
Abstract
Somatostatin receptor 2 (SSTR2) is highly expressed in neuroendocrine tumors and represents as a therapeutic target. Several peptide analogs mimicking the endogenous ligand somatostatin are available for clinical use, but poor therapeutic effects occur in a subset of patients, which may be correlated with subtype selectivity or cell surface expression. Here, we clarify the signal bias profiles of the first-generation peptide drug octreotide and a new-generation small molecule paltusotine by evaluating their pharmacological characteristics. We then perform cryo-electron microscopy analysis of SSTR2-Gi complexes to determine how the drugs activate SSTR2 in a selective manner. In this work, we decipher the mechanism of ligand recognition, subtype selectivity and signal bias property of SSTR2 sensing octreotide and paltusotine, which may aid in designing therapeutic drugs with specific pharmacological profiles against neuroendocrine tumors.
Similar content being viewed by others

Introduction
Neuroendocrine tumors (NETs) are rare and serious tumors that are commonly caused by the excessive secretion of hormones, which result in complicated clinical syndromes and disorders1,2. For example, pituitary adenoma is a tumor of the pituitary gland that produces too much growth hormone (GH) and can lead to acromegaly3. Treatment of NETs include strategies that inhibit excess hormones and block hormone action4,5.
Somatostatin receptors (SSTRs) are members of the G protein-coupled receptor (GPCR) superfamily and contain five subtypes (SSTR1–5). SSTRs, especially SSTR2, are expressed at high levels in NETs and have emerged as important biomarkers4,6. SSTRs and their endogenous ligands, somatostatin (SST) peptides, exert important physiological regulation of neuroendocrine functions7. Previous studies have shown that SSTs and analogs are linked to decreased GH secretion, suggesting that the SSTR family may be a major therapeutic target for NETs1. However, the use of SST for disease treatment is limited owing to its short half-life in the body. A series of long-acting SST analogs have been developed and explored in clinical trials8. The first-generation drug octreotide is a biologically stable synthesized SST analog used for the treatment of acromegaly7,9. However, ~30% of the acromegalic patients do not respond well to octreotide10. Second-generation drugs have been developed and were recently reported to exhibit high safety profile11. Notably, agents with subtype selectivity have always been pursued in drug discovery targeting SSTR2, which could facilitate to improve the efficacy of potential drugs and avoid of side effects caused by off-target4,7,12. After much effort has been made over years, an SSTR2 subtype-selective agonist paltusotine has recently been developed by Crinetics, which is an orally bioavailable non-peptide small molecule and is undergoing phase 3 trials for the clinical treatment of acromegaly and other NETs.
SSTR2 transmits extracellular signals into cells through Gαi/o coupling and β-arrestin recruitment pathways. cAMP inhibition causes decreased hormone secretion and cell proliferation13,14, while the β-arrestin recruitment results in subsequent receptor internalization thus influence the cell membrane distribution level and desensitization of SSTR2, which may lead to drug resistance and poor clinical curative effect15,16,17,18. Previous clinical evidence showed that the expression level of SSTR2 occurred down-regulated during the administration of SST analog octreotide for acromegalic patients19, and lower of β-arrestin 1 in pituitary adenomas was associated with better drug efficacy20. It appears that available agents represent diverse therapeutic windows, probably due to their different pharmacological profiles. Therefore, understanding of the pharmacological characteristics and molecular mechanism of different ligands, including peptide and small-molecule drugs, may help improve therapeutic strategies involving SSTR2 signaling. Recently, the structures of SSTR2 bound to multiple ligands have been reported, however the subtype selectivity of ligand for distinguishing group 2 SSTRs and the bias property remain elusive21,22,23,24,25.
Here, we explore the pharmacological features and molecular mechanisms of different SST ligands, including peptide and small-molecule drugs, with SSTR2. We find that the small molecule paltusotine possesses a better Gi-biased property compared with octreotide. The structures of SSTR2-Gi signaling complexes bind to octreotide or paltusotine are determined by cryo-electron microscopy (cryo-EM). From these results combined with the mutagenesis and cell-based functional assays, we elucidate the mechanism of how different drugs activate SSTR2 signaling and uncover a minor pocket in the paltusotine-bound structure. Furthermore, the subtype selectivity against SSTRs and bias property towards SSTR2 of paltusotine are deciphered. Our findings provide insights into the molecular mechanism of the promising clinical small-molecule drug for the treatment of NETs.
Results
Pharmacological characterization of octreotide and paltusotine
Octreotide is a cyclic octapeptide with pharmacophore mimicking SST14 (cyclic tetradecapeptide somatostatin-14), and the new-generation paltusotine is a selective non-peptide agonist that is apparently smaller than octreotide in size (Fig. 1a). We explored the pharmacological features of octreotide and paltusotine using HEK293 cells expressing SSTR2. cAMP accumulation inhibition assays revealed that the drugs displayed a similar ability to activate G protein-dependent signaling via SSTR2 (Fig. 1b). In β-arrestin recruitment assays, paltusotine exhibited a lower efficacy of β-arrestin recruitment, resulting in less receptor internalization, compared with octreotide (Fig. 1c–f). These results demonstrate that paltusotine is a better G-protein-biased agonist of SSTR2 compared with octreotide. These findings are consistent with a previous report showing that paltusotine may have better clinical application than octreotide and suggest that the level of receptor internalization may be associated with the therapeutic efficacy of the drugs16,26. Therefore, understanding the mechanism of ligand recognition and activation would help determining the bias feature of SSTR2 in response to diverse ligands.

a The chemical structures of octreotide and paltusotine; similar moieties are highlighted in pink. b, c The efficacy of octreotide- and paltusotine-induced SSTR2 Gi signaling (b) and β-arrestin recruitment (c). Data represent mean ± SEM from three independent experiments. d Internalization of SSTR2 in HEK293 cells treated with octreotide and paltusotine measured by ELISA assays by detecting the expression of SSTR2 on the cell surface. Data represent mean ± SEM from three independent experiments. e, f eGFP-tagged SSTR2 plasmid was transfected in HEK293 cells and then cells were treated with octreotide (e) or paltusotine (f) for 30 min. The cells were analyzed by confocal fluorescence microscopy (green, eGFP-SSTR2; blue, DAPI). Scale bar, 50 μm.
Recognition mechanism of octreotide by SSTR2
We next assembled the SSTR2-Gi1 complex by co-expression of the SSTR2 receptor with human Gαi1 and human Gβ1γ2 in Sf9 cells. The complex structures of octreotide-bound and paltusotine-bound SSTR2 coupled to the Gi1 heterotrimer were determined by cryo-EM with a resolution of 3.37 and 3.24 Å, respectively (Fig. 2a–d, Supplementary Figs. 1–2 and Supplementary Table 1). The high-quality density enabled us to build the structural models accurately, including key residues for ligand recognition (Supplementary Fig. 3). The overall structures of the two complexes were very similar, with a root mean square deviation value of 0.76 Å for the Cα within the receptor and 1.25 Å for the receptor complex. Compared with the inactive structure of SSTR2 (PDB code: 7UL5), the structure of agonists bound SSTR2 exhibit obvious rearrangements of the key switches and TM5/TM6 required for G-protein coupling (Supplementary Fig. 4a), suggesting that the SSTR2-Gi1 signaling complexes with agonists adopt an active conformation.

a, b The three-dimensional map (a) and overall structural model (b) of octreotide-bound SSTR2 in complex with Gi1 and scFv16. c, d The three-dimensional map (c) and overall structural model (d) of paltusotine-bound SSTR2 in complex with Gi1 and scFv16. e, f Structural details of octreotide bound to SSTR2. SSTR2 is shown in a cartoon diagram and colored in green-cyan; octreotide is shown as sticks and colored in light pink. Octreotide occupied the orthosteric binding pocket of SSTR2; the electron density map of octreotide is shown in tv-blue. g–i The detailed interaction network between octreotide and SSTR2. The detailed interaction network within (D)-Phe1-Cys2-Phe3 (g), Thr6-Cys7-Thr8-ol (h) and the core region (i) are shown. Amino acid residues involved in octreotide binding are shown as sticks. Hydrogen bonds are indicated as gray dashed lines. j The effects of SSTR2 mutations in the core region on octreotide-induced cAMP inhibition. Data represent mean ± SEM from three independent experiments for wild-type SSTRs and its mutants. WT: wild-type. k Effects of mutations involved in octreotide binding of SSTR2 on Gi signaling. Bars represent the ΔpEC50 relative to wild-type. Statistical differences between wild-type and mutants were determined by one-way of variance ANOVA with Dunnett’s test. **P < 0.02; ***P < 0.001; ns, not significant; n.d., not detected (P = < 0.001, n.d., n.d., 0.002, <0.001, <0.001, 0.002, <0.001, <0.001, <0.001, 0.946, 0.961, >0.999, <0.001, <0.001, <0.001, from top to bottom). Data represent mean ± SEM from three independent experiments. l Sequence alignment of the residues around the core binding pocket of SSTR2 with other SSTR members.
The well-defined density in the orthosteric binding pocket allowed us to model octreotide unambiguously. Octreotide is a cyclic peptide that is bridged by a disulfide bond formed by cysteine at position 2 and position 7 (Fig. 2e, f). The main region of octreotide occupies the orthosteric binding site comprised by TM2, TM3, TM5, TM6, and TM7; the terminal region of octreotide is located in the extracellular vestibule formed by ECL2 and ECL3 (Fig. 2e–i). The binding of SSTR2 with octreotide exhibits the same recognition mechanism compared with previous reporting21,22 (Supplementary Fig. 4b, c). (D)-Phe1 of octreotide forms hydrophobic interactions with I284 and P286 in ECL3 of SSTR2. The disulfide bond forms van der Waals forces with the side chain of F2947.35, while the main chain of Cys2 makes polar contacts with the side chains of N2766.55 and S2796.58. Phe3 forms hydrophobic interactions with I19545.52 and Y2055.35 in the extracellular end of TM5 (Fig. 2g). Octreotide contains the same pharmacological core region ((D)-Tyr4 and Lys5) as that in SST14, the side chains of (D)-Tyr4 and Lys5 insert into the bottom of the orthosteric site of the receptor and are necessary for receptor activation (Supplementary Fig. 4d). Furthermore, (D)-Tyr4 is accommodated in a hydrophobic pocket formed by residues F1273.37, F2085.38 and F2726.51, and Lys5 makes direct interactions with the side chains of D1223.32, Q1263.36, and Y3027.43 via a polar network (Fig. 2i). Position 3.32 (D or E) was reported to be a key conserved residue involved in ligand binding and activation in aminergic receptors or peptide receptors27,28. Notably, (D)-Tyr4 and Lys5 in octreotide or SST analogs are the core region of the cyclic peptide. Alanine replacement of the residues F2085.38, F2726.51, Y3027.43, Q1263.36, and D1223.32 in SSTR2 significantly decreased the cAMP inhibition potency (Fig. 2j, k, Supplementary Fig. 5 and Supplementary Table 2), suggesting that the interactions of (D)-Tyr4 and Lys5 from the core region with the receptor are essential for the biological activity of the hormone SST or octreotide.
Octreotide bound to the orthosteric binding pocket by extensive interactions with SSTR2; this ligand retains high binding affinity and efficacy with the SSTR family members, including SSTR2, SSTR3, and SSTR5. Combined with findings from the cell-based functional assays, the results of sequence alignment and structural analysis reveal that conserved residues 3.32-3.36-7.43 constitute a common motif for the pharmacophore of peptide agonist binding (Fig. 2l).
Recognition mechanism of paltusotine by SSTR2
The small molecule paltusotine is a highly selective agonist of SSTR2 relative to other SSTR subtypes. The cryo-EM structure of the SSTR2-Gi complex bound to paltusotine showed that paltusotine occupies the bottom of the orthosteric binding pocket (OBP) (Figs. 2c, d and 3a). Notably, paltusotine largely resembles the core region of octreotide, with an aminopiperidin moiety and an aromatic ring of hydroxybenzonitrile mimicking Lys5 and (D)-Trp4, respectively, and makes extensive contacts with the major pocket of the OBP (Fig. 3b). In particular, the amide group of aminopiperidin forms a similar salt bridge with D1223.32 or hydrogen bond with Y3027.43 as that in the structure of the octreotide-bound receptor, whereas the hydroxybenzonitrile group of paltusotine is projected into a hydrophobic cavity that can accommodate the side chain of tryptophan from octreotide. The additional amine group makes a hydrogen bond with Q1263.36 (Fig. 3c). We further performed mutagenesis studies and measured the cellular cAMP level induced by paltusotine using HEK293 cells expressing SSTR2. The results demonstrate that the common motif D3.32-Q3.36-Y7.43 plays a critical role in diverse ligand recognition or receptor activation in SSTR2 (Fig. 3d and Supplementary Table 3).

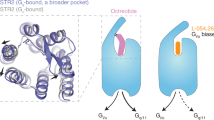
a Structural details of paltusotine bound to SSTR2. SSTR2 is shown in cartoon diagram and colored in violet; paltusotine is shown as sticks and colored in cyan. Paltusotine occupied the bottom of the orthosteric binding pocket of SSTR2; the electron density map of paltusotine is shown in tv-blue. b Three-dimensional structural comparison of octreotide (light pink) and paltusotine (cyan). c The detailed interaction network between paltusotine and the SSTR2 core binding pocket. Amino acid residues involved in paltusotine binding are shown as sticks. Hydrogen bonds are indicated as gray dashed lines. d The effects of the SSTR2 mutations in the core region on paltusotine-induced cAMP inhibition. Data represent mean ± SEM from three independent experiments for wild-type SSTR2 and its mutants. WT: wild-type. e The detailed interaction network within the paltusotine-binding minor pocket. Amino acid residues within the minor pocket are shown as sticks and colored in green-cyan (octreotide-bound SSTR2) and violet (paltusotine-bound SSTR2). f–h Conformational comparison of residues involved in divergent parts of ligand binding are shown in f (T19445.51 and F2947.35 residue pair), g (core region) and h (downstream). Key residues are shown in sticks and colored in green-cyan (octreotide-bound SSTR2) and violet (paltusotine-bound SSTR2); octreotide is colored in light pink and paltusotine is colored in cyan.
A notable feature of paltusotine binding is that the large 3,5-difluorophenyl moiety is placed toward the extracellular end of TM2, creating a minor pocket formed by L992.60, Q1022.63, and V1032.64 in SSTR2. In addition, the residues Y501.39 and D2957.36 from TM1 and TM7 are engaged with paltusotine recognition through van der Waals forces (Fig. 3e). The substitution of D2957.36 with a bulkier side chain reduced the activation potency of the ligand (Supplementary Fig. 6).
Comparison of binding modes of paltusotine and octreotide with SSTR2
The two structures of the SSTR2-Gi signaling complex bound to paltusotine or octreotide reveal a similar configuration of TM bundles on the extracellular side (Supplementary Fig. 6c). However, some significant differences are observed at the ligand binding pocket. Paltusotine lacks the extended region of octreotide (Fig. 3b), resulting in the loss of extensive contacts with the extracellular ends of TM5, TM6, ECL3, and TM7. Notably, the side chains of residues Y2055.35, F2947.35, T19445.51, and I19545.52 have notable displacements when the receptor senses paltusotine. Specifically, F2947.35 slightly moves towards the difluorophenyl group of paltusotine, shortening the distance between T19445.51 in ECL2 and F2947.35 in TM7 by ~1.3 Å relative to the residue pair in octreotide-bound structure. The side chain of I19545.52 projected further into the major pocket of the OBP, strengthening the contact with paltusotine. Moreover, the significant rearrangement of Y2055.35 narrows the major pocket for paltusotine binding in SSTR2 (Fig. 3f). These observations from the structural comparison indicate different binding modes of SSTR2 with paltusotine and octreotide, and provide more evidence for the plasticity of GPCRs in response to diverse ligands.
Another difference lies in the common recognition motif (3.32-3.36-7.43) and classic microswitches in diverse ligand-bound structures. Compared with the amide group in the octreotide-bound receptor structure, the amide group of aminopiperidin in paltusotine is far away from Q1263.36 but still retains direct contact with D1223.32 and Y3027.43 (Fig. 3g), altering the polar network important for receptor activation transition. Adjacent to the network, replacements of L962.57 and L992.60 in the minor pocket occur upon paltusotine binding, which subsequently drives the downstream benzene ring of F922.53 to flip down. Thus, these rearrangements cause obvious conformational change of the surrounding hydrophobic core of TM bundle in SSTR2 within the two complexes, particularly the microswitches W2696.48 and P2205.50-I1303.40-F2656.44 motif (Fig. 3h and Supplementary Fig. 6d).
Selectivity of paltusotine for SSTR subtypes
The SSTR family comprises five members and is divided into two groups based on sequence homology, ligand binding characteristics, and pharmacological properties. SSTR1 and SSTR4 are classified into group 1, whereas SSTR2, SSTR3, and SSTR5 belong to group 2. Identification of selective ligands toward specific subtypes is challenging, especially for developing orally active small-molecule ligand. The selective mechanisms of octreotide and the small molecule L-054,264 for activating group 2 SSTRs over group 1 members have been reported22,24. The structure of SSTR2 in complex with paltusotine enabled the opportunity to examine ligand selectivity for distinguishing group 2 SSTRs, as paltusotine exerts higher selectivity and efficacy towards SSTR2 relative to other group 2 SSTRs (Fig. 4a and Supplementary Fig. 7a). In particular, L-054,264 is another small-molecule agonist of SSTR2 with distinct scaffold (Supplementary Fig. 7b), the residues F7.35, F6.54, and N6.55 in TM6 and TM7 have been reported to participate in subtype selectivity of L-054,264 for activating group 2 SSTRs over group 1 members (SSTR1/4)24, however, paltusotine lacks the direct contacts with the extended binding pocket involved in L-054,264 binding, due to different binding pose in SSTR2 (Supplementary Fig. 7c). Therefore, we tended to focus on the minor binding site engaged in paltusotine binding, sequence alignment reveals that the residues V1032.64, T19445.51, I19545.52, Y2055.35, F2947.35, and D2957.36 are divergent among group 2 SSTRs (Fig. 4b). Superposition of the active state of SSTR2 with SSTR3 (predicted active model from GPCRdb)29,30 allowed us to inspect these differences in 3D structures (Supplementary Fig. 7d). We therefore speculate that these residues may play critical roles in the selectivity of paltusotine to SSTR2.

a Gi signaling of SSTR1–5 activated by paltusotine. Data represent mean ± SEM from three independent experiments. b Sequence alignment of the residues around the paltusotine binding pocket in SSTR2 with sequences of other SSTR subtypes. c, d The effects of the residue substitution of N1032.64V or T19445.51H in SSTR2 (c) and V1012.64N or H19245.51T in SSTR3 (d) on the paltusotine-induced cAMP inhibition. Data represent mean ± SEM from three independent experiments.
To further examine the role of the divergent residues in the activation of SSTRs, we examined cAMP accumulation using the cell-based GloSensor assay. The results revealed that T19445.51 and V1032.64 markedly affected the activation of SSTRs by paltusotine and octreotide. The T19445.51H substitution in SSTR2 significantly reduced receptor activation (Fig. 4c, Supplementary Fig. 7 and Supplementary Table 4); we speculate that the side chain of histidine caused steric hindrance, impairing the contact between both ligands with SSTR2. Additionally, the V1032.64 residue in the minor pocket makes van der Waals forces contacts with the difluorophenyl moiety in paltusotine, and the V1032.64N mutation in SSTR2 reduced receptor activation induced by paltusotine. Both H19245.51T and N1012.64V substitutions in SSTR3 increased paltusotine-induced SSTR3 activation (Fig. 4d). In contrast, the V1032.64N mutant of SSTR2 and H19245.51T mutant of SSTR3 retained a similar EC50 as wild-type receptor upon octreotide stimulation (Supplementary Fig. 7j, k and Supplementary Table 5). Together, these results reveal that the residues at positions 45.51 and 2.64 may not engaged in the selectivity of peptide agonist recognition, but they can determine subtype selectivity of paltusotine for SSTR2 among group 2 SSTRs.
Signal bias properties of SSTR2 with different ligands
Paltusotine induced lower β-arrestin recruitment compared with octreotide (Fig. 1c and Supplementary Fig. 8a). To further investigate the signal bias of paltusotine and octreotide, we performed a comprehensive comparison to identify the divergent contacts in the two ligand-bound structures. An extended region of octreotide interacts with the extracellular ends of TM6/7 and ECL3, whereas paltusotine is placed into a minor pocket in SSTR2. We next explored whether the corresponding contact residues were involved in signal bias of the receptor.
We first generated a range of mutations of the residues within the extended binding pocket occupied by octreotide and minor pocket by paltusotine; substitution of I284ECL3 and K2917.32 with alanine resulted in reduced potency and efficacy of octreotide induced β-arrestin signal, while the impact on the Gi protein pathway was relatively small (Supplementary Fig. 8b and Supplementary Table 2). Notably, the N2766.55A mutation of SSTR2 nearly abolished the recruitment of β-arrestin, but the receptor still exerted Gi signaling activation induced by octreotide (Fig. 5a, b and Supplementary Table 2). By contrast, paltusotine loses the interaction with the extended region, thus the I284ECL3A, K2917.32A, and N2766.55A mutants of SSTR2 exhibited slightly enhanced recruitment of β-arrestin when activated by paltusotine (Fig. 5c, d, Supplementary Fig. 8b and Supplementary Table 3). In addition, residue F2947.35 in the orthosteric binding pocket forms hydrophobic interaction with the disulfide bond of octreotide, the side chain of F2947.35 is closer to octreotide than paltusotine. Alanine replacement of F2947.35 nearly impaired the octreotide-induced β-arrestin recruitment (Fig. 5e, f), yet slightly influenced the β-arrestin pathway activated by paltusotine (Fig. 5g, h and Supplementary Fig. 8b). These results suggest that N2766.55, I284ECL3A, K2917.32A, and F2947.35 may be key residues that contribute to the signal bias of SSTR2 when sensing octreotide. On the other hand, V1032.64 from minor pocket makes van der Waals forces with paltusotine, alanine substitution of V1032.64 markedly reduced the paltusotine-induced β-arrestin signal while had less impact on Gi signaling (Supplementary Fig. 8c and Supplementary Tables 2, 3), which indicates that avoiding the interaction with V1032.64 may further reduce β-arrestin recruitment.

a, b The effects of SSTR2 N2766.55A mutation on cAMP inhibition (a) and β-arrestin recruitment (b) induced by octreotide. Data represent mean ± SEM from three independent experiments. c, d The effects of SSTR2 N2766.55A mutation on cAMP inhibition (c) and β-arrestin recruitment (d) induced by paltusotine. Data represent mean ± SEM from three independent experiments. e, f The effects of SSTR2 F2947.35A mutation on cAMP inhibition (e) and β-arrestin recruitment (f) induced by octreotide. Data represent mean ± SEM from three independent experiments. g, h The effects of SSTR2 F2947.35A mutation on cAMP inhibition on cAMP inhibition (g) and β-arrestin recruitment (h) induced by paltusotine. Data represent mean ± SEM from three independent experiments. i, j Bias factors of the substitution of the microswitch residues in SSTR2 relative to wild-type induced by octreotide (i) and paltusotine (j). Statistical differences between wild-type and mutants were determined by one-way of variance ANOVA with Dunnett’s test. *P < 0.033, **P < 0.02; ***P < 0.001; n.d., not detected. Octreotide (P = 0.022, 0.012, <0.001, 0.012, n.d., n.d., from top to bottom). Paltusotine (P = 0.002, <0.001, <0.001, n.d., n.d., n.d., from top to bottom). Data represent mean ± SEM from three independent experiments. k Residues that markedly affect β-arrestin signaling of SSTR2 are shown in sphere sticks. Residues involved in both octreotide- and paltusotine-induced arrestin recruitment are colored in light pink; residues mainly affect the octreotide-induced β-arrestin recruitment are colored in cyan; residues mainly affect the paltusotine-induced β-arrestin recruitment are colored in lime green; residues that mainly affect Gi protein signaling are colored in bright orange.
In addition, we measured the function of the residues located in the core region and the common ligand binding pocket. The Y3027.43A substitution resulted in markedly decreased G-protein pathway activity, but it retained β-arrestin recruitment, while most of the mutants (e.g., D1223.32 and Q1263.36) markedly affected both β-arrestin and Gi signal under the treatment of whether octreotide or paltusotine. Residues like Q1022.63A mainly influenced the β-arrestin recruitment but without ligand dependence. Such residues were observed to participate in signal bias regulation, which is presumably a general feature of the receptor (Supplementary Fig. 8c and Supplementary Tables 2, 3). Moreover, we carried out the β-arrestin recruitment assays with co-transfection of GRK2, no significant differences for the efficacy of ligand-induced β-arrestin recruitment were observed in the presence or absence of GRK216,31.
The class A peptide receptors angiotensin II type 1 receptor (AT1R) and opioid receptor μOR are prototypical GPCRs that have been studied for their ligand binding and signal bias features. We found that the ligand-binding pattern of SSTR2 is distinct from those reported GPCR complex structures. In SSTR2, octreotide or paltusotine locates in a relative shallow orthosteric binding pocket. In μOR, the ligands BU72 and FH210 insert deeply into the ligand binding pockets, making direct interactions with the W6.48 microswitch residue32,33. Accordingly, the AT1R arrestin-biased ligand TRV023 promoted the rearrangement of the residues within the bottom of the binding site34 (Supplementary Fig. 8d). We wondered whether these residues located at the bottom of the orthosteric pocket of SSTR2 were involved in the biased signal transition. Therefore, we conducted mutagenesis in the core of the TM bundle, especially in critical microswitch residues that are required for receptor activation propagation, and tested the effects on the G-protein and β-arrestin signal pathway. Mutations of the conserved microswitches C6.47W6.48xP5.50, P5.50I3.40F6.44, D3.49R3.50Y3.51, and N7.49P7.50xxY7.53 motifs decreased the potency of β-arrestin recruitment more markedly than the activation ability of Gi signaling compared with wild-type SSTR2 when sensing both octreotide and paltusotine, and the microswitch residues involved signal bias property is not ligand dependent (Fig. 5i–k).
Influence of SSTR2 internalization by the ligand-induced β-arrestin signal
We further explored the impact of the residues affecting the β-arrestin signal on the internalization of SSTR2. Wild-type SSTR2 was obviously internalized in response to octreotide, leading to desensitization, by contrast, internalization and endosomal trafficking of SSTR2 were reduced under paltusotine treatment at the same concentration (Fig. 1d–f and Supplementary Fig. 9a).
Consistent with our cell-based signaling experiments, the results of ELISA assay, Bystander BRET based endosomal trafficking assay using FYVE, an early endosome marker35,36 or confocal assay reveal that SSTR2 mutants bearing I284ECL3A, K2917.32A, N2766.55A, and F2947.35A substitutions showed diminished internalization when treated with octreotide, while exhibited slightly change in the internalization under paltusotine treatment (Fig. 6, Supplementary Fig. 9b–c, d, g, j, k and Supplementary Tables 6–7). Different from these residues, alanine substitution of the microswitch residues mentioned above all resulted in reduced receptor internalization when sensing both octreotide and paltusotine (Supplementary Fig. 9e, f, h, i). Therefore, our results suggest that these residues should contribute to the octreotide-induced β-arrestin signal and the internalization of SSTR2.

a, b Effects of N2766.55A or F2947.35A mutation on the internalization of SSTR2 in HEK293 cells treated with octreotide (a) and paltusotine (b) measured by ELISA assays by detecting the expression of SSTR2 on the cell surface. Data represent mean ± SEM from three independent experiments. c, d Effects of N2766.55A or F2947.35A mutation on the internalization of SSTR2 in HEK293 cells treated with octreotide (c) and paltusotine (d) indicated by confocal fluorescence microscopy. eGFP-tagged SSTR2 wild-type or mutant plasmid was transfected in HEK293 cells and then cells were treated with 10 μM octreotide or paltusotine for 30 min and analyzed by confocal fluorescence microscopy (green, eGFP-SSTR2; blue, DAPI). Scale bar, 50 μm.
Collectively, we mapped the signaling bias–related residues in the structure and found that within the octreotide extended region, I284ECL3, K2917.32, N2766.55, and F2947.35 in the extended region of octreotide contributed to activation of the β-arrestin signal and the internalization of SSTR2 activated by octreotide. Whereas the downstream microswitch residues involved in the efficacy of β-arrestin recruitment induced by both ligands (Fig. 5k).
Octreotide and paltusotine promote apoptosis of pituitary tumor cell
Treatment of NETs by targeting SSTR2 signaling pathway was previously demonstrated to induce hormone secretion and tumor apoptosis37,38. Furthermore, we inferred whether small molecule paltusotine or peptide drug octreotide play a role in apoptosis process on pituitary tumor cell. Subsequently, the apoptotic rate of the rat pituitary-derived GH3 cells that overexpress SSTR2 was measured by flow cytometry Annexin V-FITC/PI analysis. The result reveals that both octreotide and paltusotine are able to activate proapoptotic pathways. Notably, increased apoptotic rate was observed when treating with paltusotine over octreotide (Fig. 7a, b and Supplementary Fig. 10), consistent with the bias profile of clinical drugs, the evidence also indicates a potential better performance of the small-molecule drug in clinical treatment of NETs through promoting tumor apoptosis.

a, b Statistic results of GH3 cell apoptosis assays of control, octreotide or paltusotine treated cells. Cells for Annexin V+/PI− and Annexin V+/PI+ were both considered apoptotic (a). Bars represent the percentage of apoptotic cells in a. b Statistical differences between control and octreotide or paltusotine treated cells were determined by one-way of variance ANOVA with T-test. Data are expressed as mean ± SEM from three independent experiments (**P < 0.02; ***P < 0.001; n.s., not significant). (Control compared with octreotide or paltusotine (P = < 0.001 under the treatment of 1 μM drug; P = < 0.001 under the treatment of 10 μM drug). Octreotide compared with paltusotine (ns, under the treatment of 1 μM drug; P = < 0.02 under the treatment of 10 μM drug)). The gating strategy of apoptotic cells was provided in Supplementary Fig. 10.
Discussion
GPCRs regulate a broad range of physiological and pathological processes by triggering different signaling pathways, including G-protein coupling and arrestin recruitment. Previous study has indicated that one of the receptor signaling pathways may be responsible for the therapeutic effects, whereas the other mediates unwanted side effects39. As most GPCR subfamilies, SSTRs distributed in different tissues and regulated divergent physiological processes, therefore, designing selective ligands that can achieve receptor subtype selectivity and specific signaling and even control on- or off-target side effects, could be safer therapeutic strategy. SSTR2 signals via activation of G-protein and engages β-arrestin to mediate distinct cellular signaling events and has been proved to be a valuable drug target for the treatment of acromegaly, however, subtype selectivity of SSTRs and signal bias properties of different types of ligands (peptide octreotide and small molecule paltusotine) hinder the development of effective drugs targeting SSTR2. Our study, to a certain extent, contributes to the understanding of the functional bias of ligands and guiding rational drug design targeting SSTRs.
The small-molecule agent paltusotine undergoing phase 3 trials for the treatment of NETs acts as a highly selective agonist of SSTR2, herein, we investigated the recognition mechanism by determining complex structure of SSTR2 bound to paltusotine, and an unusual minor pocket in SSTR2 was demonstrated to play an important role in ligand selectivity. In particular, the substitution of T19445.51 and V1032.64 in SSTR2 with corresponding residues (H19245.51 and V1032.64) in SSTR3 reduced receptor activation induced by paltusotine (Fig. 4c and Supplementary Fig. 7). Moreover, T19445.51 interacted with Thr6 in octreotide, and it was replaced by histidine or asparagine in SSTR3 or R5, which might be responsible for the higher octreotide efficacy for SSTR2 (Supplementary Fig. 7j) and reduced affinity for SSTR3/5. During the preparation of our manuscript, several individual groups reported the structures of SSTR2 bound to octreotide and other different types of ligands21,22,23,24,25. Subtype selectivity of peptide ligands at SSTR2 is multifaceted because of extensive contacts, especially divergent ECL regions in SSTRs which were suggested to participate in the selectivity of peptide agonist21. Together with previous reporting, our findings further profile the subtype selectivity of ligand to group 2 SSTRs (SSTR2/3/5) over group 1 SSTRs (SSTR1/4). Emerging of the structures and pharmacological studies of SSTRs will be helpful to understand subtype selective mechanism and paves a way to design effective small molecules toward subtype receptor.
Moreover, clinical studies have shown that lower expression of β-arrestin 1 in acromegalic patients may be associated with decreasing recycling rate of SSTR2 and better SST analog response20,40. Understanding of the bias properties of SSTR2 would help pharmacologists to design or optimize small molecules with biased signaling. Interestingly, we characterized that paltusotine acts as more G protein-biased ligand than octreotide, and provided structural insights to understand the bias property of paltusotine. In the paltusotine-bound structure, we found that paltusotine loses the interactions with residues I284ECL3, K2917.32, N2766.55, and F2947.35 that participated in the recruitment of β-arrestin induced by octreotide (Fig. 5, Supplementary Figs. 8 and 9). Moreover, V1032.64 contributed to the β-arrestin recruitment under the treatment of paltusotine. Designing specific drugs target the core region and with minimum contact with the extended binding pocket of SSTR2 would facilitate the development of G protein-biased ligand of SSTR2. Future studies would be highly informative to solve the SSTR2-β-arrestin complex structure for precise pharmacological characterization of novel drugs targeting SSTRs. Collectively, designing selective ligands or modulators that control specific subtype and functional signaling could be safer therapeutic strategy in GPCR drug discovery field.
Methods
SSTRs Gi-mediated cAMP inhibition assay
The levels of the intracellular 3′, 5′-cyclic AMP (cAMP) were measured by the GloSensor cAMP assay (Promega) as described in the previous studies28,41 which has been widely used. The human full-length SSTRs and mutants were cloned into the pcDNA3.1+ vector with a HA signal sequence followed by a Flag tag at the N-terminus. HEK293 cells (ATCC, CRL-1573) were seeded into 6-well microplates at a density of 1.0 × 106 cells/well and cultured for 24 h at 37 °C with 5% CO2. Cells were then co-transfected with SSTR wild-type or mutants and GloSensor plasmids. After 24 h of transfection, the cells were dissociated with TrypLE™ Express Enzyme (1X) (Thermo Fisher Scientific) and collected by centrifugation at 500 × g for 5 min. Then the collected cells were suspended in Hank’s balanced salt solution [HBSS (Thermo Fisher Scientific)] supplemented with D-fluorescein potassium salt solution (YEASEN), and then the suspension cells were seeded into 96-well plates with 90 μl/well. After incubating at room temperature for 40 min, 5 μM forskolin and ligands diluted in HBSS were added to each well and the plates were incubated at room temperature for 15 min. Fluorescence signals were measured using the Synergy H1 microplate reader (BioTek) and the data presented are means of at least three independent experiments.
NanoBiT β-arrestin 1 recruitment assay
The NanoBiT β-arrestin 1 recruitment assays were carried out according to the method described previously42. Human full-length β-arrestin 1 was cloned into pcDNA3.1+ vector with the large fragment (LgBiT) of the NanoBiT system and a 15-amino acid flexible linker (GGSGGGGSGGSSSGG) at the N-terminus. Human full-length SSTR wild-type and mutant vectors were C-terminally fused with a 15-amino acid flexible linker and the small fragment (SmBiT). HEK293 cells were seeded into six-well microplates at a density of 1.0 × 106 cells/well and cultured for 24 h at 37 °C with 5% CO2. After that, cells were co-transfected with pcDNA3.1+ vectors encoding SSTR wild-type or mutants and human β-arrestin 1. After transfecting for 24 h, the transfected cells were suspended and seeded into white 96-well plates, and then cultured at 37 °C overnight. After that the medium in each well was exchanged for 90 μl of 0.01% bovine serum albumin containing 20 μl of 50 μM coelenterazine h (Carbosynth) diluted in the HBSS assay buffer and incubated for 40 min at room temperature. Then, ligands diluted in the assay buffer was added to each well and the luminescence intensity was measured using the Synergy H1 microplate reader and detected at room temperature in a time period. All data presented are means of at least three independent experiments.
Detection of surface expression of SSTR wild-type and mutants
HEK293 cells were seeded into 96-well microplates at a density of 4 × 104 cells/well and cultured for 24 h at 37 °C with 5% CO2 and were transfected using the same procedure as the cAMP accumulation assay. The plates were washed with washing buffer (PBS) the day after transfection, and were fixed with 4% formalin at room temperature for 30 min. Cells were washed with washing buffer and blocked with PBS supplemented with 5% BSA for 1 h at room temperature. After that, cells were incubated with anti-Flag antibody (1:1000 diluted with PBS containing 5% BSA, Proteintech: 66008-4-Ig) for 1 h at room temperature. After 1 h incubation, cells were washed with washing buffer and incubating with HRP-conjugated secondary antibody for 2 h at room temperature. After incubating and washing, 50 μl of chromogenic solution was added to each well, and reacted at room temperature for 10 min, and then 0.5 M HCl were added to stop the reaction. Optical density (OD) was measured at 455 nm in Synergy H1 microplate reader and all data presented are means at least three independent experiments.
Quantification of SSTR2 internalization by ELISA assay
The quantification of SSTR2 internalization was performed following the procedure of the “Detection of surface expression of SSTR wild-type and mutants” as described in the last paragraph41, except that octreotide (MCE, HY-17365) or paltusotine (MCE, HY-109155) need to be added and incubated at 37 °C for 30 min before being fixed with 4% formalin. The absorbance at 455 nm was measured using Synergy H1 microplate reader and was compared with untreated cells. All data presented are means at least three independent experiments.
Internalization confocal assay
The internalization assay was performed as previously reported43. Briefly, HEK293 cells were transfected with plasmids encoding eGFP-tagged wild-type or mutant SSTR2. The transfected cells were plated on poly-D-lysine (Beyotime) coated coverslips in 24-well plates and grew overnight. The cells were washed with PBS, and then octreotide or paltusotine was added at the final concentration of 1–100 μM. After incubation for 30 min at 37 °C, the cells were fixed and permeabilized with methanol (−20 °C) for 5–10 min. Finally, the coverslips were mounted onto glass slides with the antifade mounting medium containing DAPI (Beyotime Biotechnology, P0131-5 ml). The confocal fluorescence images were aquired by Zeiss LSM 880 microscope with the ZEN imaging software (Zeiss).
Cell apoptosis assay
The rat pituitary GH3 cells (ATCC, CCL-82.1) were seeded in 12-well plates at 1.5 × 105/well. Octreotide or paltusotine were added to each well at the indicated concentrations, and incubated for 48 hours. Then, the cells were collected for the apoptosis assay according to the instruction of the Annexin V-FITC/PI apoptosis detection kit (BD Biosciences). Briefly, the cells were washed with cold PBS and resuspended in 1 × binding buffer. Then, 5 μl of Annexin V-FITC was added and incubated for 10 min and stained with 5 μl of PI for 5 min in the dark at room temperature. All samples were acquired by a Novocyte flow cytometer (ACEA Biosciences, China), and data were analyzed with NovoExpressTM software.
Constructs for structure determination
For the octreotide-bound SSTR2-Gi1 complex, the full-length of wild-type human SSTR2 (SSTR2FL) cDNA (UniProt: P30874) was cloned into the pFastBac1 vector with the haemagglutinin (HA) signal sequence and a Flag epitope tag at the N-terminus. To purify the paltusotine-bound SSTR2-Gi1 complex, the amino acid residues 1–30 at the N-terminus and residues 339–369 at the C-terminus of SSTR2 were truncated (SSTR231–338). The wild-type human Gαi1 (UniProt: P63096) without any modification (Gαi1WT) and the human Gαi1 with four dominant-negative mutations (Gαi14M; S47N, G203A, E245A, A326S) were subcloned into pFastBac1 vector44. The human Gβ1 containing an N-terminal 10 x His tag and the human Gγ2 were cloned into a pFastBac-dual vector for co-expression.
Expression and purification of SSTR2-Gi complex
The octreotide-bound SSTR2-Gi complex was expressed by co-infecting the HA-Flag-SSTR2FL, Gαi1WT, and Gβ1Gγ2 baculovirus into Spodoptera frugiperda (Sf9, Expression system, 94011 S) cells at a density of 3 × 106 cells/ml. After 56 h, cells were collected and lysed with 20 mM Hepes (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, Protease Inhibitor Cocktail (160 μg/ml benzamidine, 100 μg/ml leupeptin), and 25 mU/ml Apyrase (NEB) and 10 µM octreotide at room temperature for 2 h. Then the membrane fractions were solubilized with 20 mM Hepes (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG; Anatrace), 0.1% (w/v) cholesteryl hemisuccinate (CHS), 10% (v/v) glycerol, Protease Inhibitor Cocktail, 10 µM octreotide, 0.02 mg/ml scFv16, and 25 mU/ml Apyrase (NEB) followed by dounce homogenization and incubated for 2 h at 4 °C. After centrifuging at 65,000 × g for 30 min, the solubilized complex was incubated with M1 anti-Flag antibody coupled Sepharose Resin at 4 °C for 2 h. Then the target complex was eluted with 20 mM Hepes (pH 7.5), 100 mM NaCl, 0.01% (w/v) LMNG, 0.002% (w/v) CHS, 10 mM EDTA, 0.2 mg/ml Flag peptide and 10 µM octreotide. The concentrated sample was loaded onto a Superose6 increase 10/300 size exclusion column (GE Healthcare) in the buffer include 20 mM Hepes (pH 7.5), 100 mM NaCl, 0.00075% (w/v) LMNG, 0.00025% glyco-diosgenin (GDN; Anatrace), 0.0002% (w/v) CHS and 5 µM octreotide. Target complex was collected and concentrated to 5 mg/ml using an Amicon Ultra Centrifugal Filter (MWCO, 100 kDa cut-off). The paltusotine-bound SSTR2-Gi complex was prepared using the same protocol, except that SSTR2 fragment (SSTR231–338), and Gαi1 with four dominant-negative mutations (Gαi14M) were used, and paltusotine was used throughout the buffers instead of octreotide.
Expression and purification of scFv16
The procedures of the expression and purification were performed as previously described45. Briefly, scFv16 was cloned into pfastBac1 with the C-terminal 6 x His tag and the N-terminal GP67 signal peptide. Protein was expressed using the Sf9 baculovirus system and purified by Ni-NTA resin. The eluted protein was further concentrated to 4 mg/ml in a buffer contain 20 mM Hepes (pH 7.5) and 100 mM NaCl. Then the protein was flash frozen in liquid nitrogen and was stored at −80 °C for further use.
Cryo-grid preparation and EM data collection
3 μl of the octreotide-bound SSTR2-Gi1 complex at ~5 mg/ml or the paltusotine-bound SSTR2-Gi1 complex at ~20 mg/ml was applied onto glow-discharged 300-mesh Au grids (Quantifoil R1.2/1.3), respectively. The grids were blotted for 2–3 s before swiftly frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher) at 4 °C and 100% humidity. The octreotide-bound SSTR2-Gi complex was loaded into a Titan Krios cryo-electron microscope (Thermo Fisher) with a Gatan K3 direct electron detector, data collection was performed at 300 keV with the magnification of 130,000 (corresponding to a calibrated sampling of 0.46 Å per physical pixel). At a dose rate of 20 e/pix/s and a total exposure period of 2.7 s, the SerialEM software46 was used to automatically gather movie stacks in super resolution mode, providing 40 frames per stack and a total dose of 65 e−/Å2. While the paltusotine-bound SSTR2-Gi-scFv16 complex sample was loaded into a Titan Krios Cryo-EM (Thermo Fisher) with a Gatan K2 direct electron detector, data collection was performed at 300 keV with the magnification of 165,000 (corresponding to a calibrated sampling of 0.85 Å per physical pixel). With a dose rate of 7.8 e/pix/s and a total exposure period of 6 s, the EPU software (Thermo Fisher) was used to automatically gather movie stacks in counting mode, producing 36 frames per stack and a total dose of 65 e−/Å2.
Image processing and 3D reconstructions
6157 and 4746 movie stacks were obtained for the octreotide-bound SSTR2-Gi1 complex and paltusotine-bound SSTR2-Gi1 complex, respectively. The movie stacks were motion corrected using Motioncor247. Contrast transfer function (CTF) parameters were estimated by GCTF48 and Micrographs with a resolution <4 Å were eliminated. For the octreotide-bound SSTR2-Gi1 complex, a total of 2,364,681 particles were auto-picked and extracted in Relion 3.049 with the box size of 256 pixels. Following two rounds of 2D classifications, bad particles were removed and 1,159,274 well-defined particles were subjected to cryoSPARC3.150 ab initio to build the initial model. The resulting particles were then subjected to a round of cryoSPARC heterogeneous refinement. The best class determined by visual examination was subjected to cryoSPARC non-uniform refinement thus producing a sharpened map of 3.37 Å. The local resolution was calculated in cryoSPARC. As for the paltusotine-bound SSTR2-Gi1 complex, a total of 1,302,282 particles were auto-picked and extracted in Relion 4.0-beta51 with a box size of 256 pixels. 1,127,812 well-defined particles were selected after 2D classification to build the initial model. Following two rounds of 3D classifications, the best class determined by eye assessment was subjected to CTF refinement, Bayesian polishing52, 3D auto-refine, post-process and produced a 3.24 Å sharpened map. The local resolution was estimated in RELION 4.0-beta. Details for each cryo-EM reconstruction could be found in Supplementary information, Table S1.
Model building and structure refinement
The initial Gi1 and scFv16 models were created using the cryo-EM structure of the S1PR1–Gi1 complex (PDB: 7EW7), and the initial SSTR2 model was created by Swiss-model server53. UCSF Chimera1.14 was used to fit all models into the EM density map54, followed by iterative rounds of manual adjustment and real-space refinement with Coot 0.8.9.255 and Phenix 1.1956, respectively. MolProbity57 was used to validate the models. The final structures had a satisfactory model geometry, and the comprehensive refining statistics could be found in Supplementary material, Table S1. UCSF ChimeraX1.158 and PyMOL were used to generate the structural figures. (https://pymol.org/2/).
Bystander BRET (bBRET)
The human full-length β-arrestin 1 was cloned into pcDNA3.1+ vector with the Nluc of the BRET system and a 15-amino acid flexible linker (GGSGGGGSGGSSSGG) at the N-terminus. FYVE, the early endosomal marker35 was cloned into pcDNA3.1+ vector with a 15-amino acid flexible linker (GGSGGGGSGGSSSGG) and C-terminally fused to a mVenus. Wild-type or mutant SSTR2, β-arrestin 1-Nluc and FYVE-mVenus were co-transfected in HEK293 cells at a ratio of 1:0.5:4 on six-well plates. The next day, the transfected cells were plated on white 96-well and incubated overnight, and then cells were incubated with 90 µL HBSS containing 20 mM HEPES and coelenterazine h (5 µM) at room temperature for 40 min. After incubation, ligand was added and the signal was measured using the Synergy H1 microplate reader (BioTek) to determine a BRET ratio.
Statistical analysis
All data in this study were analyzed using GraphPad Prism 7 and were expressed as the error bars and the standard error of means ± SEM from at least n = 3 biologically independent experiments performed in triplicate. All curve data were normalized to the maximal wild-type and processed using a sigmoidal dose response curve in GraphPad Prism 7. Statistical differences between conditions were determined by one-way of variance ANOVA with Dunnett’s test or T-test. The bias factors (β value) were determined as described in the previous reports59 as the following equation:
where P1 is SSTRs Gi-mediated cAMP inhibition assay; P2, NanoBiT β-arrestin recruitments; β value > 0 denotes Gi protein biased, while β value < 0 indicates β-arrestin biased.
Parameters used in this equation were based on the curve fits of the combined datasets described above.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM density maps and structural data of SSTR2/octreotide (EMDB ID: EMD-33710; PDB ID: 7YAE) and SSTR2/paltusotine (EMDB ID: EMD-33708; PDB ID: 7YAC) are deposited in EMDB and PDB. The PDB codes and hyperlinks used in our structural alignment were listed below: AT1R-TRV023 (PDB code: 6OS1); μOR-Bu72 (PDB code: 5C1M) or FH210 (PDB code: 7SCG); the inactive SSTR2 (PDB code: 7UL5, colored in wheat); SSTR2-SST14(PDB code: 7T10). Other data are available in the main text or the supplementary materials. Source data underlying Figs. 1–6 and Supplementary Figs. 5–9 are provided with this paper. Source data are provided with this paper.
References
Ishida, A. et al. Discovery and SAR studies of orally active somatostatin receptor subtype-2 (SSTR2) agonists for the treatment of acromegaly. ACS Chem. Neurosci. 11, 1482–1494 (2020).
Vilar, L., Vilar, C. F., Lyra, R., Lyra, R. & Naves, L. A. Acromegaly: clinical features at diagnosis. Pituitary 20, 22–32 (2017).
AlDallal, S. Acromegaly: a challenging condition to diagnose. Int. J. Gen. Med. 11, 337–343 (2018).
Casarini, A. P. et al. Acromegaly: correlation between expression of somatostatin receptor subtypes and response to octreotide-lar treatment. Pituitary 12, 297–303 (2009).
Muhammad, A. et al. Pasireotide responsiveness in acromegaly is mainly driven by somatostatin receptor subtype 2 expression. J. Clin. Endocrinol. Metab. 104, 915–924 (2019).
Brzana, J., Yedinak, C. G., Gultekin, S. H., Delashaw, J. B. & Fleseriu, M. Growth hormone granulation pattern and somatostatin receptor subtype 2A correlate with postoperative somatostatin receptor ligand response in acromegaly: a large single center experience. Pituitary 16, 490–498 (2013).
Sun, L. & Coy, D. H. Somatostatin and its analogs. Curr. Drug Targets 17, 529–537 (2016).
Colao, A., Auriemma, R. S., Pivonello, R., Kasuki, L. & Gadelha, M. R. Interpreting biochemical control response rates with first-generation somatostatin analogues in acromegaly. Pituitary 19, 235–247 (2016).
Lamberts, S. W. J. & Hofland, L. J. ANNIVERSARY REVIEW: Octreotide, 40 years later. Eur. J. Endocrinol. 181, R173–r183 (2019).
Paragliola, R. M., Corsello, S. M. & Salvatori, R. Somatostatin receptor ligands in acromegaly: clinical response and factors predicting resistance. Pituitary 20, 109–115 (2017).
Gadelha, M. R. et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): a randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2, 875–884 (2014).
Gunther, T. et al. International union of basic and clinical pharmacology. CV. Somatostatin receptors: structure, function, ligands, and new nomenclature. Pharm. Rev. 70, 763–835 (2018).
Dent, P. et al. S49 cells endogenously express subtype 2 somatostatin receptors which couple to increase protein tyrosine phosphatase activity in membranes and down-regulate Raf-1 activity in situ. Cell Signal 9, 539–549 (1997).
Liguz-Lecznar, M., Urban-Ciecko, J. & Kossut, M. Somatostatin and somatostatin-containing neurons in shaping neuronal activity and plasticity. Front. Neural Circuits 10, 48 (2016).
Nagel, F. et al. Structural determinants of agonist-selective signaling at the sst(2A) somatostatin receptor. Mol. Endocrinol. 25, 859–866 (2011).
Treppiedi, D. et al. Somatostatin receptor type 2 (SSTR2) internalization and intracellular trafficking in pituitary GH-secreting adenomas: role of scaffold proteins and implications for pharmacological resistance. Horm. Metab. Res. 49, 259–268 (2017).
Gatto, F. et al. Low beta-arrestin expression correlates with the responsiveness to long-term somatostatin analog treatment in acromegaly. Eur. J. Endocrinol. 174, 651–662 (2016).
Cuevas-Ramos, D. & Fleseriu, M. Somatostatin receptor ligands and resistance to treatment in pituitary adenomas. J. Mol. Endocrinol. 52, R223–R240 (2014).
Fougner, S. L. et al. The clinical response to somatostatin analogues in acromegaly correlates to the somatostatin receptor subtype 2a protein expression of the adenoma. Clin. Endocrinol. 68, 458–465 (2008).
Gatto, F. et al. beta-Arrestin 1 and 2 and G protein-coupled receptor kinase 2 expression in pituitary adenomas: role in the regulation of response to somatostatin analogue treatment in patients with acromegaly. Endocrinology 154, 4715–4725 (2013).
Robertson, M. J., Meyerowitz, J. G., Panova, O., Borrelli, K. & Skiniotis, G. Plasticity in ligand recognition at somatostatin receptors. Nat. Struct. Mol. Biol. 29, 210–217 (2022).
Bo, Q. et al. Structural insights into the activation of somatostatin receptor 2 by cyclic SST analogues. Cell Discov. 8, 47 (2022).
Heo, Y. et al. Cryo-EM structure of the human somatostatin receptor 2 complex with its agonist somatostatin delineates the ligand-binding specificity. Elife 11, e76823 (2022).
Chen, L. N. et al. Structures of the endogenous peptide- and selective non-peptide agonist-bound SSTR2 signaling complexes. Cell Res. 32, 785–788 (2022).
Zhao, W. et al. Structural insights into ligand recognition and selectivity of somatostatin receptors. Cell Res. 32, 761–772 (2022).
Madan, A. et al. Paltusotine, a novel oral once-daily nonpeptide SST2 receptor agonist, suppresses GH and IGF-1 in healthy volunteers. Pituitary 25, 328–339 (2022).
Tikhonova, I. G., Gigoux, V. & Fourmy, D. Understanding peptide binding in class A G protein-coupled receptors. Mol. Pharm. 96, 550–561 (2019).
Xiao, P. et al. Ligand recognition and allosteric regulation of DRD1-Gs signaling complexes. Cell 184, 943–956.e918 (2021).
Kooistra, A. J. et al. GPCRdb in 2021: integrating GPCR sequence, structure and function. Nucleic Acids Res. 49, D335–D343 (2021).
Pandy-Szekeres, G. et al. The G protein database, GproteinDb. Nucleic Acids Res. 50, D518–D525 (2022).
Gurevich, V. V. & Gurevich, E. V. GPCR Signaling regulation: the role of GRKs and arrestins. Front Pharm. 10, 125 (2019).
Wang, H. et al. Structure-based evolution of G protein-biased mu-opioid receptor agonists. Angew. Chem. Int. Ed. Engl. 61, e202200269 (2022).
Huang, W. et al. Structural insights into micro-opioid receptor activation. Nature 524, 315–321 (2015).
Wingler, L. M. et al. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 367, 888–892 (2020).
Namkung, Y. et al. Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 7, 12178 (2016).
Smith, J. S. et al. C-X-C motif chemokine receptor 3 splice variants differentially activate beta-arrestins to regulate downstream signaling pathways. Mol. Pharm. 92, 136–150 (2017).
Barbieri, F. et al. Peptide receptor targeting in cancer: the somatostatin paradigm. Int. J. Pept. 2013, 926295 (2013).
Gadelha, M. R., Wildemberg, L. E., Bronstein, M. D., Gatto, F. & Ferone, D. Somatostatin receptor ligands in the treatment of acromegaly. Pituitary 20, 100–108 (2017).
Rankovic, Z., Brust, T. F. & Bohn, L. M. Biased agonism: an emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 26, 241–250 (2016).
Melmed, S. Acromegaly pathogenesis and treatment. J. Clin. Invest. 119, 3189–3202 (2009).
Yuan, Y. et al. Structures of signaling complexes of lipid receptors S1PR1 and S1PR5 reveal mechanisms of activation and drug recognition. Cell Res. 31, 1263–1274 (2021).
Huang, W. et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 579, 303–308 (2020).
Chen, H. et al. Structure of S1PR2-heterotrimeric G13 signaling complex. Sci. Adv. 8, eabn0067 (2022).
Liang, Y. L. et al. Dominant negative G proteins enhance formation and purification of agonist-GPCR-G protein complexes for structure determination. ACS Pharm. Transl. Sci. 1, 12–20 (2018).
Zhao, C. et al. Structural insights into sphingosine-1-phosphate recognition and ligand selectivity of S1PR3-Gi signaling complexes. Cell Res. 32, 218–221 (2022).
Mastronarde, D. N. SerialEM: a program for automated tilt series acquisition on tecnai microscopes using prediction of specimen position. Microsc. Microanal. 9, 1182–1183 (2003).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Kimanius, D., Dong, L., Sharov, G., Nakane, T. & Scheres, S. H. W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 478, 4169–4185 (2021).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–w303 (2018).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Shao, Z. et al. Identification and mechanism of G protein-biased ligands for chemokine receptor CCR1. Nat. Chem. Biol. 18, 264–271 (2022).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (32100988 to W.Y.; 32200986 to J.Z.; 31972916 to Z.S.), Science and Technology Department of Sichuan Province (2021ZYD0080 to W.Y.), National Key R&D Program of China (2019YFA0508800 to Z.S.), the National Science Foundation for Excellent Young Scholars (32122052 to X.W.), National Natural Science Foundation Regional Innovation and Development (No. U19A2003 to X.W.), the China Postdoctoral Science Foundation (BX20220219 to Z.X.; 2022M712275 to J.Z.) and Post-Doctor Research Project, West China Hospital, Sichuan University (2021HXBH064 to J.Z.). We would like to thank the staff of Sichuan University West China Cryo-EM Center and Cryo-EM Center in Southern University of Science and Technology for cryo-EM data collection and the staff of the National Center for Protein Science in Shanghai for technical support. We thank the Sichuan University State Key Laboratory of Biotherapy Microscope platform for confocal assay. This work used resources from the Duyu High Performance Computing Center, Sichuan University, and Big Data Platform at West China Hospital of Sichuan University (WCH-BDP).
Author information
Authors and Affiliations
Contributions
H.F. designed the expression constructs and purified the SSTR2–Gi complex under the supervision of L.C.; J.Y., W.H., and J.Q. performed the cell signaling assays with the assistance of C.W., H.F., and S.S.; W.H. performed Internalization confocal assay. H.F. prepared the final samples for cryo-EM data collection with the assistance of C.Z., X.T., and Z.X.; C.Z. and X.T. prepared the cryo-EM grids, collected cryo-EM images, and performed map calculations under the supervision of Z.S.; C.Z., J.Z., and X.T. built and refined the structure models under the supervision of J.Z.; J.Z. analyzed the results and wrote the manuscript. R.C., W.Y., X.W., and Z.S. supervised the overall project and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Fu, H., Yu, J. et al. Prospect of acromegaly therapy: molecular mechanism of clinical drugs octreotide and paltusotine. Nat Commun 14, 962 (2023). https://doi.org/10.1038/s41467-023-36673-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36673-z
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
