during the first discharge was described in Eq. (3). A specific capacity of 3,264 mAh g−1 (based on the mass of graphite here and throughout this paper) was delivered with a discharge voltage of ~2.8 V at 100 mA g−1 (Supplementary Fig. 3). The formation of SO2 was verified by differential electrochemical mass spectrometry (DEMS, Supplementary Fig. 4).
The formed CaCl2 on the graphite cathode was then oxidized to Cl2 during the subsequent charge step and reduced back to CaCl2 during discharge according to Eq. (4), while the deposition and stripping process of Ca metal at the anode was shown in Eq. (5), resulting in a high discharge plateau at ~3.5 V (Fig. 1b). The overall reaction of the subsequent charging and discharging processes was shown in Eq. (6). A minor discharge plateau was observed at ~2 V, which might correspond to the reduction of other charge products, such as SO2Cl222.
We found that LiDFOB played a key role in enabling the rechargeability of the Ca/Cl2 battery. For instance, the battery prepared with a LiDFOB-free electrolyte did not deliver any charge/discharge capacity, and no plateau was observed, in stark contrast to the pronounced charge/discharge behavior observed in the presence of LiDFOB (Fig. 2a), which inspired us to investigate the origin of the rechargeability of our Ca/Cl2 battery. We hypothesized that the unique chemical structure of DFOB− with two B−F and two C=O functional groups could effectively mediate the dissociation and distribution of Cl-based species (e.g., AlCl3 and SOCl2) and Ca2+, thus facilitating their immigration and improving the kinetics of the redox reaction (Fig. 2b). To test this hypothesis, we acquired Raman spectra of different electrolytes (Fig. 2c). The spectrum of bare SOCl2 showed peaks attributed to the characteristic symmetric Cl−S−Cl deformation (δs(Cl−S−Cl), 342.8 cm−1), asymmetric S−Cl stretching (νa(S−Cl), 441.1 cm−1) and symmetric S−Cl stretching (νs(S−Cl), 487.7 cm−1) vibrations23,24. The addition of AlCl3 resulted in two new peaks at 384.9 and 524.6 cm−1, corresponding to the formation of Cl2SO…AlCl3 adducts. Subsequent addition of CaCl2 showed two new Raman peaks at 363.8 and 504.5 cm−1, which could be attributed to the Ca−O vibration (Ca2+…SOCl2). In addition, the coordination peaks of ν(Al−O) and ν(S−Cl) were redshifted and weakened, indicating a weakened interaction between AlCl3 and SOCl2. Further introduction of DFOB− significantly changed the Raman profile. On the one hand, the Al−O stretching vibration peak significantly redshifted from 382.6 to 376.9 cm−1 with a significant intensity decrease. On the other hand, the coordination S−Cl stretching (AlCl3…SOCl2) peak at 524.6 cm−1 almost disappeared, confirming the sufficient dissociation between AlCl3 and SOCl2 mediated by DFOB−. This evidence indicated that the introduction of DFOB− could effectively facilitate the dissociation of the strong interaction between AlCl3 and SOCl2, thus enabling reversible and fast CaCl2/Cl2 redox reaction. In addition, the solvation structure of the CALS electrolyte remained almost unchanged before and after gelation, as verified by the highly consistent Raman spectra in Supplementary Fig. 5. Thermogravimetric analysis and differential scanning calorimetry tests indicate that the gelation of CALS electrolyte may be related to the intermolecular interaction between different substances such as Ca2+ and DFOB− (Supplementary Fig. 6)25,26.

a Galvanostatic charge–discharge curves of rechargeable Ca/Cl2 batteries using a CALS electrolyte with and without LiDFOB. The specific charge capacity and current density are 500 mAh g−1 and 100 mA g−1, respectively. b Schematic illustration of the ‘dissociation–distribution’ effect; i.e., the DFOB− anion can facilitate the dissociation of various ions, such as AlCl4−, SOCl+, and Ca2+, and benefit the distribution of Ca2+ to achieve fast migration and improve the kinetics. c Raman spectra of SOCl2-based electrolytes with different compositions. The marks in black color represent Ca−O vibrations from Ca2+…SOCl2 interaction. d, e MD simulations and the corresponding solvation structure of the CALS electrolyte without LiDFOB, respectively. f, g MD simulations and the corresponding solvation structure of the CALS electrolyte, respectively. h, i RDFs of the main ion pairs in the CALS electrolyte without and with LiDFOB, respectively. j, k RDFs of the Ca2+–Ca2+ ion pair and MSD calculations of Ca2+ in the CALS electrolyte with and without LiDFOB, respectively.
The distribution and immigration of Ca2+ were also significantly improved with the introduction of DFOB−. We performed molecular dynamics (MD) simulations to investigate the solvation structure of Ca2+ mediated by DFOB− (Fig. 2d–g; see details in the Methods of Supplementary Information), which was further confirmed by the Ca K-edge X-ray absorption spectra (XAS) (Supplementary Fig. 7). In the absence of DFOB−, the solvation shell of Ca2+ ions was only composed of Cl-based species such as Cl− and AlCl4−(Fig. 2d, e). In contrast, the introduction of DFOB− resulted in the formation of a new Ca2+–DFOB− ion pair, which significantly improved the distribution of Ca2+, as observed in the MD simulation snapshots (Fig. 2f, g). This was attributed to C=O…Ca2+ and B−F…Ca2+ coordination originating from the unique chemical structure of DFOB− (Fig. 2g and Supplementary Fig. 8). Radial distribution functions (RDFs) further confirmed the role of DFOB− in the solvation structure. The Ca2+–DFOB− ion pair distance (2.63 Å) was smaller than that of Ca2+–Cl− (3.62 Å) (Fig. 2h, i), indicating that DFOB− participated in a stronger interaction with Ca2+ that could compete with the interaction between Ca2+ and Cl-based species, thus improving their distribution (Fig. 2j). In addition, the interactions of AlCl4−–SOCl+ and Cl−–Cl− were both weakened in the presence of DFOB− (Supplementary Figs 9 and 10), which was in good agreement with the decreased intensity of the Al−O and S−Cl vibration peaks in the Raman spectra (Fig. 2c). Mean square displacement (MSD) calculations showed that the Ca2+ migration rate increased 5-fold after the introduction of DFOB− (1.21 × 10−6 vs. 0.24 × 10−6 cm2 s−1) (Fig. 2k), which was consistent with the trend of ion diffusion coefficient in electrochemical impedance spectroscopy (EIS) (Supplementary Fig. 11). Notably, the possibility that Li ions were responsible for the increased conductivity or decreased impedance could be ruled out, based on the comparison of ionic conductivity and Nyquist plot based on the CaCl2/AlCl3/SOCl2 electrolyte with and without LiCl (see details in Supplementary Fig. 11). Therefore, the improved Ca2+ distribution and ion dissociation mediated by DFOB− were verified to benefit the rechargeability of our Ca/Cl2 batteries.
Understanding cathode chemistry has important implications for improving battery performance. We thus analyzed the cathode reaction products at a variety of charge/discharge states (Fig. 3a–c). After the first discharge of the battery, the formation of CaCl2, SO2, and S was verified by X-ray photoelectron spectroscopy (XPS), consistent with Eq. (1) (Supplementary Fig. 12). The subsequent charge step, which corresponded to the conversion from CaCl2 to Cl2, exposed the edges of the graphite flakes (Fig. 3b and Supplementary Fig. 13), and the following discharge resulted in the formation of CaCl2 nanoflakes on the graphite (Fig. 3c). The high-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) profiles further confirmed the formation of CaCl2 after the first discharge (Fig. 3d). X-ray diffraction (XRD) also revealed the formation and removal of CaCl2 on graphite at the fully discharged and charged states, respectively (Fig. 3e). An air-isolating chamber was used to avoid hygroscopicity of CaCl2, and the weak CaCl2 peaks could be still observed at the fully charged state because not all the CaCl2 on cathode was consumed due to a limited charge capacity of 500 mAh g−1. We suppose that Li+ does not play a critical role in the rechargeability of Ca/Cl2 batteries, as verified by the battery performance based on the CaCl2/AlCl3/SOCl2 electrolyte with the addition of LiCl, which could not deliver any charge/discharge capacity or plateau (Supplementary Fig. 14).

a Galvanostatic charge–discharge curves of a rechargeable Ca/Cl2 battery using the CALS electrolyte. b, c SEM images of the graphite cathode at the fully charged and discharged states, respectively. Scale bars, 1 µm. The current density and specific charge capacity are 100 mA g−1 and 800 mAh g−1, respectively. d HRTEM image of the discharge products formed on the graphite cathode after the first discharge. Scale bar, 5 nm. The inset shows the SAED patterns of CaCl2. Scale bar of the inset, 2 nm−1. e XRD patterns of the pristine, the first discharged, and the second charged graphite cathodes, respectively. f, g Depth distributions of CaCl2−, Cl2−, and C− secondary ion fragments derived from TOF-SIMS depth scans of the fully discharged and charged graphite cathodes, respectively. Analysis areas, 50 × 50 μm2. The current density and specific charge capacity in e–g are 100 mA g−1 and 500 mAh g−1, respectively. h DEMS profile of the rechargeable Ca/Cl2 battery during continuous charge and discharge. i, j High-resolution Cl 2p XPS spectra of the graphite cathodes at the fully discharged and charged states, respectively.
We next performed time-of-flight secondary ion mass spectrometry (TOF-SIMS) to visualize the three-dimensional distribution of CaCl2 and Cl2 on graphite at the fully discharged and charged states, and the results were in good agreement with the cathode reaction in Eq. (1)27 (Fig. 3f, g). The formation and consumption of Cl2 during battery charge and discharge were detected by DEMS (Fig. 3h). The high-resolution Cl 2p XPS spectra further confirmed the formation of Cl2/C–Cl bond at the fully charged cathode28,29 (Fig. 3i, j). In addition, the Ca/Cl2 pouch cell showed no significant volume change at the fully charged state, which suggested that the generated Cl2 might be adsorbed/trapped within the graphite cathode (Supplementary Fig. 15). These systematic characterizations of the cathode products confirmed the reversibility of the CaCl2/Cl2 redox reaction in our rechargeable Ca/Cl2 batteries.
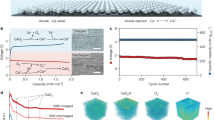
Our CALS electrolyte also improves the electrochemical stability of the Ca metal anode, which is important for battery rechargeability30,31. For instance, Ca metal that was immersed in CALS electrolyte without DFOB− became dark immediately, whereas the surface remained shiny in CALS electrolyte with DFOB− (Supplementary Fig. 16a, b). The XPS profile also showed metallic Al on Ca in the electrolyte without DFOB−, indicating that DFOB− could suppress the parasitic reaction between AlCl4− and Ca metal, thus preventing the replacement of Al metal that inhibited Ca2+ transfer across the electrode/electrolyte interface (Supplementary Fig. 16c). The high-resolution F 1 s XPS spectra of the Ca metal anode after the first discharge showed a B-F peak at ~686.6 eV, which was preserved after 40 cycles (Supplementary Fig. 17). It could be assigned to DFOB− which was adsorbed on the surface of Ca metal, and its strong interaction with Ca2+ could weaken the interaction between Ca2+ and Cl−, which suppressed the parasitic chlorination of the Ca metal anode, thus promoting the electrochemical reversibility (Supplementary Fig. 18). Additionally, DFOB− decomposition and Li deposition on the Ca metal anode might not occur, as suggested by the B 1 s and Li 1 s spectra (Supplementary Fig. 17).
We further verified the reversible morphological evolution of the Ca metal anode at a variety of charge/discharge states, without observation of ‘dead’ or dendritic Ca metal (Supplementary Figs. 19 and 20). The formation of CaCl2 on the surface of the Ca metal anode was verified by the Ca 2p and Cl 2p XPS spectra (Supplementary Fig. 16). In a Ca/Au half cell containing a CALS electrolyte, the reversibility of Ca metal plating and stripping was determined to be 90.2% by cyclic voltammetry (Supplementary Fig. 21a). In contrast, in the CALS electrolyte without DFOB−, no stripping behavior was observed (Supplementary Fig. 21b), which confirmed the critical role of DFOB− in the rechargeability of the Ca/Cl2 battery. The scanning electron microscopy (SEM) and XRD results showed the uniform deposition of Ca metal on the Au foil in the presence of our CALS electrolyte (Supplementary Fig. 22). These results suggested that the parasitic reaction between the Ca metal anode and the electrolyte was suppressed, as mediated by LiDFOB, which is critical for the rechargeability of our Ca/Cl2 battery.
We further investigated the electrochemical performance of our rechargeable Ca/Cl2 battery. It demonstrated high electrochemical reversibility over a variety of specific capacities ranging from 200 to 1000 mAh g−1 (Fig. 4a), as well as an impressive rate capability of 500 mA g−1 (Fig. 4b), demonstrating the fast kinetics of the CaCl2/Cl2 redox reaction. We also verified the high cycling stability of our rechargeable Ca/Cl2 battery over 100 cycles, whereas no rechargeability was observed when using the LiDFOB-free electrolyte (Fig. 4c). We sought to translate the impressive electrochemical performance into practical Ca metal batteries. For instance, retention performance (shelf life) is a critical parameter that determines the practical use of a battery32. To the best of our knowledge, however, current Ca metal batteries cannot afford sufficient retention capability, owing to the poor electrochemical stability of both electrodes used in conventional Ca-based electrolytes33. Our Ca/Cl2 battery demonstrated remarkable retention capability, e.g., when maintained in a fully charged state for 1, 3, and 5 days, the rechargeability was well retained (Fig. 4d), indicating the high electrochemical stability of the charging product in our Ca/Cl2 battery. For an as-prepared battery held at the open-circuit voltage for 30 days, 96.5% of the original specific capacity was retained, and highly consistent charge–discharge profiles and impedances were observed (Fig. 4e and Supplementary Fig. 23). These results suggested that our Ca/Cl2 battery exhibited excellent retention performance in various charge/discharge states, which is highly attractive for practical applications.

a Galvanostatic charge–discharge curves of a rechargeable Ca/Cl2 battery using the CALS electrolyte with various specific charge capacities of 200, 500, and 1000 mAh g−1. Current density, 100 mA g−1. b Rate performance of the rechargeable Ca/Cl2 battery with various current densities from 100 to 500 mA g−1. Specific charge capacity, 200 mAh g−1. c Cycling performance of rechargeable Ca/Cl2 batteries using the CALS electrolyte with and without LiDFOB. d Electrochemical performance of the rechargeable Ca/Cl2 battery with various retention durations of 1, 3, and 5 days. e Electrochemical performance of rechargeable Ca/Cl2 batteries using the CALS electrolyte immediately and after 30 days. f Galvanostatic charge−discharge curves of rechargeable Ca/Cl2 batteries with and without the use of a separator. The inset shows the solid-state CALS electrolyte in an inverted glass vial. Scale bar, 2 cm. g Galvanostatic charge–discharge curve of the rechargeable Ca/Cl2 battery working at 0 °C. The inset shows a fully charged Ca/Cl2 coin cell lighting up a commercial lamp at a low temperature. Scale bar, 5 cm. The specific charge capacity and current density in c–g are 200 mAh g−1 and 100 mA g−1, respectively.
The solid-state CALS electrolyte enables the production of separator-free Ca/Cl2 batteries, which can effectively reduce the volume and weight of batteries for practical applications (Fig. 4f). In addition, the high ionic conductivities of our CALS electrolyte (e.g., 3.6 and 5.3 mS cm−1 at 0 and 25 °C, respectively) allow us to explore low-temperature battery performance, which remains a major challenge for current Ca metal batteries. Remarkably, our Ca/Cl2 battery functioned normally at 0 °C, retaining ~92.2% of its room-temperature energy efficiency (Fig. 4g), thus revealing fast ion transfer and reaction kinetics even at low temperatures. In addition, the high cycling stability was well maintained at 0 °C (Supplementary Fig. 24). To the best of our knowledge, this is the first rechargeable Ca metal battery with acceptable low-temperature capability to date11,34,35, which represents an important step toward practical application.
Discussion
In conclusion, we report the rechargeable Ca/Cl2 battery based on the reversible cathode redox reaction between CaCl2 and Cl2, which introduces a new paradigm for Ca metal batteries with comprehensively high energy densities and rate capabilities. LiDFOB, a key electrolyte mediator with unique B−F and C=O functional groups, can facilitate the dissociation and distribution of Ca2+ and Cl-based species, thus enabling reversible and fast CaCl2/Cl2 redox reaction. The prepared rechargeable Ca/Cl2 battery delivers discharge voltages of 3 V and remarkable specific capacity (up to 1000 mAh g−1) and rate capability (up to 500 mA g−1). The retention and low-temperature performance also overcome the key bottlenecks that have hindered practical applications of Ca metal batteries to date. Our findings can not only benefit Ca metal batteries by enabling highly desirable cathode chemistry but also, in a broader context, revive the once overlooked Cl-based cathode chemistry for multivalent metal batteries, and harness them for sustainable, low-cost, and high-performance energy storage.
Methods
Preparation of the electrodes and electrolytes
Graphite (99%, Aladdin) was used for cathode preparation. It was mixed with polytetrafluoroethylene (PTFE, 60% aqueous dispersion, Fuel Cell Store) at a mass ratio of 9:1 in ethanol solution. The obtained suspension was then subjected to ultrasonic treatment for 1 h to obtain the cathode slurry. A Ni foam was cut into a 14 mm diameter (1.54 cm2) circular using a manual disk cutter (MSK-T-10, MTI), followed by dropping 50 μL cathode slurry onto it. After evaporation of the ethanol at 80 °C, the above slurry coating and drying processes were repeated until the mass loading of graphite reached 1.0−1.5 mg cm−2. The obtained cathodes were further dried at 80 °C in a vacuum oven for 2 h. The Ca metal anodes were prepared by polishing the Ca flakes (99%, Sigma Aldrich, working area of 0.8 cm× 0.8 cm, thickness of 0.5 ± 0.04 mm) in an argon-filled glovebox with the contents of H2O and O2 below 1 ppm. All the electrolytes were also prepared in the same glovebox. AlCl3 (99%, anhydrous, Aladdin) and SOCl2 (99%, Aladdin) were used as received. LiDFOB (99%, anhydrous, Aladdin) and CaCl2 (99.9%, anhydrous, Meryer) were dried at 120 °C for 12 h in a vacuum chamber before use. Typically, 6 M AlCl3 and 1.3 M LiDFOB were dissolved in 1 mL SOCl2, followed by the addition of 1.2 M CaCl2 under continuous stirring for 1 h to obtain the CALS electrolyte. The gelation of the CALS electrolyte generally took 2 h, which allowed us to assemble batteries using the electrolyte in the liquid state.
Electrochemical measurements
All the batteries were made inside an argon-filled glovebox with the contents of H2O and O2 below 1 ppm. The prepared cathode and Ca metal anode were separated by one piece of glass fiber membrane (GF/D, Whatman, 16 mm in diameter) as the separator and 150 μL electrolyte was added to each coin cell (CR2032). All the electrochemical measurements were performed at 25 °C unless otherwise stated. The charge–discharge performance of the batteries was characterized using Neware battery testing systems (CT-4008-5V50mA-164-U). Electrochemical impedance spectroscopy (EIS) was conducted on a CHI600E electrochemical workstation. The electrochemical impedance spectroscopy measurement was conducted in a CR2032 coin cell, using a graphite cathode as the working electrode, and a Ca metal foil as the counter and reference electrode. The frequency ranged from 0.1 to 105 Hz with an amplitude of 5 mV.
Characterization
XRD, SEM, TOF-SIMS, XPS, Raman, DEMS, TEM, TGA, DSC, and XAS were performed, and detailed information was provided in Supplementary Information.
Molecular dynamics (MD) simulations
All MD simulations were conducted using the GROMACS 2019.3 to investigate the solvation structure of electrolytes. The detailed information was provided in Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon request.
Code availability
The codes that support the findings of this study are available from the corresponding author upon request.
References
Armand, M. & Tarascon, J. M. Building better batteries. Nature 451, 652 (2008).
Tarascon, J. M. & Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 414, 359 (2001).
Meng, J. et al. A solution-to-solid conversion chemistry enables ultrafast-charging and long-lived molten salt aluminium batteries. Nat. Commun. 14, 3909 (2023).
Mao, M. et al. Anion-enrichment interface enables high-voltage anode-free lithium metal batteries. Nat. Commun. 14, 1082 (2023).
Muldoon, J., Bucur, C. B. & Gregory, T. Quest for nonaqueous multivalent secondary batteries: magnesium and beyond. Chem. Rev. 114, 11683–11720 (2014).
Deng, X. et al. Anode chemistry in calcium ion batteries: a review. Energy Stor. Mater. 53, 467–481 (2022).
Nielson, K. V. & Liu, T. L. Dawn of calcium batteries. Angew. Chem. Int. Ed. 59, 3368–3370 (2020).
Chen, C., Shi, F. & Xu, Z. L. Advanced electrode materials for nonaqueous calcium rechargeable batteries. J. Mater. Chem. A 9, 11908–11930 (2021).
Kim, S. et al. Investigation of Ca insertion into α‑MoO3 nanoparticles for high capacity Ca-ion cathodes. Nano Lett 22, 2228–2235 (2022).
Ponrouch, A., Frontera, C., Bardé, F. & Palacín, M. R. Towards a calcium-based rechargeable battery. Nat. Mater. 15, 169–172 (2016).
Gummow, R. J., Vamvounis, G., Kannan, M. B. & He, Y. Calcium-ion batteries: current state-of-the-art and future perspectives. Adv. Mater. 30, 1801702 (2018).
Wang, M. et al. Reversible calcium alloying enables a practical room-temperature rechargeable calcium-ion battery with a high discharge voltage. Nat. Chem. 10, 667–672 (2018).
Li, L. et al. A covalent organic framework for high-rate aqueous calcium-ion batteries. J. Mater. Chem. A 10, 20827–20836 (2022).
Torres, A., Casals, J. L. & Arroyo-de Dompablo, M. E. Enlisting potential cathode materials for rechargeable Ca batteries. Chem. Mater. 33, 2488–2497 (2021).
Staniewicz, R. J. A study of the calcium-thionyl chloride electrochemical system. J. Electrochem. Soc. 127, 782 (1980).
Meitav, A. & Peled, E. Calcium‐Ca(AlCl4)-thionyl chloride cell: performance and safety. J. Electrochem. Soc. 129, 451 (1982).
Liang, Y. et al. Current status and future directions of multivalent metal-ion batteries. Nat. Energy 5, 646–656 (2020).
Li, M. et al. Design strategies for nonaqueous multivalent-ion and monovalent-ion battery anodes. Nat. Rev. Mater. 5, 276–294 (2020).
Hou, S. et al. Solvation sheath reorganization enables divalent metal batteries with fast interfacial charge transfer kinetics. Science 374, 172–178 (2021).
Li, Z. et al. Rechargeable calcium–sulfur batteries enabled by an efficient borate-based electrolyte. Small 16, 2001806 (2020).
Tohru, S., Yuichi, K. & Yoko, H. Coupling of nitroxyl radical as an electrochemical charging catalyst and ionic liquid for calcium plating/stripping toward a rechargeable calcium–oxygen battery. J. Mater. Chem. A 5, 13212 (2017).
Zhu, G. et al. Rechargeable Na/Cl2 and Li/Cl2 batteries. Nature 596, 525–530 (2021).
Bedfer, Y. & Corset, J. Raman spectroscopic studies of the structure of electrolytes used in the Li/SOCl2 battery. J. Power Sources 9, 267–272 (1983).
Mosier-Boss, P. A. et al. Raman and infrared spectroscopy of the AlCl3-SOCl2 system. J. Chem. Soc. Faraday Trans. 1 85, 11–21 (1989).
Zheng, J. et al. Directed self-assembly of herbal small molecules into sustained release hydrogels for treating neural inflammation. Nat. Commun. 10, 1604 (2019).
Judith, M., César, S. & David, D. Release of small bioactive molecules from physical gels. Chem. Soc. Rev. 47, 1484–1515 (2018).
Zhu, G. et al. High-capacity rechargeable Li/Cl2 batteries with graphite positive electrodes. J. Am. Chem. Soc. 144, 22505–22513 (2022).
Piao, H., Adib, K. & Barteau, M. A. A temperature-programmed X-ray photoelectron spectroscopy (TPXPS) study of chlorine adsorption and diffusion on Ag (111). Surf. Sci. 557, 13–20 (2004).
Zhu, G. et al. Shedding light on rechargeable Na/Cl2 battery. Proc. Natl. Acad. Sci. USA 120, e2310903120 (2023).
Hou, Z. et al. Correlation between electrolyte chemistry and solid electrolyte interphase for reversible Ca metal anodes. Angew. Chem. Int. Ed. 50, e202214796 (2022).
Song, H., Su, J. & Wang, C. Hybrid solid electrolyte interphases enabled ultralong life Ca-metal batteries working at room temperature. Adv. Mater. 33, e2006141 (2021).
Xu, Q. et al. A low-cost and recyclable Mg/SOCl2 primary battery via synergistic solvation and kinetics regulation. Adv. Funct. Mater. 5, 2210343 (2022).
Yan, L. et al. Recent progress in rechargeable calcium-ion batteries for high-efficiency energy storage. Energy Stor. Mater. 60, 102822 (2023).
Dong, X., Wang, Y. G. & Xia, Y. Promoting rechargeable batteries operated at low temperature. Acc. Chem. Res. 54, 3883–3894 (2021).
Arroyo-de Dompablo, M. E. et al. Achievements challenges, and prospects of calcium batteries. Chem. Rev. 120, 6331–6357 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22209108) and the Fundamental Research Funds for the Central Universities (23X010301599). The authors acknowledge the 4B7A station at the Beijing Synchrotron Radiation Facility.
Author information
Authors and Affiliations
Contributions
H.S. conceived and designed the research project. S.G. and X.Z. performed electrode preparation, electrolyte synthesis, battery production, and electrochemical characterization. B.Y. and Y.W. performed Raman spectroscopy. S.G., Q.X., Z.O., S.W., L.Y., and M.L. performed scanning electron microscopy, transmission electron microscopy, X-ray photoelectron spectroscopy, time-of-flight secondary ion mass spectrometry, thermogravimetric analysis, and differential scanning calorimetry. L.W. and S.G. analyzed molecular dynamics calculations. Yo. W., C.M., and X.J.Z performed Ca K-edge X-ray absorption experiments and Athena analysis. H.S., S.G., and X.Z. prepared the manuscript. All the authors participated in the data analysis and discussion.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jun Haruyama and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Geng, S., Zhao, X., Xu, Q. et al. A rechargeable Ca/Cl2 battery. Nat Commun 15, 944 (2024). https://doi.org/10.1038/s41467-024-45347-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-45347-3
This article is cited by
-
A rechargeable Ca/Cl2 battery
Science China Chemistry (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
