
Abstract
Traditional models of hybrid zones have assumed relatively low hybrid fitness, and thus focussed more on interspecific gene flow than on hybrid dispersal. Therefore, when hybrids have high fitness and the potential for autonomous dispersal, we have limited understanding of whether hybrid dispersal or repeated local hybrid formation is more important for maintaining hybrid zones. The invasive hybrid cattail Typha × glauca occupies an extensive hybrid zone in northeastern North America where it is sympatric with its progenitors T. latifolia and T. angustifolia. We characterized genetic diversity and genetic structure of the three taxa across a broad spatial scale where the maternal parent is relatively rare, and tested the hypothesis that the hybrid shows stronger evidence of gene flow than its progenitor species, particularly among disturbed sites (ditches) compared with established wetlands. Support for this hypothesis would suggest that dispersal, rather than repeated local formation, is more important for maintaining hybrid zones. Within each taxon, genetic differentiation among ditches was comparable to that among wetlands, although clonal richness was consistently greater in ditches, suggesting more frequent seed establishment. Genetic structure across sites was more pronounced in the hybrid compared with either progenitor species. Overall, our data reflect relatively low gene flow in hybrids, and suggest that hybrids are more likely to be created in situ than to be introduced from other sites. Despite the high fitness of invasive T. × glauca and its potential for autonomy, local processes appear more important than dispersal in maintaining this hybrid zone.
Similar content being viewed by others


Introduction
Interspecific hybridization is common across many plant taxa, and can have a variety of important ecological and evolutionary consequences; these can include greater phenotypic diversity, extinction of progenitor species and speciation (Whitney et al. 2010, Todesco et al. 2016, Goulet et al. 2017). Some outcomes of hybridization increase diversity, for example, through increased heterozygosity, the formation of stable hybrid zones, and adaptive introgression (Barton and Hewitt 1985, Stelkens et al. 2014, Norris et al. 2015). Of growing concern to conservation biologists, however, are those outcomes that decrease species diversity and potentially lead to species extinctions. Threats to the survival of parent species following ongoing hybridization have most commonly been attributed to genetic swamping (Todesco et al. 2016), which occurs when local parental genotypes are replaced by hybrid genotypes. An additional threat is demographic swamping, which arises when hybrids with relatively low fitness are formed repeatedly, and thus one or both parent species waste reproductive effort to an extent that limits population growth below the rate required for ongoing replacement (Wolf et al. 2001). The likelihood of demographic or genetic swamping resulting in the extinction of progenitor species depends on factors that influence the size and stability of hybrid zones.
Hybrid zones are typically described as either clinal zones, which in many cases are maintained by a balance between dispersal of progenitors into the zone and selection against hybrids within the zone, or mosaic zones, in which the fitness of progenitors and hybrids varies according to environmental conditions (Barton and Hewitt 1985; Harrison 1993, Burns et al. 2019). Models of hybrid zones typically incorporate patterns of dispersal (e.g., dispersal-independent and dispersal-dependent clines, Barton and Hewitt 1985), but because these models assume that hybrids have relatively low fitness in many or all environments, dispersal in this context refers to the parental genotypes. Numerous studies have therefore investigated ways in which parental dispersal and interspecific gene flow can influence hybrid zones (e.g., Dodd and Afzal-Rafii 2004, Lexer et al. 2005, Arnold et al. 2010, De La Torre et al. 2015). Meanwhile, it has become increasingly apparent that hybrids are not always less fit than their parental species (e.g., Burke et al. 1998, Fritz et al. 2006, Marques et al. 2011), and under these conditions, hybrid zones could theoretically expand through autonomous hybrid dispersal. However, this possibility has seldom been investigated (although see Bailey et al. 2009, Sloop et al. 2011), and as a result, we have a fairly limited understanding about the importance of hybrid dispersal among sites versus repeated hybrid formation within sites when zones include hybrids with relatively high fitness. These two processes could have profoundly different impacts on both the long-term sustainability and the expansion rates of hybrid zones: the former should allow for expansion, regardless of the distribution of one or both parent species, whereas the latter should mean that hybrid establishment within a site largely relies on the presence of both parent species.
The hybridizing species complex of cattails (Typha spp.; Typhaceae) in northeastern North America includes T. latifolia L., which is native to North America (Grace and Harrison 1986), T. angustifolia L., which was historically introduced from Europe (Ciotir et al. 2013a, Ciotir and Freeland 2016), and their hybrid, T. × glauca (Grace and Harrison 1986). Although there are large geographical regions in China and Europe where T. angustifolia and T. latifolia are sympatric, but rarely or never hybridize (Zhou et al. 2016, Ciotir et al. 2017), the hybrid zone occupied by T. × glauca in North America is extensive, covering a large area that includes the Upper Midwest region of the United States, southern Ontario and Quebec in Canada and Atlantic Canada (Galatowitsch et al. 1999, Freeland et al. 2013, Shih and Finkelstein 2008). Here, F1 hybrids arise following asymmetric hybridization in which T. angustifolia is pollinated by T. latifolia (Ball and Freeland 2013, Pieper et al. 2017). Hybrids can backcross bidirectionally with T. angustifolia and asymmetrically with T. latifolia, and advanced-generation hybrids are fertile although they have reduced fertility relative to F1 hybrids and progenitors (Pieper et al. 2017). All three taxa also reproduce asexually, leading to clonal spread within sites.
Typha × glauca is of conservation concern because it shows evidence of heterosis in areas where it dominates wetlands (Bunbury-Blanchette et al. 2015, Zapfe and Freeland 2015, Szabo et al. 2018), and appears to be displacing its parental species (Freeland et al. 2013, Pieper et al. 2017). In addition, these invasive hybrids reduce native plant and macroinvertebrate diversity (Tuchman et al. 2009, Lawrence et al. 2016), and alter nutrient cycling and community structure (Tuchman et al. 2009, Larkin et al. 2012, Geddes et al. 2014, Lishawa et al. 2014, Lawrence et al. 2017). These far-reaching effects highlight the need to understand mechanisms that may help to explain the maintenance and expansion of hybrid cattail. Typha angustifolia, the maternal parent of the hybrids, is the rarest of the three taxa across much of the hybrid zone (Kirk et al. 2011, Freeland et al. 2013, Pieper et al. 2018), and because it experiences genetic swamping through hybridization and backcrossing (Pieper et al. 2017), the ongoing local recruitment of F1 hybrids may become increasingly limited by a shortage of maternal plants. Hybrids could nevertheless persist through clonal reproduction, or, in the absence of hybrid breakdown (the decreased fitness of advanced-generation hybrids relative to F1s due to genome incompatibilities), through advanced-generation hybrids. In addition, if F1 hybrid seeds from maternal T. angustifolia disperse to new sites prior to germination, F1 hybrids could be maintained throughout the hybrid zone even if the distribution of T. angustifolia becomes increasingly patchy. This, in turn, could have important implications for the persistence and geographical extent of the T. × glauca hybrid zone because it would provide some autonomy for hybrid dispersal and establishment.
We investigated Typha hybrid zone dynamics at both local and landscape scales by conducting a population genetics study of Typha spp. from sites in northern Michigan, USA, and southeastern Canada, spanning a distance of ~1700 km. The prevalence of hybrids combined with the paucity of T. angustifolia (the maternal progenitor) throughout a large portion of this area led us to hypothesize that the range expansion of T. × glauca has been facilitated by ongoing dispersal of F1 hybrid seeds. We predicted that this in turn should lead to (1) less genetic differentiation and spatial genetic structure in hybrids compared with progenitors, and (2) greater clonal richness within sites in hybrids compared with progenitors, reflecting relatively high levels of seed dispersal and subsequent establishment. This study will help us to understand whether effective dispersal across the landscape has facilitated the spread of invasive T. × glauca hybrids, or whether local-scale events, such as repeated local hybrid formation and competitive superiority of hybrids, seem more important in maintaining the hybrid zone.
Methods
Sampling
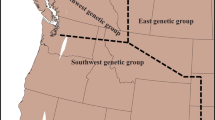
In June 2013, we sampled 22 stands along an ~1700-km longitudinal transect from Cheboygan, Michigan to Halifax and Nova Scotia (Fig. 1, Table S1). Typha flourish in a range of habitats that include wetlands plus relatively disturbed areas such as roadside ditches. To allow for the possibility of hybrids being more successful at colonizing disturbed habitats, as is often the case for invasive species (Hansen and Clevenger 2005, Alston and Richardson 2006), we sampled one wetland and one ditch site in each of 11 regions separated by ~200 km (Fig. 1). The ditch and wetland at Montmagny, Quebec, were 130 km apart, but in all other regions, the wetland and ditch site were within 20 km of each other. Wetland sites occurred at the edges of lakes in open-marsh habitats, and ditches were adjacent to highways and backed by fields. Although stand selection was largely opportunistic, i.e., accessible stands in each target region, stands ideally contained a variety of Typha morphotypes likely to include at least two Typha taxa, and had to be large enough to permit sampling of at least 100 ramets (shoots) at 3-m intervals (see below). The first criterion was not always met, particularly in eastern Canada where T. angustifolia and T. × glauca are relatively rare (Freeland et al. 2013).

Sizes of circles indicate different site types rather than reflecting sample size. Wetland and ditch sites that represent one sampling region are connected by a solid black line, and paired wetland and ditch sites are located within 20 km of each other.
At each site, transects were laid out at 3-m intervals running perpendicular to the shoreline (wetlands) or road (ditches). Leaves were collected every 3 m along each transect when possible, although some transects contained gaps of up to 10 m where cattails were not present. In wetlands, sampling began at a non-random point at the edge of a continuous cattail patch. In ditch sites, we sampled both the channel of the ditch and the adjacent areas where Typha grew in exposed soil. Ditch sites were usually quite narrow, and therefore plants were sampled from several short (up to 10 m) transects, whereas wetland sites often allowed for fewer, longer (up to 60 m) transects. All samples were collected on foot, which limited transect length in wetland sites if the water was too deep to continue sampling. In some ditch stands, it was necessary to sample several small stands as a single stand. In these instances, all stands from which samples were pooled were within 500 m of each other. Where the stand was less than 6-m wide (only at ditch sites), ramets were sampled at a random distance (0–6 m) from the edge of the stand to avoid repeatedly sampling from the same elevation relative to the bottom of the ditch on each transect (e.g., not always directly in the middle or always at the edge).
Genotyping and assignment to the taxon
A leaf section ~5 cm in length was collected from the youngest leaf of each sampled ramet. Each leaf sample was placed in an individual paper envelope and then stored in zippered plastic bags filled with Sorbead orange silica beads (eCompressedair, Oklahoma) for desiccation. On return to the lab, leaves were dried at 60 °C for 8 h to ensure complete desiccation before being stored at −20 °C. Approximately 3 cm of dried leaf tissue from each sample was ground using a Retsch® MM300 mixer mill (Haan, Germany). DNA was extracted by using either E.Z.N.A Plant DNA Kit (Omega Bio-Tek, Inc., Georgia, USA) or GeneJET Plant Genomic DNA Purification Kit (Thermo Scientific) following the manufacturer’s instructions for dried material, and eluted in deionized water to a final volume of 100 μl. Ramets were genotyped at seven microsatellite loci: TA3, TA5, TA7, TA8, TA20 (Tsyusko-Omeltchenko et al. 2003), TL213 and TL305 (Ciotir et al. 2013b). See Pieper et al. (2018) for detailed genotyping methods.
Ramets were assigned to taxon based on the alleles at loci TA3, TA5, TA8 and TA20, which are specific to either T. latifolia or T. angustifolia (Snow et al. 2010, Kirk et al. 2011). Ramets containing only alleles specific to either T. latifolia or T. angustifolia at all four loci were assigned to that species, whereas ramets containing a mixture of alleles from both species were considered hybrids. Hybrids were provisionally assigned to F1s, backcrosses to one of the progenitor species or advanced-generation hybrids using criteria following Kirk et al. (2011), although, unless otherwise noted, all hybrid classes were grouped together for analyses. Assignments of ramets to genets were based on all seven loci. Ramets with the same multilocus genotype (MLG) within a site were considered to be from the same clone (genet), and duplicate MLGs from within a site were removed before conducting all analyses, except for clonal richness (see below). The total dataset comprised 1926 ramets and 682 MLGs; however, for most analyses, only sites with ≥10 MLGs for a particular taxon were included (see below; Table S1). We used this relatively low cut-off because although we sampled an average of 87.5 plants from each site, these yielded an average of only 31 MLGs per site (Table S1, and see below) despite sampling plants that were separated by a minimum of 3 m.
We investigated potential deviations from Hardy–Weinberg equilibrium (HWE) using exact tests (Guo and Thompson 1992) for each locus × site combination, and tested for linkage disequilibrium (LD) between each pair of loci in each site using log-likelihood-ratio tests (Goudet et al. 1996). Analyses of HWE and LD were conducted using GenePop on the web (Raymond and Rousset 1995), using a dememorization number of 1000, 100 batches and 1000 iterations per patch as the Markov chain parameters for each test. Bonferroni corrections were applied to multiple comparisons when assessing the statistical significance of deviations from HWE (# sites × 7 loci) and LD (# sites × 21 locus pairs) (Rice 1989).
Genetic differentiation and spatial genetic structure
We used a negative binomial generalized linear mixed-effect model (GLMM) to determine whether there were differences in taxonomic composition between ditches and wetlands. The number of MLGs of a particular taxon was the response variable (although the conclusions were the same when the model was run using the number of ramets as the response variable), and the fixed effects were site type (ditch or wetland), taxon and the interaction between site and taxon. To evaluate whether taxonomic composition differed between site types, the model included the interaction between site type and taxon. Geographic region was included as a random effect to account for possible variation in the distribution of the taxa. We used a negative binomial distribution in the GLMM because of overdispersion in the response variable (Zuur et al. 2009). The model was run using the ‘glmer.nb’ function in the ‘lme4’ package (v. 1.1-13; Bates et al. 2016) in R (v. 3.4.0; R Core Development Team 2016). Significance tests for fixed effects were conducted using likelihood-ratio tests with type III sums of squares as implemented in the ‘Anova’ function in the ‘car’ package (v. 3.0-5; Fox et al. 2019).
We compared genetic differentiation and spatial genetic structure between hybrids and progenitors, using data only from sites that had ≥10 MLGs of the relevant taxon. These comprised 13 sites (6 wetlands and 7 ditches) with T. latifolia, 4 sites (2 wetlands and 2 ditches) with T. angustifolia and 11 sites (5 wetlands and 6 ditches) with T. × glauca (Table S1). The distributions of T. latifolia and T. × glauca sites are comparable: the maximum and average distances were 1660 and 708 km between T. latifolia sites, and 1587 km and 621 between T. × glauca sites. The relative scarcity of T. angustifolia (see ‘Results’) meant smaller maximum and average distances among sites (761 and 410 km), and comparisons of genetic differentiation estimates should take into account the different sampling patterns (see below). Different approaches to measuring pairwise genetic differentiation capture different aspects of the processes that contribute to differentiation between subpopulations (Meirmans and Hedrick 2011, Verity and Nichols 2014), and we therefore estimated genetic differentiation among sites by using both a fixation index (G′ST; Hedrick 2005) and an index of differentiation (DEST; Jost 2008). G′ST is an appropriate index to use when comparing among hybrids and progenitor species because it is standardized, and thus does not depend on the amount of genetic variation within populations (Hedrick 2005). DEST is based on allelic differentiation between subpopulations, and may be more appropriate when analyzing variable loci such as microsatellites with relatively high numbers of alleles, because it does not depend on heterozygosity (Hedrick 2005, Jost 2008, Meirmans and Hedrick 2011). For each set of metrics, we tested for statistical significance of genetic differentiation between sites using 999 permutations of MLGs across sites in GenAlEx (v. 6.503; Peakall and Smouse 2006, 2012).
For each taxon, we used GenAlEx to test for patterns of isolation by distance (Wright 1943, 1946) using Mantel tests (i.e., the correlation between genetic and (log + 1) geographic distance matrices; Mantel 1967) for all sites collectively, and also separately for each site type (wetland and ditch), except for T. angustifolia, which was present in too few sites to analyze by site type. We used both G′ST and DEST as the genetic distances, and tested for statistical significance using 999 permutations of individuals across sites. We used spatial autocorrelation to determine the spatial scales across which genetic structure was evident for each taxon. We predicted that if T. × glauca dispersed more widely than its progenitor species, it should show little evidence of spatial autocorrelation, whereas the progenitor species would show spatial autocorrelation over relatively short distances. This was investigated in GenAlEx using both G′ST and DEST, based on comparisons of genetic and (log + 1) geographic distances up to a maximum distance of 1000 km for T. latifolia and T. × glauca, and 750 km for the more restricted T. angustifolia, using 50-km distance classes. We used the method of Smouse and Peakall (1999) to generate the correlation coefficient r, which measures the correlation between genetic and geographic distances of sites over different distance classes. Spatial autocorrelation was not assessed separately by site type because that division would have generated an insufficient number of site pairs at short-distance classes. Statistical significance was assessed using 999 permutations of individuals across sites.
FLOCK was used as a non-a priori method for evaluating genetic structure because of its ability to signal uncertainty when genetic differentiation is low; in addition, because it does not try to optimize HWE or linkage equilibrium within clusters, FLOCK can reliably infer the correct number of genetic groups when gene flow is relatively high or sampling is uneven among groups (Duchesne and Turgeon 2009, 2012). FLOCK is a non-Bayesian, non-Monte Carlo Markov Chain (MCMC) method of clustering individuals through iterative reallocation of genotypes to increasingly differentiated reference groups based on maximum likelihood scores. Separate analyses were carried out for each taxon; in each case, we compared the output for k values of 2–10 using the default settings: 20 iterations for each of 50 runs, a minimum log-likelihood difference of 0 to assign an individual to a cluster, and without creating isolated reference groups. Bar plots of FLOCK output were generated using DISTRUCT (Rosenberg 2004).
Clonal richness and genetic diversity
Clonal richness for each taxon within each site was calculated as R = (G − 1)/(n − 1), in which G is the number of MLGs and n is the number of ramets (Dorken and Eckert 2001). The smallest possible value of R is 0, independent of sample size (all ramets have the same genotype), and the largest value is 1 (all ramets have unique genotypes). Within each taxon, we also tested for a correlation between clonal richness and the total number of ramets across sites to determine whether local expansion is partially driven by clonal reproduction; under these conditions, we would expect to see an inverse correlation between clonal richness and the number of ramets.
Previous studies have found higher levels of genetic diversity in T. angustifolia compared with T. latifolia in Europe (Tsyusko et al. 2005) and China (Zhou et al. 2016), and we were interested to see if this pattern was upheld over a broad spatial scale in North America. In addition, we wanted to compare genetic diversity within taxa between wetlands and ditches, because ditches typically experience higher levels of disturbance, and therefore may have higher genetic diversity due to ongoing recruitment. We therefore quantified genetic diversity for each taxon/site combination using rarefied allelic richness (AR), calculated in the package ‘hierfstat’ (v. 0.04-22; Goudet and Jombart 2015). In each case, the number of alleles was rarified to the minimum number of MLGs (≥10 in all cases) multiplied by two. Genetic diversity for each site/taxon combination was also quantified using GenAlEx as observed and expected heterozygosity (Ho and He), and the inbreeding coefficient (FIS).
Linear mixed-effect models (LMEs) were used to compare clonal richness and genetic diversity measures between taxa or site types. Models were constructed with clonal richness or a diversity metric as the response variable; taxon (T. angustifolia, T. latifolia or T. × glauca), site type (ditch or wetland) and the interaction between taxon and site type as fixed effects; and site region as a random effect. Linear mixed-effect models of clonal richness were analyzed using R values from all sites, regardless of the number of MLGs present (all ≥2), while LMEs of the genetic diversity metrics were conducted using only those sites that had at least ten MLGs of the relevant taxon. Clonal richness was square-root transformed prior to analyses to meet model assumptions. Where taxon was a significant factor, post hoc t tests were used to compare least-squares means among taxa. LMEs were run using the ‘lmer’ function in the package ‘lme4’ (Bates et al. 2016), and statistical significance of fixed effects was assessed using ‘lmerTest’ (v. 2.0-33; Kuznetsova et al. 2016) in R.
Results
We genotyped a total of 1924 plants from 22 sites across a transect of ~1700 km. Of these, 882 ramets (305 MLGs) from 18 sites were identified as T. latifolia, 842 ramets (299 MLGs) from 18 sites were identified as T. × glauca (a combination of F1s and advanced-generation or backcrossed hybrids) and 200 ramets (77 MLGs) from eight sites were identified as T. angustifolia. Broadly consistent with earlier studies (Kirk et al. 2011, Travis et al. 2010, Freeland et al. 2013), the majority of T. latifolia (~76%) were from the ten sites that we sampled from longitudes east of 71° (Montmagny, Quebec and points east), the majority of T. × glauca (~88%) were from the 12 sites that we sampled from longitudes west of 74° (Cornwall, Ontario and points west) and more than half (57%) of the relatively rare T. angustifolia were found in five sites that we sampled in the lower peninsula of Michigan (Fig. 1, Table S1).
Observed heterozygosity deficits meant that 8% of the 91 locus × site combinations for T. latifolia were not in HWE. In T. angustifolia, all of the locus × site combinations were in HWE. For T. × glauca, 34% of 77 locus × site combinations deviated from HWE with Ho > He in most cases (88%). The same locus deviated from HWE in a maximum of 3 of 13 sites for T. latifolia, and a maximum of 5 of 11 sites for T. × glauca. Linkage disequilibrium was very rare in both progenitor species (<1% of 273 and <1% of 84 locus pair × site combinations for T. latifolia and T. angustifolia, respectively), and occurred in 14% of 231 locus × site combinations for T. × glauca, with no more than 15% of those occurrences attributed to a single-locus pair. Therefore, levels of Hardy–Weinberg disequilibrium and linkage disequilibrium did not present a substantial violation of the assumptions underlying our analyses.
Stand composition
The GLMM revealed that there was an unbalanced distribution of MLGs of each taxon within stands (χ21 = 7.81; p < 0.05), but that this variation was not associated with site type (χ21 = 0.23; p = 0.63). In particular, T. latifolia and T. × glauca were each more abundant than T. angustifolia (T. angustifolia vs. T. latifolia: parameter estimate = 1.28 ± 0.58 SE, Wald’s Z = 2.21, p = 0.03; T. angustifolia vs. T. × glauca: parameter estimate = 1.51 ± 0.58 SE, Wald’s Z = 2.61, p = 0.01) (Fig. 1). Occupancy of wetlands versus ditches was similar for each taxon, leading to no interaction between region and site type (χ21 = 0.61; p = 0.74).
Spatial genetic structure
All three taxa exhibited significant differentiation across all sites. G′ST values were higher in T. angustifolia than either T. latifolia or T. × glauca, but DEST was greater in T. × glauca and T. angustifolia than in T. latifolia (Table 1). When comparisons were possible (T. latifolia and T. × glauca), G′ST and DEST values among ditches versus wetlands were comparable, although they were consistently higher among ditches (Table 1). All three taxa exhibited spatial genetic structure as evidenced by significant isolation by distance across sites, with strong IBD in T. angustifolia and T. × glauca, and weaker IBD in T. latifolia (Table 2). Typha × glauca exhibited significant spatial autocorrelation across all distance classes based on G′ST and DEST, with the strength of the autocorrelation decreasing with increasing distance class size (0.06 ≤ r ≤ 0.35; 0.001 ≤ P ≤ 0.10). There was some evidence of spatial autocorrelation in T. angustifolia at 400 –700 km for both G′ST and DEST (0.28 ≤ r ≤ 0.36; P ≤ 0.10), though this was likely reflective of relatively few T. angustifolia sites, which tended to be separated by relatively large distances, plus an outlier population (see ‘Discussion'). Spatial autocorrelation was not significant at any distance class for T. latifolia based on either genetic differentiation metric (P > 0.10).
FLOCK analysis revealed different patterns of genetic structure for each taxon. Typha latifolia did not exhibit substructure, with FLOCK identifying an ‘undecided’ number of clusters for T. latifolia (interpreted as K = 1; Duchense and Turgeon 2012). Three clusters were identified for T. angustifolia (Fig. 2a): two of these (blue and orange) have genotypes scattered across the sampling area, and one (yellow) is comprised almost entirely of MLGs from a ditch in southern Ontario (site COD). The T. angustifolia in the orange and blue clusters was distinguished by alleles at TA8 (274 common in the orange cluster and 287 in blue) and TL213 (231 common in orange and 229 common in blue). The T. angustifolia in site COD was distinguished by a private allele at locus TA7 (208, frequency = 0.18) and high frequencies of an allele at each of TA7 (210, frequency = 0.68) and TL305 (321, frequency = 0.41); all of these alleles had much lower frequencies (<0.14) in T. angustifolia from other sites. Two clusters were identified for T. × glauca: the blue cluster mostly comprised F1s (43%) and MLGs provisionally assigned as backcrosses to T. latifolia (38%), or advanced- generation hybrids (18%). The orange cluster mostly comprised F1s (75%) and MLGs provisionally assigned as backcrosses to T. angustifolia (17%) (Fig. 2b).

Each bar is a multilocus genotype that is partitioned into k-colored segments that represent the proportion of membership in each cluster. a T. angustifolia arranged by site and b T. × glauca arranged by site. Color coding in (a) and (b) are independent of each other. Sites are arranged from west to east. Site codes ending in ‘D’ represent ditches and those ending in ‘W’ represent wetlands.
Clonal richness and genetic diversity
Clonal richness ranged from 0.06 to 1 across taxa and sites (Table 3, S3). Clonal richness did not vary significantly between taxa (F2,29.07 = 0.001, P > 0.99; overall mean ± SE: 0.41 ± 0.04), but overall clonal richness was significantly greater in ditches (0.475 ± 0.044) than in wetlands (0.347 ± 0.055) (F2,21.65 = 5.50, P < 0.05), with no significant interaction between taxa and site type (F2,29.07 = 0.88, P > 0.40). Clonal richness was inversely correlated with the number of sampled ramets for T. latifolia (r = −0.51, n = 18, P < 0.05) and T. × glauca (r = −0.48, n = 18, P < 0.5), but not for T. angustifolia (r = −0.54, n = 8, P > 0.10).
Hybrids had greater genetic diversity than either progenitor species; however, there were no differences in genetic diversity metrics between site types (Table 3). Taxon was a significant factor for each diversity metric (P ≤ 0.001), whereas site type and the interaction between site type and taxon were not (P ≥ 0.35) (Table S2). Post hoc tests revealed that all genetic diversity metrics were significantly greater in T. × glauca than in either T. latifolia or T. angustifolia (T. × glauca–T. angustifolia: −4.48 ≤ t ≤ −4.14, P < 0.001; T. × glauca–T. latifolia: −10.70 ≤ t ≤ −8.14, P < 0.001), and that metrics were not different between T. latifolia and T. angustifolia (0.74 ≤ t ≤ 2.27; P ≥ 0.34). Observed heterozygosity of T. latifolia was significantly lower than expected in nine sites, whereas there were no significant differences between Ho and He for T. angustifolia (Table S3). Heterozygosity was higher than expected in T. × glauca in all but two sites (EDW and SAD, Table S3), each of which had a high proportion of MLGs provisionally identified as backcrosses to T. latifolia or advanced-generation hybrids. Site-level genetic diversity indices and FIs for each taxon are in Table S3.
Discussion
This study represents the most extensive population genetics investigation to date of Typha spp., and is the first to investigate some of the contributions of local versus landscape-level processes towards the maintenance of a Typha hybrid zone. Over a broad spatial scale spanning ~1700 km, we found that T. angustifolia is the least common cattail, and while both T. latifolia and T. × glauca occur at most sites across the sampling region, there remains an east–west variation in their relative abundance. The abundance of each taxon was similar between wetlands and ditches. Genetic differentiation and spatial genetic structure were most evident in T. × glauca, and least evident in T. latifolia. Genetic diversity was greater in T. × glauca than in either progenitor, but comparable between T. latifolia and T. angustifolia. Clonal richness was similar across all three taxa, was higher in ditches than in wetlands and for T. × glauca and T. latifolia, it was inversely correlated with local abundance. Below, we discuss what these findings suggest about gene flow in hybrids, and how processes across local and landscape scales can affect the spread of invasive hybrids.
Gene flow
Broadly consistent with an earlier study based on a smaller sample size (Freeland et al. 2013), the non-a priori analysis of genetic structure using FLOCK found very little evidence to suggest that genetic lineages clustered according to geographic region: there was no clustering in T. latifolia, and with the exception of the anomalous COD, T. angustifolia stand (see below), each of the genetic clusters identified in T. angustifolia and T. × glauca were distributed across the sampled region. This reflects ongoing dispersal and gene flow, which is often high in aquatic plants (Santamaría 2002). Typha follows the ‘Phragmites-type’ seed dispersal strategy that includes anemochory (dispersal by wind), hydrochory (dispersal by water), autochory (discharge dispersal) and endozoochory (dispersal by animals) (Sádlo et al. 2018). Waterbirds can disperse Typha seeds over long distances (Green et al. 2008), and Typha seeds can successfully germinate following passage through duck digestive tracts (Soons et al. 2008). In addition, Typha pollen is dispersed by wind, and vegetative fragments are dispersed by water following events such as spring flooding. Collectively, these dispersal strategies can be very effective in aquatic plants, for example, wind pollination and seed dispersal by wind, water or waterbirds explained high levels of genetic connectivity among stands of Triglochin procera and Phragmites australis across an extensive wetlands region in Australia (James et al. 2013). In another example, three foundational bulrush species (Schoenoplectus acutus, S. americanus and Bolboschoenus maritimus) regularly dispersed hundreds of kilometers, likely as a result of avian-mediated dispersal (Kettenring et al. 2019).
Although genetic connectivity among Typha stands is relatively high over broad spatial scales, Typha × glauca has overall higher genetic differentiation, stronger isolation by distance and stronger spatial autocorrelation at all distance classes than either progenitor species. This could reflect morphological and/or physiological differences that make seed dispersal less effective, for example, dispersal can be more effective for plants that produce relatively small seeds with relatively long viability (Alpert et al. 2000). Other studies have found greater dispersal and establishment potential in hybrids as a result of smaller, lighter seeds (e.g., Navarro-Cano et al. 2017), and future studies could investigate whether T. × glauca hybrids tend to produce larger seeds with shorter viability compared with their progenitors.
Typha angustifolia exhibited greater genetic differentiation, stronger isolation by distance, and stronger spatial genetic structure than T. latifolia, although spatial patterns of T. angustifolia should be interpreted with some caution. A stand of T. angustifolia sampled from a ditch in southern Ontario (site COD) comprised a distinct genetic cluster in the FLOCK analysis because at two loci, this population harbored high-frequency alleles that were rare or absent in T. angustifolia from other sites. Although the source of this genetic lineage is unknown, garden centers frequently sell multiple Typha species in North America, and these are sometimes sourced from outside North America (Ciotir and Freeland 2016). It is therefore possible that this COD site is an escaped cultivated lineage that is not native to North America. When T. angustifolia from site COD was excluded from analyses of genetic differentiation and spatial genetic structure, T. angustifolia showed no more evidence of genetic structure than T. latifolia. Additional investigations will be necessary to clarify spatial genetic patterns in T. angustifolia, although that endeavor may require sampling from a geographic area where T. angustifolia is more abundant. In contrast to the other two taxa, Typha latifolia exhibited insignificant spatial autocorrelation, lower genetic differentiation and weaker IBD. Together, these results indicate that gene flow is overall most restricted in T. × glauca, and that the native progenitor, T. latifolia, shows the highest degree of connections between sites.
Genetic diversity and clonal richness
The amount of sexual versus vegetative reproduction can influence gene flow among populations of facultatively sexual plants, because higher levels of sexual reproduction should yield a greater capacity for gene flow via seeds and pollen. In Sparganium emersum, for example, gene flow is mediated primarily through seed dispersal, leaving little genetic differentiation between stands that reproduce sexually, but substantial genetic structure among stands that are exclusively clonal (Pollux et al. 2007). However, we found no evidence in Typha that reproductive strategies influence gene flow: despite inter-taxon differences in gene flow, all three taxa exhibited comparable levels of clonal richness, suggesting that each is engaging in similar amounts of sexual and clonal reproduction. There was considerable variation in clonal richness between sites for all taxa, but our data suggest that an important predictor of clonal richness is the frequency of the relevant taxon. When either T. latifolia or T. × glauca was more dominant at a site, the clonal richness estimate was lower, suggesting that clonal expansion has played an important role in the local expansion of both taxa. The number of sampled T. angustifolia sites was too small to determine whether a comparable trend exists in that species. In addition, clonal richness across taxa was higher in ditches than in wetlands. Ditches could experience frequent turnover of individuals from ongoing disturbance (e.g., from vehicular traffic on adjacent roads, mowing, run-off and debris from roads and more frequent water-level drawdown), which in turn could yield greater clonal richness by introducing open spaces for new recruits. In comparison with wetlands, ditches showed neither lower estimates of genetic differentiation nor higher estimates of genetic diversity other than clonal richness, and it is therefore likely that many new recruits were from seeds that originated within the stand. We therefore suggest that local processes, including site disturbance and clonal expansion, are more important than landscape-level processes for determining clonal richness in Typha.
Implications for Typha hybrid zones
Our study suggests that the T. × glauca hybrid zone in regions around the Laurentian Great Lakes and St. Lawrence Seaway of eastern North America can be explained more by the repeated local production and clonal expansion of hybrids than by the ongoing dispersal of hybrids. This raises the possibility that hybrid persistence will be limited in the long term by the relative rarity of the seed parent T. angustifolia. Presently, most hybrids in northeastern North America are first-generation hybrids (Travis et al. 2010, Kirk et al. 2011, Freeland et al. 2013; this study), but future hybrid formation may be increasingly skewed towards advanced- generation hybrids, because hybrids can act as both pollen and seed parents (Pieper et al. 2017). The implications of this for the invasiveness of the species complex are not well understood. In general, advanced-generation hybrids may either be less fit than first-generation hybrids due to genome incompatibilities (‘hybrid breakdown’, reviewed in Burton et al. 2013), or they may yield opportunities for more invasive genotypes due to the increasingly varied gene combinations in advanced-generation hybrids compared with F1s (Arnold 1997). Some preliminary investigations suggest that advanced-generation hybrids are less robust (Zapfe and Freeland 2015) and less fertile (Pieper et al. 2017) than F1s, but large-scale investigations into this question are lacking. In addition, even if advanced-generation hybrids have lower fitness than F1 hybrids following the loss of heterosis and disruption of co-adapted gene complexes (Oakley et al. 2015), backcrossing between hybrids and progenitor species can recreate gene complexes, and can also create new, beneficial genotypes through recombination (Prentis et al. 2008, Hufford and Mazer 2003). Either of these processes could potentially restore fitness to backcrossed hybrids (Pratt et al. 1985, Mejía-Jiménez et al. 1994).
One other consideration potentially relevant to the possibility of long-term hybrid dominance comes from the fact that although we found clonal richness to be comparable between hybrids and progenitor species, hybrids unsurprisingly had the highest levels of genetic diversity according to all other measures (i.e., those based on the numbers of different alleles), and this may bestow an evolutionary advantage. Greater standing genetic variation in hybrids compared with progenitors can generate novel genotypes with adaptive potential (Buhk and Thielsch 2015), and increases in heterozygosity can elevate fitness in hybrids via heterosis (Ellstrand and Schierenbeck 2006). This could give hybrids an advantage over progenitors at local scales, despite a seemingly restricted potential for dispersal. For example, hybrid elms (Ulmus pumila × rubra) have greater genetic diversity than their progenitors at local scales, which may contribute to their invasiveness (Zalapa et al. 2010). Finally, the competitive superiority of Typha hybrids over progenitors that has been reported at local scales for a variety of traits (Bunbury-Blanchette et al. 2015, Zapfe and Freeland 2015, Szabo et al. 2018) means that even if dispersal leads to the introduction of very few hybrids into novel sites, local processes may lead to site domination by hybrids within a relatively short period of time, which in turn could allow T. × glauca to invade sites from which T. angustifolia is absent. We therefore conclude that although local processes have likely played a more important role than landscape-level processes in the establishment of T. × glauca across a broad geographical area, even low levels of dispersal combined with local competitive success could be sufficient for the long-term maintenance of this invasive hybrid.
Data availability
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.6hdr7sqx5.
References
Alpert P, Bone E, Holzapfel C (2000) Invasiveness, invasibility and the role of environmental stress in the spread of non-native plants. Perspect Plant Ecol Evol Syst 3:52–66
Alston KP, Richardson DM (2006) The roles of habitat features, disturbance, and distance from putative source populations in structuring alien plant invasions at the urban/wildland interface on the Cape Peninsula, South Africa. Biol Conserv 132:183–198
Arnold ML (1997) Natural Hybridization and Evolution. Oxford University Press, New York
Arnold ML, Tang S, Knapp SJ, Martin NH (2010) Asymmetric introgressive hybridization among Louisiana Iris species. Genes 1:9–22
Bailey JP, Bímová K, Mandák B (2009) Asexual spread versus sexual reproduction and evolution in Japanese Knotweed s.l. sets the stage for the ‘battle of the Clones’. Biol Invasions 11:1189–1203
Ball D, Freeland JR (2013) Synchronous flowering times and asymmetrical hybridization in Typha latifolia and T. angustifolia in northeastern North America. Aquat Bot 104:224–227
Barton NH, Hewitt GM (1985) Analysis of hybrid zones. Annu Rev Ecol Syst 16:113–148
Bates D, Maechler M, Bolker B (2016) lme4: Linear mixed-effects models using Eigen and S4. R package version 1.1-13, http://CRAN.R-project.org/package-lme4
Buhk C, Thielsch A (2015) Hybridisation boosts the invasion of an alien species complex: Insights into future invasiveness. Perspect Plant Ecol, Evol Syst 17(4):274–283
Bunbury-Blanchette AL, Freeland JR, Dorken ME (2015) Hybrid Typha × glauca outperforms native T. latifolia under contrasting water depths in a common garden. Basic Appl Ecol 16:394–402
Burke JM, Carney SE, Arnold ML (1998) Hybrid fitness in the Louisiana Irises: analysis of parental and F 1 performance. Evolution 52:37–43.
Burns I, James PMA, Coltman DW, Cullingham CI (2019) Spatial and genetic structure of the lodgepole × jack pine hybrid zone. Can J For Res 49(7):844–853
Burton RS, Pereira RJ, Barreto FS (2013) Cytonuclear genomic interactions and hybrid breakdown. Annu Rev Ecol Evol Syst 44:281–302
Ciotir C, Dorken M, Freeland J (2013a) Preliminary characterization of Typha latifolia and T. angustifolia from North America and Europe based on novel microsatellite markers identified through next-generation sequencing. Fundam Appl Limnol 182:247–252
Ciotir C, Freeland J (2016) Cryptic intercontinental dispersal, commercial retailers, and the genetic diversity of native and non-native cattails (Typha spp.) in North America. Hydrobiologia 768:137–150
Ciotir C, Kirk H, Row JR, Freeland JR (2013b) Intercontinental dispersal of Typha angustifolia and T. latifolia between Europe and North America has implications for Typha invasions. Biol Invasions 15:1377–1390
Ciotir C, Szabo J, Freeland J (2017) Genetic characterization of cattail species and hybrids (Typha spp.) in Europe. Aquat Bot 141:51–59
De La Torre A, Ingvarsson PK, Aitken SN (2015) Genetic architecture and genomic patterns of gene flow between hybridizing species of Picea. Heredity 115:153–164
Dodd RS, Afzal-Rafii Z (2004) Selection and dispersal in a multispecies oak hybrid zone. Evolution 58:261–269
Dorken ME, Eckert CG (2001) Severely reduced sexual reproduction of a clonal plant, Decodon verticillatus populations (Lythraceae). J Ecol 89:339–350
Duchesne P, Turgeon J (2009) FLOCK: a method for quick mapping of admixture without source samples. Mol Ecol Resour 9:1333–1344
Duchesne P, Turgeon J (2012) FLOCK provides reliable solutions to the ‘number of populations’ problem. J Hered 103:734–743
Ellstrand NC, Schierenbeck KA (2006) Hybridization as a stimulus for the evolution of invasiveness in plants? Euphytica 148(1–2):35–46
Fox J, Wesiberg S, Price B (2019) Companion to applied regression. R package version 3.0-5. http://CRAN.R-project.org/package=car
Freeland J, Ciotir C, Kirk H (2013) Regional differences in the abundance of native, introduced, and hybrid Typha spp. in northeastern North America influence wetland invasions. Biol Invasions 15:2651–2665
Fritz RS, Hochwender CG, Albrectsen BR, Czesak ME (2006) Fitness and genetic architecture of parent and hybrid willows in common gardens. Evolution 60:1215–1227
Galatowitsch SM, Anderson NO, Ascher PD, Hall A (1999) Invasiveness in wetland plants in temperate north america. Wetlands 19:733–755
Geddes P, Grancharova T, Kelly JJ, Treering D, Tuchman NC (2014) Effects of invasive Typha × glauca on wetland nutrient pools, denitrification, and bacterial communities are influenced by time since invasion. Aquat Ecol 48:247–258
Goudet J, Jombart T (2015) Hierfstat: estimation and tests of hierarchical F-statistics. R package version 0.04–22 10
Goudet J, Raymond M, De Meeiis T, Roussett F (1996) Testing differentiation in diploid populations. Genetics 144:1933–1940
Goulet BE, Roda F, Hopkins R (2017) Hybridization in plants: old ideas, new techniques. Plant Physiol 173:65–78
Grace J, Harrison J (1986) The biology of Canadian weeds: 73. Typha latifolia L., Typha angustifolia L. and Typha x glauca Godr. Can J Plant Sci 66:361–379
Green AJ, Jenkins KM, Bell D, Morris PJ, Kingsford RT (2008) The potential role of waterbirds in dispersing invertebrates and plants in arid Australia. Freshw Biol 53:380–392
Guo SW, Thompson EA (1992) A Monte Carlo method for combined segregation and linkage analysis. Am J Hum Genet 51:1111–1126
Hansen MJ, Clevenger AP (2005) The influence of disturbance and habitat on the presence of non-native plant species along transport corridors. Biol Conserv 125:249–259
Harrison R (1993) Hybrid zones and the evolutionary process. Oxford University Press, Oxford
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Hufford KM, Mazer SJ (2003) Plant ecotypes: genetic differentiation in the age of ecological restoration. Trends Ecol Evol 18:147–155
James EA, Jordan R, Griffin PC (2013) Spatial genetic analysis of two polyploid macrophytes reveals high connectivity in a modified wetland. Freshw Biol 58:2102–2113
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kettenring KM, Mossman BN, Downard R, Mock KE (2019) Fine-scale genetic diversity and landscape-scale genetic structuring in three foundational bulrush species: implications for wetland revegetation. Restor Ecol 27:408–420
Kirk H, Connolly C, Freeland JR (2011) Molecular genetic data reveal hybridization between Typha angustifolia and Typha latifolia across a broad spatial scale in eastern North America. Aquat Bot 95:189–193
Kuznetsova A, Brockhoff PB, Chistensen RHB (2016) lmerTest: Tests for random and fixed effects for linear mixed effect models (lmer objects of lme4 package). R package version 2.0-33. http://CRAN.R-project.org/package=lmerTest
Larkin DJ, Freyman MJ, Lishawa SC, Geddes P, Tuchman NC (2012) Mechanisms of dominance by the invasive hybrid cattail Typha × glauca. Biol Invasions 14:65–77
Lawrence B, Bourke K, Lishawa SC, Tuchman NC (2016) Typha invasion associated with reduced aquatic macroinvertebrate abundance in northern Lake Huron coastal wetlands. J Gt Lakes Res 42:1412–1419
Lawrence BA, Lishawa SC, Hurst N, Castillo BT, Tuchman NC (2017) Wetland invasion by Typha × glauca increases soil methane emissions. Aquat Bot 137:80–87
Lexer C, Fay MF, Joseph JA, Nice M-S, Heinze B (2005) Barrier to gene flow between two ecologically divergent Populus species, P. alba (white poplar) and P. tremula (European aspen): the role of ecology and life history in gene introgression. Mol Ecol 14:1045–1057
Lishawa SC, Jankowski K, Geddes P, Larkin DJ, Monks AM, Tuchman NC (2014) Denitrification in a Laurentian Great Lakes coastal wetland invaded by hybrid cattail (Typha × glauca). Aquat Sci 76:483–495
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Marques I, Nieto Feliner G, Martins-Loução MA, Fuertes Aguilar J (2011) Fitness in Narcissus hybrids: Low fertility is overcome by early hybrid vigour, absence of exogenous selection and high bulb propagation. J Ecol 99:1508–1519
Meirmans PG, Hedrick PW (2011) Assessing population structure: FST and related measures. Mol Ecol Resour 11:5–18
Mejía-Jiménez A, Muñoz C, Jacobsen HJ, Roca WM, Singh SP (1994) Interspecific hybridization between common and tepary beans: increased hybrid embryo growth, fertility, and efficiency of hybridization through recurrent and congruity backcrossing. Theor Appl Genet 88:324–331
Navarro-Cano JA, Schwienbacher E, Sánchez-Balibrea J, Erschbamer B (2017) The role of seed traits as segregation factors of hybrids in wild populations of Cistus (Cistaceae). Plant Biosyst 151:530–538
Norris LC, Main BJ, Lee Y, Collier TC, Fofana A, Cornel AJ et al. (2015) Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proc Natl Acad Sci 112:815–820
Oakley CG, Ågren J, Schemske DW (2015) Heterosis and outbreeding depression in crosses between natural populations of Arabidopsis thaliana. Heredity 115:73–82
Peakall R, Smouse PE (2006) GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinforma Appl 28:2537–2539
Pieper SJ, Freeland JR, Dorken ME (2018) Coexistence of Typha latifolia, T. angustifolia (Typhaceae) and their invasive hybrid is not explained by niche partitioning across water depths. Aquat Bot 144:46–53
Pieper SJ, Nicholls AA, Freeland JR, Dorken ME (2017) Asymmetric hybridization in cattails (Typha spp.) and its implications for the evolutionary maintenance of native Typha latifolia. J Hered 108:479–487
Pollux BJA, Jong MDE, Steegh A, Verbruggen E, Van Groenendael JM, Ouborg NJ (2007) Reproductive strategy, clonal structure and genetic diversity in populations of the aquatic macrophyte Sparganium emersum in river systems. Mol Ecol 16:313–325
Pratt RC, Bressan RA, Hasegawa PM (1985) Genotypic diversity enhances recovery of hybrids and fertile backcrosses of Phaseolus vulgaris L. × P. acutifolius A. Gray. Euphytica 34:329–334
Prentis PJ, Wilson JRU, Dormontt EE, Richardson DM, Lowe AJ (2008) Adaptive evolution in invasive species. Trends Plant Sci 13:288–294
R Core Development Team (2016) R: A language and environment for statistical computing v. 3.4.0. R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org/
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49:1280–1283
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Sádlo J, Chytrý M, Pergl J, Pyšek P (2018) Plant dispersal strategies: a new classification based on the multiple dispersal modes of individual species. Preslia 90:1–22
Santamaría L (2002) Why are most aquatic plants widely distributed? Dispersal, clonal growth and small-scale heterogeneity in a stressful environment. Acta Oecologica 23(3):137–154
Shih JG, Finkelstein Sa (2008) Range dynamics and invasive tendencies in Typha latifolia and Typha angustifolia in eastern North America derived from herbarium and pollen records. Wetlands 28:1–16
Sloop CM, Ayres DR, Strong DR (2011) Spatial and temporal genetic structure in a hybrid cordgrass invasion. Heredity 106:547–556
Smouse PE, Peakall R (1999) Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 82:561–573
Snow AA, Travis SE, Wildova R, Fer T, Sweeney PM, Marburger JE et al. (2010) Species-specific SSR alleles for studies of hybrid cattails (Typha latifolia x T. angustifolia; Typhaceae) in North America. Am J Bot 97:2061–2067
Soons MB, Van Der Vlugt C, Van Lith B, Heil GW, Klaassen M (2008) Small seed size increases the potential for dispersal of wetland plants by ducks. J Ecol 96:619–627
Stelkens RB, Brockhurst MA, Hurst GDD, Greig D (2014) Hybridization facilitates evolutionary rescue. Evol Appl 7:1209–1217
Szabo J, Freeland JR, Dorken ME (2018) The effects of leaf litter and competition from hybrid cattails (Typha × glauca) on the seed germination and seedling performance of its parental species. Aquat Bot 145:29–36
Todesco M, Pascual MA, Owens GL, Ostevik KL, Moyers BT, Hübner S et al. (2016) Hybridization and extinction. Evol Appl 9:892–908
Travis SE, Marburger JE, Windels S, Kubatova B (2010) Hybridization dynamics of invasive cattail (Typhaceae) stands in the Western Great Lakes Region of North America: a molecular analysis. J Ecol 98:7–16
Tsyusko-Omeltchenko OV, Schable NA, Smith MH, Glenn TC (2003) Microsatellite loci isolated from narrow-leaved cattail Typha angustifolia. Mol Ecol Notes 3:535–538
Tsyusko OV, Smith MH, Sharitz RR, Glenn TC (2005) Genetic and clonal diversity of two cattail species, Typha latifolia and T. angustifolia (Typhaceae), from Ukraine. Am J Bot 92:1161–1169
Tuchman NC, Larkin DJ, Geddes P, Wildova R, Jankowski K, Goldberg DE (2009) Patterns of environmental change associated with Typha × glauca Invasion in a Great Lakes Coastal Wetland. Wetlands 29:964–975
Verity R, Nichols RA (2014) What is genetic differentiation, and how should we measure it—Gst, D, neither or both? Mol Ecol 23:4216–4225
Whitney KD, Ahern JR, Campbell LG, Albert LP, King MS (2010) Patterns of hybridization in plants. Perspect Plant Ecol Evol Syst 12:175–182
Wolf DE, Takebayashi N, Rieseberg LH (2001) Predicting the Risk of Extinction through Hybridization. Conserv Biol 15(4):1039–1053
Wright S (1943) Isolation by distance. Genetics 28:114–138
Wright S (1946) Isolation by distance under diverse systems. Genetics 31:39–59
Zalapa JE, Brunet J, Guries RP (2010) The extent of hybridization and its impact on the genetic diversity and population structure of an invasive tree,Ulmus pumila (Ulmaceae). Evol Appl 3:157–168
Zapfe L, Freeland JR (2015) Heterosis in invasive F-1 cattail hybrids (Typha × glauca). Aquat Bot 125:44–47
Zhou B, Yu D, Ding Z, Xu X (2016) Comparison of genetic diversity in four Typha species (Poales, Typhaceae) from China. Hydrobiologia 770:117–128
Zuur AF, Ieno EN, Walker NJ, Saveliev AA, Smith GM (2009) Mixed Effects Models and Extensions in Ecology with. R. Springer Science, New York, NY, USA
Acknowledgements
We thank Michelle Scriver and Sage Fleming for assistance in the field, and David Fearis for assistance in the lab. This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) via a Postgraduate Scholarship to SJP, and Discovery Grants to MED (RGPIN 355954-2013) and JRF (RGPIN 229737-2012).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pieper, S., Dorken, M. & Freeland, J. Genetic structure in hybrids and progenitors provides insight into processes underlying an invasive cattail (Typha × glauca) hybrid zone. Heredity 124, 714–725 (2020). https://doi.org/10.1038/s41437-020-0307-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-0307-y
This article is cited by
-
Self-fertilization does not lead to inbreeding depression in Typha parent species or hybrids
Evolutionary Ecology (2024)
-
Undetected but Widespread: the Cryptic Invasion of Non-Native Cattail (Typha) in a Pacific Northwest Estuary
Estuaries and Coasts (2023)
-
Tracking Vegetation Transitions Due to Invasion of Cattail (Typha) in Lake Superior Coastal Peatlands
Wetlands (2023)
-
Evidence of hybrid breakdown among invasive hybrid cattails (Typha × glauca)
Heredity (2022)
-
Distributions of native and invasive Typha (cattail) throughout the Prairie Pothole Region of North America
Wetlands Ecology and Management (2022)
