
Abstract
Estimating total narrow-sense heritability in admixed populations remains an open question. In this work, we used extensive simulations to evaluate existing linear mixed-model frameworks for estimating total narrow-sense heritability in two population-based cohorts from Greenland, and compared the results with data from unadmixed individuals from Denmark. When our analysis focused on Greenlandic sib pairs, and under the assumption that shared environment among siblings has a negligible effect, the model with two relationship matrices, one capturing identity by descent and one capturing identity by state, returned heritability estimates close to the true simulated value, while using each of the two matrices alone led to downward biases. When phenotypes correlated with ancestry, heritability estimates were inflated. Based on these observations, we propose a PCA-based adjustment that recovers the true simulated heritability. We use this knowledge to estimate the heritability of ten quantitative traits from the two Greenlandic cohorts, and report differences such as lower heritability for height in Greenlanders compared with Europeans. In conclusion, narrow-sense heritability in admixed populations is best estimated when using a mixture of genetic relationship matrices on individuals with at least one first-degree relative included in the sample.
Similar content being viewed by others

Introduction
Heritability is the fraction of phenotypic variance attributed to genetics. More specifically, assuming that the variance \(\sigma _{\mathrm{P}}^2\) of a phenotype equals the sum of its genetic \(\sigma _{\mathrm{G}}^2\) and environmental variance \(\sigma _{\mathrm{e}}^2\), heritability in its broad sense (H2) is expressed by the ratio \(\sigma _{\mathrm{G}}^2\)/\(\sigma _{\mathrm{P}}^2\) (Fisher 1918; Wright 1921). The genetic variance \(\sigma _{\mathrm{G}}^2\) can be further broken down into its additive (\(\sigma _{\mathrm{A}}^2\)), dominant (\(\sigma _{\mathrm{D}}^2\)), and epistatic (\(\sigma _{\mathrm{I}}^2\)) components (Visscher et al. 2008; Zaitlen and Kraft 2012). Most of the existing literature focuses on the fraction of phenotypic variance \({{\upsigma }}_{\mathrm{P}}^2\) owing to additive effects alone (\(\sigma _{\mathrm{A}}^2\)/\(\sigma _{\mathrm{P}}^2\))—the so-called narrow-sense heritability (h2).
Heritability is interesting in its own right, but it is also pivotal in quantitative genetic studies with many practical uses. Because, by definition, heritability measures the contribution of genetics to a phenotype, it allows us to gain insights into the genetic architecture of a trait. Moreover, knowledge about the heritability of a trait helps us evaluate the effectiveness of a genome-wide association study (GWAS), as the so-called SNP heritability \(h_g^2\) of a trait informs us about the maximum discovery potential of a given genotyping platform. Similarly, heritability estimates provide an upper bound to the accuracy of polygenic predictions with predictors potentially having higher performance in more heritable traits.
There exist many ways to estimate narrow-sense heritability h2 and they usually boil down to estimating the variance owing to additive effects (\(\sigma _{\mathrm{A}}^2\)). Assuming an additive model, a classical approach is to use the phenotypic correlation between related individuals (Fisher 1918; Wright 1921; Yang et al. 2010), which for a pair j and k is
Kcausal is an idealized genetic relationship matrix (GRM) that reflects genetic relationships between individuals at an unknown set of causal variants.
Because the set of causal variants is unknown, Kcausal has been approximated by the expected relatedness in the pedigree matrix KPED, which equals twice the kinship matrix Φ
The entries in the kinship matrix Φ are known as kinship coefficients. Kinship coefficient φ is the probability that a random allele from subject j is identical by descent (IBD) to an allele at the same locus from subject k. As an example, for a pair of full siblings j and k, this expected probability equals ¼ and therefore KPED[j,k] = ½. For a design that focuses exclusively on sib pairs and assuming no dominance contribution, an estimate of the additive genetic variance \({\hat{\mathrm \upsigma }}_{\mathrm{A}}^2\) is therefore two times the phenotypic correlation rP of sib pairs
When extended pedigrees are available, the entire KPED can be leveraged in a linear mixed-model (LMM) framework. In this case, the phenotype vector y is modeled as
and the genetic variance \({\hat{\mathrm \upsigma }}_{\mathrm{A}}^2\) is estimated with restricted maximum likelihood (Shaw 1987; Blangero and Almasy 1997; Lange 2002; Kang et al. 2010).
When genetic data are also available, the total IBD fraction of the genome KIBD[j,k], also referred to as \({\hat{\mathrm \uppi }}\) (\({\hat{\mathrm \uppi }}\) = 2φ), can be estimated for j and k and used in the above LMM instead of its expected value from KPED[j,k]. In other words, instead of being approximated with KPED, Kcausal is now approximated with KIBD (Visscher et al. 2006, 2007).
The advent of SNP chips resulted in the use of thousands of markers in the computation of the GRM, thus allowing to estimate heritability in samples without pedigree information (Yang et al. 2010; Lee et al. 2011). In this case, assuming an N × M genotype matrix \({\bar{\mathbf{G}}}\) with zero-column mean and unit-column variance, Kcausal is approximated with an identity-by-state (IBS) genetic covariance matrix
Given that the set of M-typed SNPs typically does not include all causal variants and/or it includes tag SNPs that are in imperfect linkage disequilibrium (LD) with said variants, the use of KIBS can lead to an underestimation of the total heritability. For unrelated individuals, this estimate reflects only the proportion of phenotypic variance captured directly or indirectly by the typed SNPs—i.e., the so-called SNP heritability \(h_{g}^{2}\) (note that SNP heritability \(h_{g}^{2}\) is smaller than total heritability h2 and is based on unrelated individuals). The GCTA software (Yang et al. 2011) uses KIBS on unrelated individuals in order to estimate \(h_{g}^{2}\). It has been shown that \(h_{g}^{2}\) can vary with minor allele frequency (MAF), LD, and genotype certainty (Speed et al. 2017). The LDAK software (Speed et al. 2012) can be used in order to accommodate these parameters in the computation of KIBS.
More recently, it has been shown that KIBD can be effectively substituted in the LMM by an IBS genetic covariance matrix KIBS>t, in which all KIBS entries below a threshold t are set to zero (Zaitlen et al. 2013). Moreover, the same authors introduced a method for the simultaneous estimation of SNP \(h_{g}^{2}\) and total heritability h2 by jointly fitting an LMM with KIBD & KIBS (or KIBS>t instead of KIBD). This way, the authors provided heritability estimates with narrower confidence intervals and showed that total heritability estimates under this approach were very similar to those under KIBD (or KIBS>t) alone.
In order for existing methods to produce meaningful heritability estimates, no population structure should be present in the studied samples (Zaitlen and Kraft 2012). Population structure can arise when individuals of different ancestry are found in the same sample and/or when individuals are admixed. Individuals from different populations tend to have different minor allele frequencies as well as different environmental exposures (Zaitlen and Kraft 2012). Because population structure correlates with environmental structure, it can inflate heritability estimates. It has been shown that, for SNP heritability (\(h_{g}^{2}\)) estimates, inclusion of principal components (PCs) as fixed effects cannot fully account for the structure bias (Browning and Browning 2011). Moreover, the differences in ancestral allele frequencies affect the computation of KIBS, which ceases to be proportional to KIBD—even for close relationships. To date, there is not a clear strategy for estimating and interpreting heritability in structured/admixed populations.
Nevertheless, an LMM that uses a relationship matrix Kγ based on local ancestry, i.e., the genetic ancestry of an individual at a particular chromosomal location, instead of genotypes has been proposed (Zaitlen et al. 2014). The authors of this method produced accurate estimates of total heritability h2 for several phenotypes in admixed African-American samples by fitting an LMM with Kγ and rescaling its regression coefficient accordingly. However, this method also has limitations, as it relies on accurate knowledge of local ancestry and assumes that samples are unrelated.
In light of the above, finding an efficient framework for total narrow-sense heritability estimates in admixed populations with high levels of relatedness remains an open question. Many experimental designs could benefit from a better understanding of how heritability estimates are affected by the simultaneous presence of population and family structure. In this work, we use extensive simulations to evaluate the performance of existing classical and LMM frameworks for estimating total (h2)—but also SNP (\(h_{g}^{2}\))—heritability in two population-based cohorts from Greenland. The Greenlanders are a population isolate with many unique characteristics, such as small census size, high levels of relatedness, and extensive population structure with ancestry from both the Inuit and Europeans (Moltke et al. 2015; Pedersen et al. 2017). Our goal is to find a way to estimate and understand narrow-sense heritability in such populations, thus gaining valuable insights into the genetic architecture of complex traits in said populations.
Materials and methods
Samples
Greenlanders
The Greenlandic subjects (N = 4659) came from two general health surveys. The first survey (Bjerregaard et al. 2003) consisted of Greenlanders living in Denmark (the BHH cohort, N = 546), recruited during 1998–1999, as well as Greenlanders living in Greenland (the B99 cohort, N = 1328), recruited during 1999–2001 as part of a general population health survey. The second survey (Jørgensen et al. 2013) consisted of Greenlanders living in Greenland (the IHIT cohort, N = 2785), recruited during 2005–2010 as part of a population health survey.
Danes
The population-based Danish sample (N = 5470) was obtained from Inter99 (Glümer et al. 2004), a randomized intervention study collected at the Research Centre for Prevention and Health. In addition, 513 (N = 1169) Danish sib pairs were identified across several available Danish cohorts, namely the population-based cohorts (i) Inter99 (Jørgensen et al. 2003) (N = 294), (ii) Helbred2006 (Thuesen et al. 2014) (N = 121), (iii) Helbred2008 (Byberg et al. 2012) (N = 38), and (iv) Helbred2010 (Aadahl et al. 2014) (N = 57), all recruited from the Research Centre for Prevention and Health, Glostrup Hospital, Denmark, as well as cohorts collected for the study of type 2 diabetes, namely (v) Vejle Diabetes Biobank (Petersen et al. 2016) (N = 570), recruited at the Vejle Hospital, Denmark, (vi) the ADDITION study (Lauritzen et al. 2000) (N = 8), recruited at the Department of General Practice at the University of Aarhus, Denmark, and (vii) SDC (Andreasen et al. 2008) (N = 81) recruited at the outpatient clinic at Steno Diabetes Center, Denmark.
Genotyping and quality control
Both the Greenlandic and the unrelated Danish samples were typed on Illumina’s Cardio-MetaboChip (Illumina, San Diego, CA, USA). The Cardio-MetaboChip includes 196,725 SNPs selected from genetic studies of cardiovascular, metabolic, and anthropometric traits (Voight et al. 2012). Moreover, the unrelated Danish samples and the Danish sib pairs were typed on Illumina’s Infinium OmniExpress chip, which includes ~710,000 markers. Standard quality control was carried out separately on each dataset with PLINK v1.9 (Chang et al. 2015) and included filtering for per-individual (‑‑mind 0.01) and per-marker (--geno 0.01) genotype missingness = 1%. The datasets passing quality control consisted of (i) 4659 Greenlanders typed on 187,181 Cardio-MetaboChip autosomal SNPs, (ii) 5470 unrelated Danes typed on 186,639 Cardio-MetaboChip and 618,037 OmniExpress autosomal SNPs, and (iii) 1169 Danes forming sib pairs typed on 609,605 OmniExpress autosomal SNPs.
Phenotype simulations
We simulated 1000 quantitative phenotypes with true total narrow-sense heritability h2 = {0.4, 0.6, 0.8} using real genetic data from 4659 Greenlanders and 6639 (5470 + 1169) Danes. Simulations were carried out separately on each dataset as follows:
First, we defined an N × C causal genotype matrix Gcausal by sampling C = 1500 SNPs from a list of all available SNPs.
Second, we sampled SNP effects, represented by a C × 1 effect vector b, from a standard normal distribution N(0,1). In order to model the relationship between the effect size of SNP i and its allele frequency fi, a genotype matrix G can be standardized according to the formula
(Speed et al. 2017). GCTA assumes a specific inverse relationship between SNP effect size and allele frequency by converting the original matrix Gcausal into a zero-column mean and unit-column variance standard score matrix \({\bar{\mathbf G}}_{{\mathbf{causal}}}\). This can be seen as a special case of the above standardizing formula for α = −1.
Third, we computed a vector of polygenic score S by multiplying \({\bar{\mathbf G}}_{{\mathbf{causal}}}\) by the corresponding effect vector b
and its additive genetic variance as
Finally, we computed the phenotype vector P by adding an environmental vector ε of i.i.d. error terms to vector S
Error terms were sampled from the distribution \({\mathrm{N}}\left( {0,{\mathrm{var}}\left( {\mathbf{S}} \right)\left( {\frac{1}{{h^2}} - 1} \right)} \right)\) (Yang et al. 2011).
In some simulations involving the Greenlanders, we also modeled the interaction between environment and ancestry by adding an interaction vector E × Anc to the sum
In particular, if \(h_{\mathrm{E} \times \mathrm{Anc}}^2\) is the proportion of phenotypic variance explained by the interaction and θInuit is the vector of proportion of Inuit ancestry, then
As a consequence, the noise terms in ε are now sampled from \({\mathrm{N}}\left( {0,{\mathrm{var}}\left( {\mathbf{S}} \right)\left( {\frac{1}{{h^2}} - 1} \right) - {\mathrm{var}}\left( {{\mathbf{E}} \times {\mathbf{Anc}}} \right)} \right)\).
In other simulations involving the Greenlandic sib pairs, we added a vector E reflecting shared environment—i.e., the household effect (Almasy and Blangero 1998)—between siblings
In particular, we drew environmental effects from a normal distribution making sure to assign the same value to all individuals belonging to the same sibling cluster. As a consequence, the noise terms in ε are now sampled from \({\mathrm{N}}\left( {0,{\mathrm{var}}\left( {\mathbf{S}} \right)\left( {\frac{1}{{h^2}} - 1} \right) - {\mathrm{var}}\left( {\mathbf{E}} \right)} \right)\).
Linear mixed model
In an LMM, the phenotype y is modeled as a mixture of fixed and random effects (i.e., the effects of the causal variants)
Assuming that b ~ N(0, \(\frac{{\upsigma}_{\mathrm{A}}^{2}}{{\mathrm{C}}}\)) and ε ~ Ν(0, \({{\upsigma }}_{\mathrm{e}}^2{\mathbf{I}}\)) under the GCTA model with α = −1, y follows a multivariate normal distribution with mean μ and variance
such that
Total narrow-sense heritability is then defined as
Because Kcausal is unknown, we approximated it with other GRMs instead, such as KIBD, KIBS, and KIBS>t.
Relationship matrices
We computed the KIBD matrix for the entire Greenlandic sample from pairwise kinship coefficients (\({\hat{\mathrm \uppi }}_{{\mathrm{j}},{\mathrm{k}}}\) = 2φj,k = ½k1,j,k + k2,j,k) using RelateAdmix (Moltke and Albrechtsen 2014) or alternatively REAP (Thornton et al. 2012) on a genotype file with MAF cutoff = 0.01. We note that the total genomic IBD estimates are generally robust to the ascertainment scheme of the array used. We subsequently identified 1465 Greenlandic sib pairs by use of empirical thresholding over k1 (IBD1, 0.3 < k1 ≤ 0.7) and k2 (IBD2, 0.1 < k2 ≤ 0.5) on the RelateAdmix output. We then recomputed the KIBD matrix for the identified sib pairs using RelateAdmix. KIBD for the Danish sib pairs was computed with the PLINK --genome flag, using MAF cutoff = 0.01. For both the Greenlandic and Danish sib pairs, we also computed a KIBD>t matrix, in which all entries below a threshold t = 0.05 were set to zero, and a \({\mathbf{K}}_{{\mathbf{IBD}}}^0\) matrix, in which all between-sib-pair values were set to zero.
KIBS and KIBS>t (t = 0.05) for both Greenlanders and Danes were computed with GCTA using a MAF cutoff = 0.01. Causal variants were removed from the computation of KIBS and KIBS>t. Because causal variants are selected from the entire list of available SNPs, we assume that they have an allele frequency distribution similar to the genotyped SNPs. We can explicitly control for this by adding --grm-adj 0 to the GCTA command line; however, this setting had no effect on our estimates and was dropped early (data not shown). For some heritability estimations, we also computed \({\mathbf{K}}_{{\mathbf{IBS}}}^ \ast\) after removing not only the causal variants, but also all variants in LD with those (i.e., applying extreme LD pruning in the vicinity of causal variants). In addition, we estimated heritability by use of a \({\mathbf{K}}_{{\mathbf{IBS}}}^{\mathbf{c}}\) matrix in which causal variants were included in the computation. Finally, household effects in Greenlandic sib pairs were captured by use of the KHH matrix, whereby 1’s were assigned to all pairs of an individual with itself and its siblings and 0’s otherwise.
Heritability estimation
Additive genetic variance \({\hat{\mathrm \upsigma }}_{\mathrm{A}}^2\) and, subsequently, total narrow-sense heritability h2 were estimated for various GRMs with the GRM-based restricted maximum likelihood (GREML) procedure implemented in GCTA and in LDAK. For the sib pairs in particular, we carried out total narrow-sense heritability estimations using (i) the IBD-based matrices (KIBD, KIBD>t, and \({\mathbf{K}}_{{\mathbf{IBD}}}^0\)) alone, (ii) the IBS-based matrices (KIBS, \({\mathbf{K}}_{{\mathbf{IBS}}}^ \ast\), and \({\mathbf{K}}_{{\mathbf{IBS}}}^{\mathbf{c}}\)) alone, (iii) KIBS together with KIBD, KIBD>t, \({\mathbf{K}}_{{\mathbf{IBD}}}^0\), or KIBS>t, and (iv) the classical sib-pair approach. When two relationship matrices were used, \({\hat{\mathrm \upsigma }}_{\mathrm{A}}^2\) was equal to the sum of the two variance components corresponding to said matrices (Zaitlen et al. 2013). In the particular case of evaluating the household effect, its variance \({\hat{\mathrm \upsigma }}_{{\mathrm{HH}}}^2\) was subtracted from the final estimation. We also estimated \({\hat{\mathrm \upsigma }}_{\mathrm{A}}^2\) after adjusting for the first 5, 10, or 20 PCs, or a proportion of Inuit ancestry (where applicable). We note that SNP array ascertainment is not expected to affect total heritability estimates, as those are dependant on robust IBD measures. Conversely, SNP heritability estimates are sensitive to the ascertainment scheme of a given genotyping platform.
Analysis settings
We ran phenotype simulations and heritability estimates on four groups of Greenlanders: (i) all samples (N = 4659), (ii) sib pairs (N = 1688), (iii) more distantly related individuals (“cousins”; N = 2615), and (iv) unrelated individuals (N = 585), as well as two separate groups of Danes: (i) unrelated individuals (N = 5470), and (ii) sib pairs (N = 1169). In both populations, the \({\hat{\mathrm \uppi }}\) threshold for identifying unrelated individuals was 0.0625. Note that we did not merge the unrelated Danes with the Danish sib pairs because, unlike the Greenlanders, they do not come from the same population-based study. We estimated heritability on the above groups using different GRMs, without and with covariates (summarized in Table 1). For the sake of simplicity, we do not show results that are nonsensical (e.g., KIBD for unrelated individuals).
Application to real data
We applied the best-performing model to real phenotypic data from the two population-based Greenlandic cohorts. All phenotypes considered were quantitative and consisted of basic anthropometric traits (height, weight, body mass index, hip circumference, waist circumference, and waist-to-hip ratio), as well as serum lipid levels (total cholesterol, HDL cholesterol, LDL cholesterol, and triglycerides). Data were rank-transformed to the quantiles of a standard normal distribution. Age and sex were included as covariates. We also carried out an empirical investigation of the impact of allele frequency and LD weighting—as defined in the LDAK model (Speed et al. 2017)—on heritability estimates. In particular, we estimated total narrow-sense heritability for the ten available traits assuming seven different genotype standardizations by setting LDAK’s parameter α = {−1.25, −1, −0.75, −0.5, −0.25, 0, 0.25}, and accounting for LD weighting. In this context, the model used by GCTA can be seen as a special case of the LDAK model by setting α = −1 and ignoring LD weighting.
Results
Admixture and relatedness in the Greenlandic and Danish data
Principal component analysis (PCA) and ADMIXTURE (Alexander et al. 2009) analysis of 4659 individuals showed that the general Greenlandic population is the result of admixture between Greenlandic Inuit and European populations, and that there is high variance in the admixture profiles of the Greenlanders (Fig. 1a) (Moltke et al. 2015; Pedersen et al. 2017). We note that assuming K = 2 ancestral components is a simplification of the admixture history of the Greenlanders as it has been previously shown that there are up to three distinct Inuit ancestral components with FST values as high as 0.04 (Moltke et al. 2015). Conversely, a sample of 5470 Danish individuals appeared largely unstructured (Fig. 1b), matching previous observations (Athanasiadis et al. 2016). We identified a large number of sib pairs in the Greenlandic sample (1465 pairs, N = 1688 individuals, Fig. S1A). We also confirmed the lack of relatedness in the 5470 unrelated Danish samples (Fig. S1B) and the presence thereof in the 513 Danish sib pairs (Fig. S1C).

a Principal component analysis of the entire Greenlandic sample (N = 4659). Individuals were colored according to their admixture proportions as estimated with ADMIXTURE assuming K = 2 ancestral components. Note that Greenlanders with a high proportion of Inuit ancestry (blue points) present further population structure along the second principal component. b Principal component analysis of the unrelated Danish sample (N = 5470). Due to persisting batch effects, the Danish data were pruned with PLINK (window size = 50; step size = 5; r2 = 0.1) before the analysis.
Identity by state in the Greenlandic and Danish data
We illustrate the intrinsic differences of IBS between admixed and unadmixed populations by plotting the IBS-based genetic covariance against the IBD-based \({\hat{\mathrm \uppi }}\) estimates from the Greenlandic and Danish sib pairs, respectively (Fig. 2). For a given kinship (e.g., full siblings), the corresponding IBS values were far more dispersed in the Greenlanders (Fig. 2a) than in the Danes (Fig. 2b). This is due to the heterogeneous admixture profiles in the Greenlanders (Fig. 1a). In other words, whereas IBS is proportional to IBD in unadmixed individuals, this does not hold for the admixed individuals.

a Scatterplot of within-pair (blue) and between-pair (black) IBS-based genetic covariance against IBD-based \({\hat{\uppi }}\) = 2φ estimates for the Greenlandic (MetaboChip) siblings. IBS was computed with GCTA and IBD was computed with RelateAdmix. b Scatterplot of within-pair (blue) and between-pair (black) IBS-based genetic covariance against IBD-based \({\hat{\uppi }}\) = 2φ estimates for the Danish (OmniExpress) siblings. IBS was computed with GCTA and IBD was computed with PLINK. A negative IBS value denotes that two individuals are related to each other less than average. An y = x dotted line is shown in red.
Heritability estimates in phenotypes with no population-specific environmental effects
We explored a number of approaches for estimating narrow-sense heritability h2 in the admixed Greenlandic and the unadmixed Danish population. We simulated quantitative traits by (i) randomly selecting 1500 causal loci with effect sizes depending on the allele frequency, such that the effect sizes of the standardized genotypes are normally distributed as assumed in the GCTA software, and (ii) adding environmental noise so that the true simulated h2 was 0.4, 0.6, or 0.8. In these simulations, all individuals were set to having the same environmental variance regardless of their ancestry. We then estimated h2 in an LMM framework for different GRMs—e.g., KIBD, KIBS, and KIBS>t and combinations thereof (Fig. 3; Figs. S2 and S3; Supplementary file). In the following paragraphs, we first report the results based on one GRM, followed by the results based on two GRMs. As a reminder, for heritability estimates to be interpreted as “total” (h2), it is required that the sample includes related individuals.

a Mean total heritability estimates and 95% confidence intervals from 1 000 simulated phenotypes with true simulated heritability of 0.4 (peach), 0.6 (yellow) and 0.8 (green) in Greenlandic sib pairs. b Mean total heritability estimates and 95% confidence intervals in Danish sib pairs. Genotype scaling parameter ɑ was set to −1 (GCTA’s standard) for both phenotype simulation and heritability estimation. KIBD: IBD-based GRM; KIBD>t: IBD-based GRM in which all entries below t = 0.05 were set to zero; KIBS: IBS-based GRM.
Total heritability estimates in sib pairs using one GRM
The use of KIBD, which captures the fraction of the genome-shared IBD (\({\hat{\mathrm \uppi }}\)), resulted in underestimates of total heritability both in the Greenlandic and the Danish sib pairs (Fig. 3). Total heritability in the Greenlandic sib pairs was also underestimated when we used KIBD>t (Fig. 3a) or \({\mathbf{K}}_{{\mathbf{IBD}}}^0\) (Fig. S2A). Conversely, using KIBD>t or \({\mathbf{K}}_{{\mathbf{IBD}}}^0\) in the Danish sib pairs did not result in any significant downward biases (Fig. 3b; Fig. S2B). These results were insensitive to the method of IBD inference: both our method of choice (RelateAdmix) and an alternative (REAP) returned similar results (data not shown).
For closely related unadmixed individuals, IBS is proportional to IBD, and therefore estimates based on KIBS will correspond to the total narrow-sense heritability as well (Hayes et al. 2009). Indeed, true simulated h2 was fully recovered in the Danish sib pairs when KIBS was used (Fig. 3b), but showed consistent downward biases across all simulated h2 values in the Greenlandic sib pairs (Fig. 3a). Removing all SNPs with any LD with the causal variants (r2 = 0) from the GRM (i.e., using the \({\mathbf{K}}_{{\mathbf{IBS}}}^ \ast\) matrix) returned lower yet still comparable h2 estimates in both the Greenlandic (Fig. S2A) and Danish sib pairs (Fig. S2B). Conversely, when causal variants were included in the computation of the GRM (i.e., using the \({\mathbf{K}}_{{\mathbf{IBS}}}^{\mathbf{c}}\) matrix), then \({\mathbf{K}}_{{\mathbf{IBS}}}^{\mathbf{c}} \cong\) Kcausal and, consequently, h2 was recovered (Fig. S4).
Total heritability estimates in sib pairs using two GRMs
The use of two GRMs (e.g., KIBD & KIBS) for heritability estimates is meant to leverage datasets in which both closely and more distantly related individuals are present (Zaitlen et al. 2013). However, when we applied this approach to the entire Greenlandic dataset (N = 4659), we observed a downward bias in total heritability estimates for the KIBD & KIBS model, while using KIBS>t & KIBS erroneously returned heritability estimates near 1.00 regardless of the true simulated value (Fig. S5). Nevertheless, when we performed the two-GRM analysis on the 1465 Greenlandic sib pairs alone, the true simulated h2 was almost perfectly recovered for all GRM combinations, with estimates showing only a minor downward bias (Fig. 3a; Fig. S2A). Interestingly, these models outperform the classical sib-pair analysis as evidenced by their lower root-mean-square deviation (Table S1; Fig. S3). Extending the sample to include more distant relatives (“cousins”; \(\hat \pi\) = [0.15, 0.67]; N = 2615) resulted in underestimates of the total heritability (Fig. S6), implying that the KIBD & KIBS model performs efficiently only on first-degree relatives when admixture is present. We therefore further examined the KIBD & KIBS model in the following section, focusing our attention on the sib pairs.
Total heritability estimates of phenotypes with shared environment between siblings
When we performed the two-GRM analysis of phenotypes that included the effect of shared environment on the 1465 Greenlandic sib pairs, the true simulated h2 was inflated (squares in Fig. S7). The stronger the household effect, the higher the inflation. Notably, the inclusion of 10 PCs or a proportion of Inuit ancestry did not have any noticeable effect on the estimates (circles and triangles in Fig. S7). However, when we performed the KIBD & KIBS & KHH analysis and subtracted the variance of the household effect, we were able to recover almost perfectly the total heritability estimates (diamonds in Fig. S7).
SNP heritability estimates in unrelated individuals
As previously mentioned, the use of KIBS on unrelated individuals yields SNP (\(h_g^2\)) rather than total heritability (h2) estimates (Yang et al. 2010). Bearing this in mind, we estimated \(h_g^2\) in both unrelated Greenlanders and unrelated Danes (Fig. S8; Table S2). In all cases, we found that \(h_g^2\) < h2, as expected. For the Danish samples in particular, the MetaboChip \(h_g^2\) was smaller than the OmniExpress \(h_g^2\).
Heritability estimates in phenotypes with population-specific environmental effects
In all of the above simulations, we assumed that the environmental component was independent of ancestry. However, when we added to the simulated phenotypes an environmental component correlating with ancestry, the use of KIBS in the unrelated Greenlanders led to overestimates of SNP heritability \(h_g^2\), despite adjusting for population structure (Table S2).
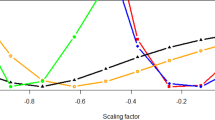
Adjusting the KIBD model for either the first 10 PCs or a proportion of Inuit ancestry produced a consistent yet uninterpretable pattern along the \(\frac{{h_{\mathrm{E} \times \mathrm{Anc}}^2}}{{h^2}}\) ratio (Fig. S9). On the contrary, adjusting the KIBD & KIBS model for the same covariates produced a predictable as well as interpretable pattern across all choices for the \(\frac{{h_{\mathrm{E} \times \mathrm{Anc}}^2}}{{h^2}}\) ratio (Fig. 4). In particular, estimates from the KIBD & KIBS model without covariates corresponded to the inflated quantity \(\frac{{\sigma _{\mathrm{A}}^2 \, + \ \sigma _{{\mathrm{E}} \times {\mathrm{Anc}}}^2}}{{\sigma _{\mathrm{A}}^2 \, + \ \sigma _{{\mathrm{E}} \times {\mathrm{Anc}}}^2 + \ \sigma _{\mathrm{e}}^2}}\) (squares in Fig. 4). After adjustment for ancestry, the resulting estimates corresponded to removing the environmental interaction component (\(\sigma _{{\mathrm{E}} \times {\mathrm{Anc}}}^2\)) from the numerator and denominator of the formula (i.e., \(\frac{{\sigma _{\mathrm{A}}^2}}{{\sigma _{\mathrm{A}}^2 \, + \ \sigma _{\mathrm{e}}^2}}\); circle and triangle points in Fig. 4). We note that adjustment for 10 PCs was equivalent to adjustment for a proportion of Inuit ancestry (Fig. 4).

The relationship between the environment-by-ancestry (E × Anc) interaction and the true simulated heritability is quantified by the \(h_{\mathrm{E} \times \mathrm{Anc}}^2\)/h2 ratio on the x axis. The models with the covariates (circles and triangles) correspond to estimates of conditional total heritability after adjusting for the E × Anc effect (dotted line). Further rescaling of the conditional heritability estimates by 1 − \(\frac{{\hat \sigma _{\mathrm{E} \times \mathrm{Anc}}^2}}{{\sigma _P^2}}\) returns the marginal simulated heritability (dashed line). \(h_{\mathrm{E} \times \mathrm{Anc}}^2\): proportion of variance captured by the E × Anc interaction; Inuit adm. :proportion of Inuit admixture. Adjustment for 5 or 20 first PCs returned virtually identical results (data not shown).
Thanks to the interpretability of the resulting “conditional” estimates, we were able to recover the true simulated heritability \(\frac{{{{\upsigma }}_{\mathrm{A}}^2}}{{{{\upsigma }}_{\mathrm{A}}^2 \, + \ {{\upsigma }}_{{\mathrm{E}} \times {\mathrm{Anc}}}^2 \, + \ {{\upsigma }}_{\mathrm{e}}^2}}\) – i.e., the “marginal” heritability (Weissbrod et al. 2018) (diamond points in Fig. 4). We achieved this by rescaling the conditional estimates by a factor of \(1 - \frac{{{\hat{\mathrm \upsigma }}_{{\mathrm{E}} \times {\mathrm{Anc}}}^2}}{{{{\upsigma }}_{\mathrm{P}}^2}}\), where \({\hat{\mathrm \upsigma }}_{{\mathrm{E}} \times {\mathrm{Anc}}}^2\) is an estimate of the environmental interaction variance computed as \({\hat{\mathrm \upsigma }}_{{\mathrm{E}} \times {\mathrm{Anc}}}^2 = {{\upsigma }}_{\mathrm{P}}^2 - {{\upsigma }}_{{\hat{\mathrm P}}}^2\). \({\hat{\mathrm P}}\) is the phenotype residuals after regressing out the effect of structure captured either by admixture proportions or the first two principal components.
Application to real phenotypes
We applied the best model (i.e., KIBD & KIBS & 10 PCs) and the follow-up PCA-based adjustment to ten quantitative traits in the 1465 Greenlandic sib pairs (Table 2). Not all phenotypes were equally sensitive to the PCA-based adjustment of their estimated conditional heritability, implying trait-specific environment-by-ancestry interactions. The GCTA model accommodates only one type of genotype standardization (α = −1), resulting in strong assumptions about the distribution of effect sizes. We therefore also used the LDAK model (Speed et al. 2017) and found that the optimal α value for genotype standardization varied across traits with most phenotypes supporting α ≥ −0.5 (Fig. S10; Table S3). Total heritability estimates under the LDAK model (Speed et al. 2017) were generally higher than under GCTA (Table 2; Table S3), with the greatest difference observed for height (0.657 ± 0.042 for GCTA against 0.786 ± 0.041 for LDAK). In addition, heritability estimates in eight out of ten real phenotypes were smaller in the Greenlanders than in their European or Mexican counterparts estimated with similar models.
Discussion
In this work, we explored the performance of existing methods for heritability estimates in the admixed Greenlandic population. Our goal was to propose a framework for unbiased heritability estimates in datasets where both population and family structure are notably present, as well as a way to interpret the resulting estimates. Even though the main focus is on total narrow-sense heritability (h2), we also report the results for SNP heritability (\(h_g^2\)), a quantity that has gained a lot of attention in the past decade due to the availability of GWAS data (Yang et al. 2010; Lee et al. 2011; Browning and Browning 2011).
Through extensive simulations, we observed that all LMMs using one GRM led to downward biases in total heritability estimates when applied to family data from Greenland. Common choices of GRM, such as KIBD and KIBS, led to underestimates of total heritability in Greenlandic sib pairs, whereas no such biases were generally observed for the Danish sib pairs, indicating that inheriting DNA from different ancestral populations (i.e., admixture) exerts a biasing effect on both IBD- and IBS-based estimates. Even though this is not surprising for the IBS-based estimates, as the IBS ~ IBD assumption does not hold for the Greenlanders, it is not very clear why IBD-based heritability estimates are also affected by admixture. One possible explanation could be that IBD estimates become less accurate for more distantly related pairs, and therefore including them in the LMM introduces noise as evidenced by the underperformance of the full KIBD matrix in the Danes.
We also observed that an LMM with two GRMs (KIBD & KIBS), a method designed to work on data with notable presence of family structure (Zaitlen et al. 2013), also led to downward biases in total heritability estimates when applied to the entire dataset from Greenland. However, when the same analysis focused on the Greenlandic sib pairs, it returned nearly unbiased heritability estimates. This could be due to the fact that, by restricting the analysis to the sib pairs, we controlled more efficiently for the noise that comes from between-sib-pair IBD estimates (Moltke and Albrechtsen 2014). The KIBD & KIBS model performs well under the assumption that shared environment among siblings has a negligible effect. We show that a nonzero household effect can potentially inflate total heritability estimates, but this effect can be accounted for with the inclusion of a shared environment matrix KHH—at least in the simulation setting. We note that the KIBD & KIBS model outperformed the KIBD model in the Danes, rendering more advisable the use of two GRMs in total narrow-sense heritability estimates in unadmixed populations too.
When there is no environmental correlation with ancestry, the KIBD & KIBS (or any other combination of one IBD- and one IBS-based GRM) model provides an accurate estimate of the true heritability matched only by the classical sib-pair analysis. However, we expect environmental structure to exert an inflating effect on heritability estimates due to its correlation with genetic structure. We found that adjusting for structure did not remove the inflation. Nevertheless, we provide a way to interpret the resulting total heritability estimates from the KIBD & KIBS & 10 PC models, as well as a way to adjust for the inflation. In particular, this inflated quantity is referred to as “conditional heritability” in a recent paper (Weissbrod et al. 2018), after adjusting for model covariates like in our case. We observed that, under the KIBD & KIBS & 10 PC models, the resulting conditional heritability estimate will be inflated by a factor of 1/(1 − \(h_{\mathrm{E} \times \mathrm{Anc}}^2\)), and we propose an adjustment that accounts efficiently for this inflation in order to retrieve the “marginal heritability” (Weissbrod et al. 2018). Finally, we note that the classical sib-pair approach will also produce inflated estimates when there is interaction with the environment, and that adjustment for PCs will not fix the issue.
As for the total narrow-sense heritability estimates of the real phenotypes obtained with the best model (KIBD & KIBS & 10 PCs), we observe that in some occasions, these are lower for the Greenlandic population than for European populations. A notable example is height, for which total heritability in the Greenlanders was estimated to be 0.656 ± 0.042 (0.611 after the PCA-based adjustment), whereas in unadmixed Europeans it was estimated at 0.860 (Visscher et al. 2007). We believe that this could be due to the reduced genetic diversity observed in the Greenlanders as a consequence of their particular population history, which included an extreme and prolonged bottleneck in recent times (Moltke et al. 2015; Pedersen et al. 2017), even though we did observe a notable increase when LD weighting was included in the estimation model according to LDAK (Speed et al. 2017).
Finally, our SNP heritability (\(h_g^2\)) estimates in unrelated Greenlanders could be inflated due to genetic structure as reported previously (Browning and Browning 2011), even though we could not assess the level of inflation. In any case, SNP heritability estimates in the Greenlanders should be interpreted with caution because, as we saw, IBS measures are affected by admixture that can lead to artificially increased levels of LD between causal and typed markers.
It is important to note that this work does not solve all problems of heritability estimates in admixed populations. Our work should be viewed as a first attempt to explore the problem, and therefore the insights and solutions we provide here might not apply in all cases. Additional work is warranted in order to, e.g., model more accurately complex patterns of environmental stratification—similar to the household effect (Almasy and Blangero 1998)—and exposure of the same genetic ancestry to different environmental backgrounds. In addition, even though there are multiple methods for improving heritability estimates using, for example, LD score regression or partitioning SNPs according to allele frequencies (Gazal et al. 2017; Evans et al. 2018), we have not explored them here as they are harder to implement in admixed populations, where LD patterns and allele frequencies can be misspecified.
In summary, we advise against the use of KIBD or KIBS alone for total narrow-sense heritability estimates in populations with substantial levels of population and family structure. Instead, KIBD & KIBS & 10 PCs on a subset with high relatedness (preferably sib pairs) are advisable, given that KIBD can now be efficiently computed for admixed populations (Thornton et al. 2012; Moltke and Albrechtsen 2014), with the caveat that the method could be capturing sizeable levels of shared environment among siblings. In any case, the resulting conditional h2 estimates should be viewed as potentially inflated by a factor that we estimated at 1/(1 − \(h_{\mathrm{E} \times \mathrm{Anc}}^2\)), and an additional PCA-based adjustment should be carried out in order to recover the marginal total heritability estimate.
Data availability
The Greenlandic MetaboChip data have been submitted to the European Genome-Phenome Archive (https://ega-archive.org/) under accession number EGAS00001002641.
References
Aadahl M, Linneberg A, Møller TC, Rosenørn S, Dunstan DW, Witte DR et al. (2014) Motivational counseling to reduce sitting time: a community-based randomized controlled trial in adults. Am J Prev Med 47:576–586
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211
Andreasen CH, Stender-Petersen KL, Mogensen MS, Torekov SS, Wegner L, Andersen G et al. (2008) Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes 57:95–101
Athanasiadis G, Cheng JY, Vilhjálmsson BJ, Jørgensen FG, Als TD, Le Hellard S et al. (2016) Nationwide Genomic Study in Denmark Reveals Remarkable Population Homogeneity. Genetics 204:711–722
Bjerregaard P, Curtis T, Borch-Johnsen K, Mulvad G, Becker U, Andersen S et al. (2003) Inuit health in Greenland: a population survey of life style and disease in Greenland and among Inuit living in Denmark. Int J Circumpolar Health 62(Suppl 1):3–79
Blangero J, Almasy L (1997) Multipoint oligogenic linkage analysis of quantitative traits. Genet Epidemiol 14:959–964
Browning SR, Browning BL (2011) Population structure can inflate SNP-based heritability estimates. Am J Hum Genet 89:191–193
Byberg S, Hansen A-LS, Christensen DL, Vistisen D, Aadahl M, Linneberg A et al. (2012) Sleep duration and sleep quality are associated differently with alterations of glucose homeostasis. Diabet Med 29:e354–e360
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7
Evans LM, Tahmasbi R, Vrieze SI, Abecasis GR, Das S, Gazal S et al. (2018) Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat Genet 50:737–745
Fisher RA (1918) The correlation between relatives on the supposition of Mendelian inheritance. Trans Roy Soc Edin 52:399–433
Gazal S, Finucane HK, Furlotte NA, Loh P-R, Palamara PF, Liu X et al. (2017) Linkage disequilibrium–dependent architecture of human complex traits shows action of negative selection. Nat Genet 49:1421–1427
Glümer C, Carstensen B, Sandbaek A, Lauritzen T, Jørgensen T, Borch-Johnsen K et al. (2004) A Danish diabetes risk score for targeted screening: the Inter99 study. Diabetes Care 27:727–733
Hayes BJ, Visscher PM, Goddard ME (2009) Increased accuracy of artificial selection by using the realized relationship matrix. Genet Res 91:47–60
Jørgensen ME, Borch-Johnsen K, Stolk R, Bjerregaard P (2013) Fat distribution and glucose intolerance among Greenland Inuit. Diabetes Care 36:2988–2994
Jørgensen T, Borch-Johnsen K, Thomsen TF, Ibsen H, Glümer C, Pisinger C (2003) A randomized non-pharmacological intervention study for prevention of ischaemic heart disease: baseline results Inter99. Eur J Cardiovasc Prev Rehabil 10:377–386
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong S-Y, Freimer NB et al. (2010) Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42:348–354
Lange K (2002) Mathematical and statistical methods for genetic analysis, 2nd edn. Springer-Verlag, New York
Lauritzen T, Griffin S, Borch-Johnsen K, Wareham NJ, Wolffenbuttel BH, Rutten G et al. (2000) The ADDITION study: proposed trial of the cost-effectiveness of an intensive multifactorial intervention on morbidity and mortality among people with Type 2 diabetes detected by screening. Int J Obes Relat Metab Disord 24(Suppl 3):S6–S11
Lee SH, Wray NR, Goddard ME, Visscher PM (2011) Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet 88:294–305
Mamtani M, Kulkarni H, Dyer TD, Almasy L, Mahaney MC, Duggirala R et al. (2014) Waist circumference is genetically correlated with incident Type 2 diabetes in Mexican-American families. Diabet Med 31:31–35
Moltke I, Albrechtsen A (2014) RelateAdmix: a software tool for estimating relatedness between admixed individuals. Bioinformatics 30:1027–1028
Moltke I, Fumagalli M, Korneliussen TS, Crawford JE, Bjerregaard P, Jørgensen ME et al. (2015) Uncovering the genetic history of the present-day Greenlandic population. Am J Hum Genet 96:54–69
Pedersen C-ET, Lohmueller KE, Grarup N, Bjerregaard P, Hansen T, Siegismund HR et al. (2017) The effect of an extreme and prolonged population bottleneck on patterns of deleterious variation: insights from the Greenlandic Inuit. Genetics 205:787–801
Petersen ERB, Nielsen AA, Christensen H, Hansen T, Pedersen O, Christensen CK et al. (2016) Vejle Diabetes Biobank—a resource for studies of the etiologies of diabetes and its comorbidities. Clin Epidemiol 8:393–413
Shaw RG (1987) Maximum-likelihood approaches applied to quanti- tative genetics of natural populations. Evolution 41:812–826
Speed D, Cai N, Consortium UCLEB, Johnson MR, Nejentsev S, Balding DJ (2017) Reevaluation of SNP heritability in complex human traits. Nat Genet 49:986–992
Speed D, Hemani G, Johnson MR, Balding DJ (2012) Improved heritability estimation from genome-wide SNPs. Am J Hum Genet 91:1011–1021
Thornton T, Tang H, Hoffmann TJ, Ochs-Balcom HM, Caan BJ, Risch N (2012) Estimating kinship in admixed populations. Am J Hum Genet 91:122–138
Thuesen BH, Cerqueira C, Aadahl M, Ebstrup JF, Toft U, Thyssen JP et al. (2014) Cohort profile: the Health2006 cohort, research centre for prevention and health. Int J Epidemiol 43:568–575
van Dongen J, Willemsen G, Chen W-M, de Geus EJC, Boomsma DI (2013) Heritability of metabolic syndrome traits in a large population-based sample. J Lipid Res 54:2914–2923
Vattikuti S, Guo J, Chow CC (2012) Heritability and genetic correlations explained by common SNPs for metabolic syndrome traits. PLoS Genet 8:e1002637
Visscher PM, Hill WG, Wray NR (2008) Heritability in the genomics era–concepts and misconceptions. Nat Rev Genet 9:255–266
Visscher PM, Macgregor S, Benyamin B, Zhu G, Gordon S, Medland S et al. (2007) Genome partitioning of genetic variation for height from 11,214 sibling pairs. Am J Hum Genet 81:1104–1110
Visscher PM, Medland SE, Ferreira MAR, Morley KI, Zhu G, Cornes BK et al. (2006) Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet 2:e41
Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS et al. (2012) The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet 8:e1002793
Weissbrod O, Flint J, Rosset S (2018) Estimating SNP-based heritability and genetic correlation in case-control studies directly and with summary statistics. Am J Hum Genet 103:89–99
Wright S (1921) Systems of mating. I. the biometric relations between parent and offspring. Genetics 6:111–123
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR et al. (2010) Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42:565–569
Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88:76–82
Zaitlen N, Kraft P (2012) Heritability in the genome-wide association era. Hum Genet 131:1655–1664
Zaitlen N, Kraft P, Patterson N, Pasaniuc B, Bhatia G, Pollack S et al. (2013) Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet 9:e1003520
Zaitlen N, Pasaniuc B, Sankararaman S, Bhatia G, Zhang J, Gusev A et al. (2014) Leveraging population admixture to characterize the heritability of complex traits. Nat Genet 46:1356–1362
Acknowledgements
We would like to thank the staff and participants of the IHIT, B99, and BBH cohorts facilitating this study. The staff and steering committees from Research Centre for Prevention and Health, Glostrup, Denmark, from the ADDITION-DK study, University of Aarhus, Denmark, from Vejle Diabetes Biobank, Vejle Hospital, Denmark, and from Steno Diabetes Center, Gentofte, Denmark are acknowledged for their contribution to collecting and characterizing the Danish cohorts.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics
The two Greenlandic surveys received ethics approval from the Commission for Scientific Research in Greenland (project 2011-13, ref. no. 2011-056978; project 2013-13, ref. no. 2013-090702), and the Danish studies were approved by the local ethics committees (protocol ref. no. H-3-2012-155). All studies were conducted in compliance with the Helsinki Declaration II, and all participants gave their written consent after being informed about the study orally and in writing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Giorgio Bertorelle
Supplementary information
Rights and permissions
About this article

Cite this article
Athanasiadis, G., Speed, D., Andersen, M.K. et al. Estimating narrow-sense heritability using family data from admixed populations. Heredity 124, 751–762 (2020). https://doi.org/10.1038/s41437-020-0311-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-0311-2
This article is cited by
-
Rare genetic variants explain missing heritability in smoking
Nature Human Behaviour (2022)
