
Abstract
V/A-ATPase is a motor protein that shares a common rotary catalytic mechanism with FoF1 ATP synthase. When powered by ATP hydrolysis, the V1 domain rotates the central rotor against the A3B3 hexamer, composed of three catalytic AB dimers adopting different conformations (ABopen, ABsemi, and ABclosed). Here, we report the atomic models of 18 catalytic intermediates of the V1 domain of V/A-ATPase under different reaction conditions, determined by single particle cryo-EM. The models reveal that the rotor does not rotate immediately after binding of ATP to the V1. Instead, three events proceed simultaneously with the 120˚ rotation of the shaft: hydrolysis of ATP in ABsemi, zipper movement in ABopen by the binding ATP, and unzipper movement in ABclosed with release of both ADP and Pi. This indicates the unidirectional rotation of V/A-ATPase by a ratchet-like mechanism owing to ATP hydrolysis in ABsemi, rather than the power stroke model proposed previously for F1-ATPase.
Similar content being viewed by others

Introduction
The proton translocation ATPase/synthase family includes F-type enzymes found in eubacteria, mitochondria, and chloroplasts, and the V/A type enzymes found in archaea and some eubacteria1,2,3,4,5 (Fig. 1A). These ATPases produce the majority of cytosolic ATP from ADP and Pi using energy derived from the transmembrane proton motive force generated by cellular respiration6. These ATPases share a common molecular architecture, consisting of a hydrophilic V1/F1 domain responsible for ATP hydrolysis or synthesis, and a hydrophobic Vo/Fo domain housing a proton translocation channel7,8,9. The chemical reaction (ATP hydrolysis/synthesis) in V1/F1 is tightly associated with proton movement through Vo/Fo using a rotary catalytic mechanism, where both reactions are coupled by rotation of the central rotor complex relative to the surrounding stator apparatus, which includes the ATPase active hexamer6,10,11 (Fig. 1B).

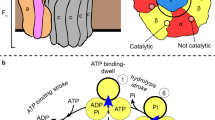
A Illustration of subunit composition of different types of rotary ATPases; prokaryotic V/A-ATPase (left), eukaryotic V-ATPase (middle), prokaryotic F-ATPase (right). The stators are represented in various colors and the rotors are represented in gray. B A schematic model of the rotary catalytic mechanism of the V/A-ATPase. When powered by ATP, the central rotor composed of D1F1d1c10 (gray) rotates against a surrounding stator composed of A3B3E2G2a1 (white), coupled with proton translocation across the membrane. C The conventional catalytic cycle of V/A-ATPase. At low ATP concentration, the ATP binding dwell time is increased. ATPγS also prolongs the ATP hydrolysis dwell.
The V/A-ATPase from the thermophilic bacterium, Thermus thermophilus (Tth) is one of the best-characterized ATP synthases3,12. The overall architecture and subunit composition of V/A-ATPase is more similar to that of the eukaryotic V-ATPase, rather than F-type ATPase. However, the Tth V/A-ATPase has a simpler subunit structure than the eukaryotic V-ATPase and shares the ATP synthase function of F-type ATPase13 (Fig. 1A). The V1 domain of Tth V/A-ATPase (A3B3D1F1) is an ATP-driven rotary motor where the central DF shaft rotates inside the hexameric A3B3 containing three catalytic sites, each composed of an AB dimer. The Vo domain (E2G2d1a1c12) is composed of stator parts including the a subunit and two EG peripheral stalks and the d1c12 rotor complex, which consists of a central rotor complex with the DF subunits of V114,15,16. When ATP hydrolysis by A3B3 powers the DF shaft, the reverse rotation of the central rotor complex drives proton translocation in the membrane-embedded Vo domain (Fig. 1B).
According to the binding change mechanism of ATP synthesis6, the three catalytic sites in ATP synthases are in different conformations but interconvert sequentially between three different conformations as catalysis proceeds. Indeed, our previous structure demonstrated that the A3B3 hexamer in the V/A-ATPase adopts an asymmetrical structure composed of three different AB dimers, termed open (ABopen), semi-closed (ABsemi), and closed (ABclosed)16,17.
Experimental studies using specific rotational probes attached to DF revealed that ATP-driven rotation of the central shaft was unidirectionally clockwise when viewed from the V1 side10. At low ATP concentrations where ATP binding is rate-limiting, rotation proceeds in steps of 120°, commensurate with the three catalytic sites of AB dimers18. When using 40 nm gold beads with almost negligible viscous resistance, V1 also pauses every 120° even at an ATP concentration around Km without a sign of substeps19. These single-molecule experiments on V1 suggest that both catalytic events, ATP hydrolysis and product (ADP and Pi) release occur at an individual ATP binding position, and imply the presence of chemo-mechanically stable catalytic intermediates (Fig. 1C and Supplementary Fig. 1).
However, single-molecule observation experiments only allow us to see the motion of the shaft to which the observation probe is bound, and do not tell us what events are occurring at each catalytic site. To elucidate the entire rotational mechanism of the V/A-ATPase, we must determine the structures of catalytic intermediates of the rotary ATPase during rotation. There are many reaction intermediates of the enzyme during turnover, and this structural heterogeneity makes successful crystallization of a specific state very challenging.
Technological breakthroughs in single-particle Cryo-EM, such as the development of direct electron detectors, and advances in image processing and automation20,21, have triggered a revolution in structural biology, making this the technique of choice for large and dynamic complexes unsuitable for crystallization. In addition, by freezing Cryo-EM grids at different time points or under different reaction conditions, it is possible to trap intermediate states and thus build up a picture of the chemo-mechanical cycle of biological macromolecular complexes step by step. To date, there are few examples of studies that have successfully captured such details of a catalytic cycle at atomic resolution using Cryo-EM22,23.
Here, we report several keys, and thus far uncharacterized, intermediate states of V/A-ATPase, obtained under different reaction conditions. Comparison of these structures provides insight into the cooperativity between the three catalytic sites and demonstrates a rotary catalytic mechanism powered by ATP hydrolysis.
Results
Sample preparation for Cryo-EM structural analysis
We previously determined the Cryo-EM structures of the wild-type V/A-ATPase containing an ADP in the catalytic site of ABclosed16,17. The V/A-ATPase bound to the inhibitory ADP exhibits no ATPase activity until the ADP is removed13,16,24. Partial ADP removal from ABclosed is possible by dialysis against an EDTA-phosphate buffer, but it is difficult to obtain a homogenous nucleotide-free V/A-ATPase after such a treatment, due to the high binding affinity of the ADP to ABclosed (Supplementary Table 1). To obtain a homogeneous ATPase active enzyme, mutant V/A-ATPase (A/S232A, T235S) with reduced nucleotide-binding affinity was purified from T. thermophilus membranes10. The mutated V/A-ATPase exhibits higher Km values for nucleotide in both the ATP hydrolysis and synthesis reactions than the wild-type enzyme, but the enzymatic and rotational properties are almost the same as those of the wild-type enzyme24. The mutated V/A-ATPase is fully activated for ATPase activity after EDTA/phosphate dialysis; no ADP or ATP was found in the enzyme by quantitative analysis of nucleotides (Supplementary Fig. 2 and Table 1). We incorporated the nucleotide-free V/A-ATPase into nanodiscs comprising the MSP1E3D1 scaffold protein and DMPC. The resulting V/A-ATPase obeys simple Michaelis–Menten kinetics and exhibits ATPase activity of 22 s−1 and the Km of 394 µM ATP (Supplementary Fig. 2b).
The nucleotide-free V/A-ATPase (Nucfree) was used for cryo-grid preparation under different ATPase reaction conditions (Supplementary Fig. 1c). Results of the structural analysis of the protein under each set of reaction conditions are summarized in Supplementary Fig. 3a–d.
The structures of V/A-ATPase without nucleotide (Vnucfree)
The flow charts showing image acquisition and reconstitution of the 3D structure of V/A-ATPase without nucleotide are summarized in Supplementary Fig. 3a. We obtained structures of three rotational states of V/A-ATPase without nucleotide; state1 at 3.1 Å, state 2 at 4.7 Å, and state 3 at 6.3 Å resolution, with the DF shaft positions differing by 120° in each case (Fig. 2). Using signal subtraction of the Vo domain, we achieved resolution of 3.0 and 4.1 Å for the V1 domain including half of the EG stalk in state1 and 2, allowing us to build atomic models of the V1 domain of these states.

A Cryo-EM density map of whole V/A-ATPase of state1 in the absence of nucleotide (Vnucfree structure). B Cryo-EM density map of V1EG of state1 without nucleotide (left). Cross-sections of the nucleotide-binding sites (right upper) and A3B3 C-terminal region (right lower) viewed from the top. C Comparison of the AB dimer structures in Vnucfree. AB dimers are shown as space-filling models and superimposed on the β barrel domain (A subunit 1–70 a.a.). Left; ABopen (solid) vs. ABsemi (semi-transparent), middle; ABopen (solid) vs. ABclosed (semi-transparent), and ABsemi (solid) vs. ABclosed (semi-transparent). D Comparison between each AB subunit in Vnucfree. The subunits are shown as wire representations. A and B subunits are superimposed on the β barrel domain.
The three AB dimers in the V1 domain adopted open (ABopen), semi-closed (ABsemi), and closed (ABclosed) states, respectively (Fig. 2B, C). The tip of the C-terminal helix bundle (CHB) of Aopen is in contact with the C-terminal helix of the D subunit, and the wide part of the CHB of Bopen is in contact with the N-terminal helix of the D subunit, respectively (Supplementary Fig. 4a–c). The ABsemi and ABclosed also interact with the coiled-coil of subunit D in specific regions of the CHB, respectively (Supplementary Fig. 4d–g).
The differences in the structures of the three AB dimers, when superimposed on the β barrel domains of both A and B subunits, are the result of the movement of the N-terminal bulge domain, the nucleotide-binding domain (NB) of the A subunit and the CHBs of both the A and B subunits (Fig. 2D). When comparing the structure of ABopen and ABsemi, both the NB and CHB of the Asemi are in closer proximity to Bsemi than Bopen, resulting in a closed structure of ABsemi (Fig. 2C, D). The structure of Bopen is very similar to Bsemi, as shown in Fig. 2D. In the ABclosed, both the CHB and NB domains of Aclosed are in closer proximity to Bclosed, and the CHB of Bclosed moves to Aclosed, resulting in the more closed structure of ABclosed compared to ABsemi (Fig. 2C, D).
In the ABclosed and ABsemi dimers, densities for the catalytic side chains are well resolved, but no density corresponding to nucleotide was observed (Fig. 3A). Hereafter we refer to the structure as the Vnucfree. The structure of Vnucfree is very similar to the previously reported ADP inhibited structure16,17. For state1, the rmsd value for the Cα chains of A3B3DF of the Vnucfree and ADP inhibited structures is 1.98 Å (Supplementary Fig. 5). In addition, the Vnucfree is also similar to the structures under the saturated-ATP condition determined in this study, with the positions of the catalytic side chains almost identical in both cases (Supplementary Fig. 6). This indicates that the V1 domain adopts the same conformation, including the arrangement of the DF shaft in the A3B3 and the geometry of the catalytic side chains, irrespective of the presence or absence of bound ATP.

Upper panels: Vnucfree (A), Vprehyd (B), V3nuc (C), and V2nuc (D) viewed from the cytosolic side. The scale bar is 20 Å. Magnified views of the three nucleotide-binding sites (ABopen, ABsemi, and ABclosed) in each structure are shown in the rows below. Cryo-EM maps are represented as semi-translucent. Bound nucleotides and Mg ions are shown in ball-and-stick and sphere representation, respectively. The scale bar is 4 Å.
In the density maps obtained for state1 of Vnucfree, the CHB of the AB dimers was slightly blurred, likely due to structural heterogeneity. To classify the probable substates of state1, we performed focused 3D classification using a mask covering ABopen and Bsemi (Supplementary Fig. 3a). We identified a cryoEM structure of the original state1 from 39,902 particles at 3.1 Å resolution and another substate from 24,101 particles at 3.1 Å resolution. We termed the substates reconstructed from these major particle classes as state1-1 and state1–2, respectively. The atomic model initially constructed as state1 is identical to the atomic model of state1–1. The structures of the sub-states are very similar, with most differences due to the movement of the CHB of the A and B subunits. Therefore, we quantified the difference in the structure of the CHB observed when the structures were superimposed on the N-barrel domain (Supplementary Tables 2 and 3). Substates were also obtained under other reaction conditions (see below) and the rmsd values shown in Supplementary Tables 2 and 3 are used to discuss which subunits are responsible for the differences in the structure of the substates obtained under different reaction conditions.
Structures obtained at a saturating ATP concentration
Cryo-grids were prepared using a reaction mixture of nucleotide-free V/A-ATPase, containing the regenerating system and ATP at a saturating concentration of 6 mM. The reaction mixture was incubated for 120 s at 25 °C and then loaded onto a holey grid, followed by flash freezing.
We determined three rotational states followed by focused refinement using a V1EG mask for each state (Supplementary Fig. 3b). In the density maps obtained for each state, the amino acid residues of the nucleotide-binding sites in both ABclosed and ABsemi were well resolved, but the CHB domains of the AB dimers were blurred due to structural heterogeneity, as with the Vnucfree. For state1, we identified an atomic resolution structure of state1-1 from 40,831 particles at 3.1 Å resolution and state1–2 from 28,801 particles at 3.2 Å resolution by further 3D classification without alignment (Supplementary Fig. 3b). The same classification analysis was performed for state2 and state3, yielding state 2–1 (3.0 Å resolution) and state 2–2 (3.4 Å resolution), and state 3–1 (3.0 Å resolution) and state 3–2 (3.4 Å resolution) respectively. In these structures, nucleotide densities have been identified in the three catalytic sites. Hereafter, we refer to the structures obtained at ATP saturating conditions as V3nuc.
The structure of ABopen of V3nuc state1–1 is almost identical to that of Vnucfree state1–1 (Supplementary Fig. 6). This is confirmed by the fact that the rmsd values in the CHB of Aopen and Bopen for V3nuc state1–1 and Vnucfree are less than 1 Å (Supplementary Tables 2 and 3). The ABopen of state1–2 adopts a slightly more closed conformation compared to that of state1–1, which results from a movement of CHB of Bopen toward the β-barrel domain (Fig. 4C). Nevertheless, the ABopen of V3nuc with bound ATP retains the interaction with the DF shaft, indicating that ATP binding to the ABopen does not move the DF shaft.

The subunits of state1–1 and 1–2 were superimposed on the β barrel domain (A: 1–70 a.a., B: 1–70 a.a.). Ribbon models are colored by the rmsd values calculated for the atoms of the main chain; gray (small changes) to red (large changes). Magnified views of the CHBs are represented in the lower panels as wire models. The models of state1–1 are represented in gray, and state1–2 are represented in different colors. A Subunits in ABclosed, B subunits in ABsemi, C subunits in ABopen.
The ABsemi in state1–2 has a more closed structure than that in state1–1 mainly due to the movement of CHB in Bsemi (Fig. 4B, Supplementary Table 3). In summary, V3nuc state1–2 has a more closed structure than state1–1 due to the movement of the CHB of both Asemi and Bsemi, but the slightly closed conformation of state1–2 is independent of ATP binding to the ABopen.
Structures of the catalytic sites at AB dimers of V3nuc
In both state1–1 and state1–2 structures obtained under ATP saturating conditions, a bound ATP molecule is clearly observed in the catalytic site of ABopen (Fig. 3B and Supplementary Fig. 7c). The catalytic sites in the ABclosed and ABsemi in both the state1–1 and state1–2 also contained density corresponding to an ATP molecule, and in these cases, the associated magnesium ions were visible (Fig. 3B and Supplementary Fig. 7a, b). In the V3nuc structure, we did not find density corresponding to nucleotides between the D and A subunits as reported in a previous paper25 (Supplementary Fig. 8).
In the catalytic site of ABsemi of V3nuc, the density of each nucleotide phosphate atom was easily identifiable (Supplementary Fig. 7b), indicating that the ATP molecule occupies the catalytic site in ABsemi. The protein structure is sufficiently clear to also provide a detailed picture of the configuration of the catalytic side chains (Fig. 5A–C). The γ-phosphate of ATP and the magnesium ion are coordinated by the A/K234 and A/S235 residues on the P-loop, which contains the conserved nucleotide-binding motif26. The aromatic ring of A/F230, not conserved in F type ATPase, is oriented away from the triphosphate moiety, allowing access of the guanidium group to the arginine finger (Supplementary Fig. 9). Considering clear EM density for the γ-phosphate of the ATP bound in ABsemi, hydrolysis of ATP is unlikely to proceed in ABsemi.

Left panels; Comparison of the three nucleotide-binding sites (ABopen (A), ABsemi (B), and ABclosed (C)) of state1–1 of V3nuc shown with colored (green, blue, and pink) atoms and bonds, and main chain, and state1–1 of Vprehyd shown with gray atoms, bonds and main chain. Right panels; schematic representations of the coordination of the ATP group in the three binding sites of V3nuc and Vprehyd in parentheses. The distances between the atoms are shown in dotted lines. All distances are in Å.
The nucleotide-binding site of the ABclosed is shown in Fig. 5B. The geometry of ATP binding in ABclosed is very similar to that found in the ABsemi, however, the carbonyl group of A/E257 is closer to the γ-phosphate by about 1 Å than ABsemi. In state1–2 of V3nuc, the γ-phosphate of ATP bound to the ABclosed appears to be separated from β-phosphate when a relatively high density threshold is used (Supplementary Fig. 10). These findings strongly suggest that ATP bound to ABclosed is either already hydrolyzed or in the process of being hydrolyzed. The state of ATP in ABclosed is discussed further in Supplementary Note.
In the nucleotide-binding site of the ABopen of V3nuc, the adenosine moiety of ATP is occluded as in the ABsemi and ABclosed, with A/F415, A/Y500, and A/V236 forming the adenine binding pocket, however, the hydrogen bonding of the ribose moiety to the side chain of B/N363 is lost due to movement of the CHB of Aopen (Fig. 5C). Unlike in the ABclosed and ABsemi, the phenyl group of A/230F in the ABopen is closer to the tri-phosphate group of ATP due to the torsion of the main chain, resulting in the formation of a hydrophobic barrier between the catalytic side chains and the triphosphate moiety of ATP (Supplementary Fig. 9). Compared with ABsemi, the side chains of catalytic residues A/E257, A/R258, and B/R360 of ABopen are much further away from the γ-phosphate of ATP (10.0, 10.0, and 7.1 Å, respectively. Consequently, the configuration of the catalytic residues in the nucleotide-binding site of ABopen is not appropriate for hydrolysis of the bound ATP. Instead, the bound ATP has the potential to zipper the AB interface via interaction with the surrounding catalytic residues, which ultimately results in the transition of ABopen to a more closed form via a typical zipper conformational change.
Structures of V/A-ATPase waiting for ATP to bind
To determine the ATP-waiting structure of V/A-ATPase, we prepared a cryo-grid with a reaction mixture containing 4 µM enzyme, 50 µM ATP, and the ATP regeneration system, pre-incubated for 300 s. We reconstructed three rotational states (state1, 2.7 Å, state2, 3.3 Å, and state3 3.6 Å resolution) from the single-particle images of the holo-complex and three rotational states of V1EG (state1, 2.8 Å, state2, 3.1 Å, and state3, 2.8 Å) by focused masked refinement. For state1, two substates (state1–1, 2.9 Å and state1–2, 3.0 Å) were separated by focused classification using ABopen and ABsemi masks (Supplementary Fig. 3c).
In both ABsemi and ABclosed of state1–1, the apparent density of ATP-magnesium was observed, but the density of the γ-phosphate at the ABclosed is weaker than that at the ABsemi (Fig. 3C and Supplementary Fig. 7a, b). In contrast, density was not observed in the nucleotide-binding site of ABopen. For state1–2, as in state1–1, nucleotides are present in both ABsemi and ABclosed, while ABopen is empty. Hereafter we refer to the structure as V2nuc.
The overall structure and geometry of the catalytic residues of state1–1 of V2nuc are largely identical to state1–1 of Vnucfree and V3nuc (Supplementary Fig. 11). This structural similarity between V2nuc, Vnucfree, and V3nuc is confirmed by the low rmsd values when comparing the CHB of the A and B subunits of these structures (Supplementary Tables 2 and 3). The similarity of the structures of these substrates indicates that the structural polymorphism of the V1 domain is independent of the binding of ATP to AB dimers.
Structures obtained at a saturating concentration of ATPγS (Vprehyd)
The V1-ATPase from T. thermophilus is capable of hydrolyzing ATPγS, however, the turnover rate of ATPγS is much lower than that of ATP due to the decrease in hydrolysis rate18 (Supplementary Fig. 2d). Thus, pre-hydrolysis structures of V/A-ATPase can be obtained at 4 mM ATPγS. The cryo-grid was prepared by blotting of the reaction mixture comprising Nucleotide-free V/A-ATPase and 4 mM of ATPγS in the absence of the regenerating system in order to exclude any effect of regenerated ATP produced from hydrolyzed ATPγS. We reconstructed three rotational states from the acquired EM images using the CRYOARM300 (JEOL). After the focused masked refinement of the V1EG domain, we obtained atomic resolution structures of each state (state1, 2.7 Å, state2, 3.4 Å, and state3, 3.6 Å), respectively (Supplementary Fig. 3d). We refer to these structures as Vprehyd. For state1, two sub-states, state1–1 and state1–2, were obtained at 2.7 and 2.9 Å resolution respectively, by focused classification using a mask with ABopen and ABsemi (Supplementary Fig. 3d). For the ABopen of Vprehyd, bound ATPγS is clearly observed in the catalytic site, which has an almost identical structure to that of the ATP bound state of V3nuc (Fig. 3B).
The nucleotide-binding sites of the ABclosed and ABsemi of Vprehyd are almost identical to those of V3nuc, respectively, as shown in Fig. 5. The γ-phosphate group of the bound ATPγS molecule in the ABsemi is well resolved as seen in for ATP in V3nuc and V2nuc (Fig. 3d and Supplementary Fig. 7b). In contrast, the density of γ-phosphate of ATPγS at the ABclosed is faint (Supplementary Fig. 7a), suggesting that the ATPγS in the ABclosed has already been hydrolyzed and the bound nucleotide in the ABclosed is ADP. This indicates that the ABsemi is in the pre-hydrolysis conformation, waiting for ATP hydrolysis.
Discussion
We have obtained catalytic intermediates of the V1 domain, Vnucfree, V3nuc, V2nuc, and Vprehyd, with different nucleotide occupancy. Despite the different nucleotide occupancy of these structures, their overall conformations are very similar. For instance, the rmsd of the Cα chains of A3B3 in Vnucfree and V3nuc state1–1 is 1.98 Å. In addition, the relative position of the central DF shaft within the asymmetric A3B3 is almost the same in the Vnucfree and V3nuc structures. These findings demonstrate that the configuration between the DF shaft and individual AB dimers is independent of the state of nucleotide occupancy of each AB dimer. In other words, the structure of the V1 domain adopts three rotational states, 1–3, during continuous ATP hydrolysis, with the conformational changes in the A3B3 hexamer driven by ATP hydrolysis, being discrete rather than continuous.
The V3nuc structure, obtained under ATP saturation conditions shows all three catalytic sites occupied by ATP or the products of hydrolysis (ADP + Pi). Since the hydrolyzed Pi is clearly visible in the ABclosed of V3nuc (Supplementary Fig. 10), it is assumed that V3nuc is the structure before dissociation of Pi from the catalytic site in ABclosed. We also obtained the V2nuc structure in which ATP and product(s) are bound to ABsemi and ABclosed, respectively, but ABopen is empty. The V2nuc is therefore assumed to be the structure of the protein awaiting ATP binding to ABopen. When using ATPγS as a substrate, which has a very slow hydrolysis rate, the high-resolution atomic structure of the V1 domain allowed visualization of ATPγS molecules bound to the catalytic sites of ABopen and ABsemi, as well as identification of the hydrolyzed ATPγS at the ABclosed. The Vprehyd reveals both that the ABclosed adopts the post-hydrolysis state where the product of phosphate (Pi) is dissociated, and that the ABsemi is awaiting ATP hydrolysis.
The structures provide important insights into the chemo-mechanical cycle of V/A-ATPase. The V/A-ATPase undergoes a unidirectional conformational change from state1 to state2 to state3 when powered by ATP. Thus, V3nuc of state1, in which three catalytic sites are already occupied by nucleotides, should change to state2 of V2nuc, following ATP hydrolysis at ABsemi, and the subsequent or simultaneously dissociation of ADP and Pi by the discrete structural transition of ABclosed to ABopen (Fig. 6). This demonstrates that rotation of the rotary ATPase proceeds via the tri-site model with the protein progressing through a two nucleotide bound state and a three-nucleotide bound state, settling the long-standing debate on whether the bi-site model or tri-site model is appropriate for rotary ATPases5,6,27,28,29,30.

The structures of V/A-ATPase viewed from the cytosolic side are shown as ribbon models. The coiled-coil of the DF subunits is shown in gray. The bound ATP molecules are highlighted in sphere representations. State1–1 and 1–2 of V2nuc are in equilibrium and are fluctuating. These structures transit to state1–1 and 1–2 of V3nuc by ATP binding to ABopen, without a 120° rotation step of the DF rotor. V3nuc in state1–1 and state1–2 are also in equilibrium. ATP hydrolysis at ABsemi and zipper motion at ABopen occur simultaneously. This triggers the transition of V3nuc in state1–2 to V2nuc state2–2 together with the 120° rotation step and simultaneous release of ADP and Pi. State2 of V2nuc returns to state1 via state3 of V2nuc by the same process. Asterisks indicate the structures which were not identified in this study.
Based on previous single molecular observation experiments for both F1- and V1-ATPase, ATP binding onto the enzyme directly triggers the first 120° rotation step of the DF shaft18,19,30 (Supplementary Fig. 1a). In light of our findings presented here, this scheme needs to be redrawn; the rotor does not immediately travel 120° as a result of ATP binding to enzyme.
The next catalytic event after ATP binding is ATP hydrolysis in ABsemi. Each conformational change, from ABopen to ABsemi, ABsemi to ABclosed, and ABclosed to ABopen occurs simultaneously, with the rotation of the shaft, and with the hydrolysis of ATP in the ABsemi and release of products (ADP and Pi) from the ABclosed (Fig. 6 and Supplementary Movie 1). This is in contrast to the classical rotary model, where catalytic events occur in sequence at the three catalytic sites, until now the broadly accepted mechanism of action of the F1-ATPase6,27,31,32.
In the V2nuc and V3nuc, two sub-states, state1–1 and state1–2 were identified (Supplementary Fig. 12). These substrates were also identified in Vnucfree, thus the conformational dynamics of the V1 domain are independent of ATP binding. In other words, state1–1 and state1–2 are in a thermal equilibrium state, irrespective of nucleotide occupancy in each catalytic site. Both ABsemi and ABopen in state1–2 adopt more closed structures than those in state1–1, suggesting that state1–2 of V3nuc is likely an intermediate structure just prior to the 120° rotation step of the DF shaft. Compared to state1–2 of V3nuc, state1–2 of Vprehyd exhibits slightly more closed structures of ABopen and ABsemi (Supplementary Fig. 13), likely to be associated with the progress of the catalytic reaction in ABclosed, i.e., the dissociation of the phosphate. In this respect, state1–2 of Vprehyd may be another reaction intermediate structure in which the Pi in the ABclosed is released prior to the 120° step (Supplementary Fig. 14).
Based on the catalytic intermediates of the V1 domain of V/A-ATPases obtained under four different reaction conditions, we propose a model for the ATP-driven rotation mechanism of V/A-ATPases (Fig. 7, Supplementary Fig. 15).

The schematic models of ABopen, ABsemi, and ABclosed are shown in green, pink, and blue, respectively. The coiled-coil region of the D subunit in contact with A3B3 is shown in gray. In the ADP inhibited state, the entrapped ADP in ABclosed hampers the structural transition of ABopen to ABsemi by binding of ATP to ABopen. The Vnucfree in the ground state is activated by the binding of ATP to the catalytic sites. In V2nuc awaiting ATP binding, binding of ATP to ABopen does not induce the 120° rotation step. In V3nuc, both zipper motion of ABopen and ATP hydrolysis in ABsemi induce unzipper motion of ABclosed accompanying the release of ADP and Pi. The catalytic events in the three AB dimers occur simultaneously with the 120˚ step of the DF shaft, resulting in the structural transition of state1 of V3nuc to state2 of V2nuc.
When ATP binds to Vnucfree, which is in a stable initial state (ground state), the enzyme transits into the steady-state for ATP hydrolysis (Fig. 7, upper row). The V3nuc structure is formed by binding of ATP to the ABopen of V2nuc but the binding of ATP itself does not cause structural transitions between AB dimers associated with the 120° step of the DF shaft.
In the V3nuc structure, where nucleotides are bound to all three AB dimers, three distinct but associated catalytic events occur at the three AB dimers simultaneously and these events are coupled to the first 120° rotation step of the DF shaft. One of the driving events for this transition is the conformational change from the ATP-bound ABopen to the more closed ABsemi, which can be explained by a zipper motion of ABopen occurring upon ATP binding. From our structures, a comparison between ABopen and ABsemi implies that new hydrogen bonds form between the triphosphate moiety of ATP and the surrounding side chain groups of B/R360, A/258R, A/257E, and A/K234 (Fig. 5).
The Vprehyd structures indicate that the ATP bound to ABsemi is awaiting hydrolysis. The conformational change from ABsemi to ABclosed should occur spontaneously because it involves ATP hydrolysis, an exergonic reaction. In contrast, ADP bound to ABclosed hampers the unzipper motion in ABclosed, thereby preventing the overall structural transition of the V1 domain. This is supported by the fact that V/A-ATPase adopts the ADP inhibited state in which ADP is entrapped in ABclosed (Fig. 7, upper line). The enzyme in the ADP-inhibited state does not show ATP hydrolysis activity even at saturated ATP concentration13,16.
In summary, the ATP-driven unidirectional rotation of V/A-ATPase proceeds by a discrete structural transition between the three rotational states, i.e., the potential barrier to the structural transition of ABclosed to ABopen, accompanied by the release of ADP and Pi, is overcome by both a zipper motion of ABopen by the bound ATP and ATP hydrolysis in ABsemi. Since the ATP hydrolysis reaction is a heat dissipation process, the structural transition of ABsemi to ABclosed associated with the ATP hydrolysis occurs spontaneously and irreversibly, resulting in a unidirectionality of the 120° steps of the rotor. In other words, our model explains the unidirectional rotation by a ratchet-like mechanism driven by ATP hydrolysis, rather than the power stroke model proposed previously for F1-ATPase5,33.
V/A-ATPase and FoF1 are molecular machines based on the same construction principle, and thus are likely to share the same rotary mechanism. In fact, observation of rotation with high time-resolved rotation analysis using a tiny gold rod showed that bacterial F-ATPase and an A-ATPase share the same rotation mechanism34. For both rotary ATPases, a 120° rotation step together with ATP hydrolysis occurs after the catalytic dwell under ATP-saturated conditions. Importantly, for the thermophilic F1, the first 120° rotation step also includes ~80° and ~40° substeps, suggesting the existence of at least one additional catalytic intermediate of F15,27,35. A recent structural study of thermophilic F1-ATPase indicated a possible intermediate structure responsible for the substeps32. In the V/A-ATPase, any intermediate structure containing phosphate after ADP or Pi release is likely to be in an unstable state, and therefore studies on the ATP-driven rotation of V/A-ATPase have failed to reveal the presence of any substep19.
Methods
Preparation of Tth V/A-ATPase for biochemical assay and cryo-EM imaging
The Tth V/A-ATPase containing His3 tags on the C-terminus of each c subunit and the TSSA mutation (S232A and T235S) on the A subunit was isolated from T. thermophilus membranes as previously described24 with the following modifications. The enzyme, solubilized from the membranes with 10% Triton X-100 was purified by Ni2+-NTA affinity with 0.03% dodecyl-β-d-maltoside (DDM). For bound nucleotide removal, the eluted fractions containing Tth V/A-ATPase were dialyzed against 200 mM Sodium phosphate, pH 8.0, 10 mM EDTA, and 0.03% DDM overnight at 25 °C with three buffer changes, followed by dialysis against 20 mM Tris-Cl, pH 8.0, 1 mM EDTA, and 0.03% DDM (TE buffer) prior to anion exchange chromatography using a 6 ml Resource Q column (GE healthcare). The Tth V/A-ATPase was eluted by a linear NaCl gradient using a TE buffer (0–500 mM NaCl, 0.03% DDM). The eluted fractions containing holo-Tth V/A-ATPase were concentrated to ~10 mg/ml using Amicon 100k molecular weight cut-off filters (Millipore). For nanodisc incorporation, the 1,2-Dimyristoyl-sn-glycero-3-phosphorylcholine (DMPC, Avanti) was used to form lipid bilayers in reconstruction as previously described16. Purified Tth V/A-ATPase solubilized in 0.03% n-DDM was mixed with the lipid stock and membrane scaffold protein MSP1E3D1 (Sigma) at a specific molar ratio VoV1:MSP:DMPC lipid = 1:4:520 and incubated on ice for 0.5 h. Then, 200 μL of Bio Beads SM-2 equilibrated with a wash buffer (20 mM Tris-HCl, pH8.0, 150 mM NaCl) was added to the 500 μL mixture. After 2 h incubation at 4 °C with gentle stirring, an additional 300 μL of Bio Beads was added and the mixture was incubated overnight at 4 °C to form the nanodiscs. The supernatant of the mixture containing nanodisc-Tth V/A-ATPase (nd-V/A-ATPase) was loaded onto the Superdex 200 Increase 10/300 column equilibrated with wash buffer. The peak fractions were collected, analyzed by SDS-PAGE, and concentrated to ~4 mg/mL. The prepared nd-V/A-ATPase was immediately used for biochemical assay or cryo-grid preparation since nd-V/A-ATPase aggregates within a few days.
Biochemical assay
The quantitative analysis of bound nucleotides of Tth V/A-ATPase was carried out using anion-exchange high-performance liquid chromatography13. Bound nucleotides were released from the enzyme by the addition of 5 μl of 60% perchloric acid to 50 μl of the enzyme solution. Thereafter, the mixture was incubated on ice for 10 min. Then, 5 μl of 5 M K2CO3 solution was added and the mixture was incubated on ice for 10 min. The resulting pellet was removed by centrifugation at 4 °C. The supernatant was applied to a Cosmopak-200 column equilibrated with 0.1 M sodium phosphate buffer (pH 7.0). The column was eluted isocratically with the same buffer at a flow rate of 0.8 ml/min. The nucleotide was monitored at 258 nm. The peak area was determined by automatic integration.
ATPase activity was measured at 25 °C with an enzyme-coupled ATP-regenerating system, as described previously13. The reaction mixture contained 50 mM Tris-HCl (pH 8.0), 100 mM KCl, different concentrations of ATP-Mg, 2.5 mM phosphoenolpyruvate (PEP), 50 μg/ml pyruvate kinase (PK), 50 μg/ml lactate dehydrogenase, and 0.2 mM NADH in a final volume of 2 ml. The reaction was started by the addition of 20 pmol nd V/A-ATPase to 2 ml of the assay mixture, and the rate of ATP hydrolysis was monitored as the rate of oxidation of NADH was determined by the absorbance decrease at 340 nm.
Cryo-EM imaging of Tth V/A-ATPase
Sample vitrification was performed using a semi-automated vitrification device (Vitrobot, FEI). For nd-V/A-ATPase that underwent nucleotide removal, hereafter referred to as nucfree nd-V/A-ATPase, 2.4 μl of sample solution at a concentration of 3 mg/ml (2 μM) was applied to glow discharged Quantifoil R1.2/1.3 molybdenum grid discharged by Ion Bombarder (Vacuum Device) for 1 min. The grid was then automatically blotted once from both sides with filter paper for 6 s blot time. The grid was then plunged into liquid ethane with no delay time.
The reaction basal buffer (RB buffer) containing 50 mM Tris-Cl, pH 8.0, 100 mM KCl, and 2 mM MgCl2 was used for different reaction conditions. For saturated ATP or ATP waiting condition, 4 µM of nucfree nd-V/A-ATPase was mixed with the same volume of ×2 RB buffer containing 10 mM PEP, 200 μg/ml of PK, 12 mM, or 100 µM of ATP-Mg. Then the mixtures were incubated for 120 or 300 s at 25 °C, followed by blotting and vitrification, respectively. For the ATPγS saturated condition, 4 µM of nucfree nd-V/A-ATPase was mixed with the same volume of ×2 RB buffer containing 8 mM ATPγS-Mg, then incubated for 300 s at 25 °C, followed by the blotting and vitrification.
With the exception of the saturated ATPγS condition, cryo-EM imaging was performed with a Titan Krios (FEI/Thermo Fisher) operating at 300 kV acceleration voltage and equipped with a direct K3 (Gatan) electron detector in electron counting mode (CDS). Data collection were carried out using SerialEM software36 at a calibrated magnification of 0.88 Å pixel−1 (×81,000) and a total dose of 50.0 e− Å−2 (or 1.0 e− Å−2 per frame) (where e− specifies electrons) with a total 5 s exposure time. The defocus range was −0.8 to −2.0 μm. The data were collected as 50 movie frames.
For the saturated ATPγS condition, Cryo-EM movie collection was performed with a CRYOARM 300 (JEOL) operating at 300 keV accelerating voltage and equipped with a K3 (Gatan) direct electron detector, in electron counting mode (CDS) using the data collection software serialEM. The pixel size was 1.1 Å/pix (×60,000) and a total dose of 50.0 e− Å−2 (1.0 e− Å−2 per frame) with a total 3.0 s exposure time (50 frames) with a defocus range of −1.0 to −3.5 μm.
Image processing
Image processing steps for each reaction condition are summarized in Supplementary Fig. 3a–d. Image analysis software, Relion 3.1 and Cryosparc 3.2, were used37,38. CTFFIND 4.1 and MotionCor2 were used for CTF estimation and movie correction in Relion39,40. Topaz software was used for machine-learning-based particle picking41. We started with 15,317 movies for the nucleotide-free enzyme (nucfree nd-V/A-ATPase), 13,164 movies for saturated ATP condition, 15,711 movies for saturated ATP condition, and 17,522 movies for ATPγS condition. The software used in the steps is indicated in the figure. Autopicking based on template matching or based on Topaz machine-learning resulted in 4,354,341 particles for the nucfree nd-V/A-ATPase, 2,300,834 particles for the ATP saturated condition, 1,671,397 particles for the ATP waiting condition, and 4,677.284 particles for the ATPγS waiting condition. Particles were extracted at 5x the physical pixel size from the movie-corrected micrographs and selected using 2D or 3D classification (nucfree nd-V/A-ATPase; 132,904 particles, saturated ATP; 188,673 particles, ATP waiting; 186,928 particles, ATPγS; 197,960 particles). The selected particles were extracted at full pixel size and subjected to 3D auto-refinement refollowed by CTF refinement by Bayesian polishing. Another round of 3D auto-refine, CTF refinement, and a final round of masked auto-refinement gave holo-V/A-ATPase maps at between 2.7 and 6.3 Å resolution. The membrane domain was visible but not particularly clear compared to the hydrophilic V1 domain in a holo-enzyme map. This seemed to be due to the structural flexibility between the membrane domain and V1 in the holo-enzyme. Focused refinement with signal subtraction targeting the V1EG region improved the map quality of the V1EG region (Supplementary Fig. 3a–d). The refinements provided the density maps for V1EG under each condition at 2.8–4.1 Å resolution. After the focused refinement, masked classification on Aopen and Bsemi subunits was carried out to classify the conformational differences. The resolution was based on the gold standard Fourier shell correlation = 0.142 criterion.
Model building and refinement
To generate the atomic model for the V1EG region of V/A-ATPase, the individual subunits of the V1EG model from the previous structure of V/A-ATPase (PDBID: 6QUM) were fitted into the density map as rigid bodies25 with particular focus on the N terminal region of EG stalk (E; 1–77 aa., G; 2–33 aa). The rough initial model was refined against the map with Phenix suite phenix.real_space_refine program42. The initial model was extensively manually corrected residue by residue in COOT42 in terms of side-chain conformations. Peripheral stalks were removed due to low resolution in this region. The corrected model was again refined by the phenix.real_space_refine program with secondary structure and Ramachandran restraints, then the resulting model was manually checked by COOT. This iterative process was performed for several rounds to correct remaining errors until the model was in good agreement with geometry, as reflected by the MolProbity score of 1.08–1.74 and EMRinger score of 1.59–3.9443,44. For model validation against over-fitting, the built models were used for calculation of FSC curves against both half maps, and compared with the FSC of the final model against the final density map used for model building by phenix.refine program. The statistics of the obtained maps and the atomic model were summarized in Supplementary Tables 4–11. RMSD values between the atomic models were calculated using UCSF chimera45. All the figures were rendered using UCSF chimeraX46.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The cryo-EM maps have been deposited in the EMDB under accession codes 31841, 31842, 31843, 31844, 31845, 31846, 31847, 31848, 31849, 31850, 31851, 31852, 31853, 31854, 31855, 31856, 31857, 31858, 31859, 31860, 31861, 31862, 31863, 31864, 31865, 31866, 31867, 31868, 31869, 31870, 31871, 31872, and 31873. The atomic models have been deposited in the Protein Data Bank under accession codes 7VAI, 7VAJ, 7VAK, 7VAL, 7VAM, 7VAN, 7VAO, 7VAP, 7VAQ, 7VAR, 7VAS, 7VAT, 7VAU, 7VAV, 7VAW, 7VAX, 7VAY, and 7VB0. The initial model for model building is accessible in PDB under accession number 6QUM. The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Kuhlbrandt, W. Structure and mechanisms of F-type ATP synthases. Annu. Rev. Biochem. 88, 515–549 (2019).
Forgac, M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929 (2007).
Yokoyama, K. & Imamura, H. Rotation, structure, and classification of prokaryotic V-ATPase. J. Bioenerg. Biomembr. 37, 405–410 (2005).
Guo, H. & Rubinstein, J. L. Cryo-EM of ATP synthases. Curr. Opin. Struct. Biol. 52, 71–79 (2018).
Yoshida, M., Muneyuki, E. & Hisabori, T. ATP synthase—a marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2, 669–677 (2001).
Boyer, P. D. The ATP synthase—a splendid molecular machine. Annu. Rev. Biochem. 66, 717–749 (1997).
Abrahams, J. P., Leslie, A. G., Lutter, R. & Walker, J. E. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 (1994).
Allegretti, M. et al. Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature 521, 237–240 (2015).
Mazhab-Jafari, M. T. et al. Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nature 539, 118–122 (2016).
Imamura, H. et al. Evidence for rotation of V1-ATPase. Proc. Natl Acad. Sci. USA 100, 2312–2315 (2003).
Noji, H., Yasuda, R., Yoshida, M. & Kinosita, K. Jr. Direct observation of the rotation of F1-ATPase. Nature 386, 299–302 (1997).
Yokoyama, K., Oshima, T. & Yoshida, M. Thermus thermophilus membrane-associated ATPase. Indication of a eubacterial V-type ATPase. J. Biol. Chem. 265, 21946–21950 (1990).
Yokoyama, K. et al. V-ATPase of Thermus thermophilus is inactivated during ATP hydrolysis but can synthesize ATP. J. Biol. Chem. 273, 20504–20510 (1998).
Toei, M. et al. Dodecamer rotor ring defines H+/ATP ratio for ATP synthesis of prokaryotic V-ATPase from Thermus thermophilus. Proc. Natl Acad. Sci. USA 104, 20256–20261 (2007).
Yokoyama, K., Nakano, M., Imamura, H., Yoshida, M. & Tamakoshi, M. Rotation of the proteolipid ring in the V-ATPase. J. Biol. Chem. 278, 24255–24258 (2003).
Kishikawa, J. I. et al. Mechanical inhibition of isolated V(o) from V/A-ATPase for proton conductance. Elife 9, e56862 (2020).
Nakanishi, A., Kishikawa, J. I., Tamakoshi, M., Mitsuoka, K. & Yokoyama, K. Cryo EM structure of intact rotary H(+)-ATPase/synthase from Thermus thermophilus. Nat. Commun. 9, 89 (2018).
Imamura, H. et al. Rotation scheme of V1-motor is different from that of F1-motor. Proc. Natl Acad. Sci. USA 102, 17929–17933 (2005).
Furuike, S. et al. Resolving stepping rotation in Thermus thermophilus H(+)-ATPase/synthase with an essentially drag-free probe. Nat. Commun. 2, 233 (2011).
Bai, X. C., McMullan, G. & Scheres, S. H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 40, 49–57 (2015).
Cheng, Y. Single-particle cryo-EM-How did it get here and where will it go. Science 361, 876–880 (2018).
Sobti, M. et al. Cryo-EM reveals distinct conformations of E. coli ATP synthase on exposure to ATP. Elife 8, e43864 (2019).
Hiraizumi, M., Yamashita, K., Nishizawa, T. & Nureki, O. Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science 365, 1149–1155 (2019).
Nakano, M. et al. ATP hydrolysis and synthesis of a rotary motor V-ATPase from Thermus thermophilus. J. Biol. Chem. 283, 20789–20796 (2008).
Zhou, L. & Sazanov, L. A. Structure and conformational plasticity of the intact Thermus thermophilus V/A-type ATPase. Science 365, eaaw9144 (2019).
Menz, R. I., Walker, J. E. & Leslie, A. G. Structure of bovine mitochondrial F(1)-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell 106, 331–341 (2001).
Adachi, K. et al. Coupling of rotation and catalysis in F(1)-ATPase revealed by single-molecule imaging and manipulation. Cell 130, 309–321 (2007).
Löbau, S., Weber, J. & Senior, A. E. Nucleotide occupancy of F1-ATPase catalytic sites under crystallization conditions. FEBS Lett. 404, 15–18 (1997).
Boyer, P. D. Catalytic site forms and controls in ATP synthase catalysis. Biochim. Biophys. Acta 1458, 252–262 (2000).
Watanabe, R. & Noji, H. Timing of inorganic phosphate release modulates the catalytic activity of ATP-driven rotary motor protein. Nat. Commun. 5, 3486 (2014).
Watanabe, R., Iino, R. & Noji, H. Phosphate release in F1-ATPase catalytic cycle follows ADP release. Nat. Chem. Biol. 6, 814–820 (2010).
Sobti, M., Ueno, H., Noji, H. & Stewart, A. G. The six steps of the complete F(1)-ATPase rotary catalytic cycle. Nat. Commun. 12, 4690 (2021).
Wang, H. & Oster, G. Energy transduction in the F1 motor of ATP synthase. Nature 396, 279–282 (1998).
Sielaff, H. et al. Power stroke angular velocity profiles of archaeal A-ATP synthase versus thermophilic and mesophilic F-ATP synthase molecular motors. J. Biol. Chem. 291, 25351–25363 (2016).
Yasuda, R., Noji, H., Yoshida, M., Kinosita, K. Jr. & Itoh, H. Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase. Nature 410, 898–904 (2001).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Barad, B. A. et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Acknowledgements
We are grateful to all the members of the Yokoyama Lab for their continuous support and technical assistance. Our research was supported by Grant-in-Aid for Scientific Research (JSPS KAKENHI) Grant Number 20H03231 to K.Y., 20K06514 to J.K., and Grant-in-Aid for JSPS Fellows Grant No. 20J00162 to A. Nakanishi, and Takeda Science Foundation to K.Y. Our research was also supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant No. JP17am0101001 (Support No. 1312), Grants-in-Aid from “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) to K.M. (Project No. 12024046), and the Research Program for Next Generation Young Scientists of “Five-star Alliance” in “NJRC Mater. & Dev.” under Grant No. 20215008 to A. Nakano.
Author information
Authors and Affiliations
Contributions
K.Y., J.K., A. Nakanishi, and A. Nakano designed, performed, and analyzed the experiments. J.K., A. Nakanishi, K.Y., A. Nakano, A.F., and S.S. analyzed the data and contributed to the preparation of the samples. T.K. and K.M. provided technical support and conceptual advice. K.Y. designed and supervised the experiments and wrote the paper. All authors discussed the results and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Stephen Muench and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kishikawa, J., Nakanishi, A., Nakano, A. et al. Structural snapshots of V/A-ATPase reveal the rotary catalytic mechanism of rotary ATPases. Nat Commun 13, 1213 (2022). https://doi.org/10.1038/s41467-022-28832-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28832-5
This article is cited by
-
Mechanism of ATP hydrolysis dependent rotation of bacterial ATP synthase
Nature Communications (2023)
-
Six states of Enterococcus hirae V-type ATPase reveals non-uniform rotor rotation during turnover
Communications Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
