
Abstract
The care for patients with serious conditions is increasingly guided by genomic medicine, and genomic medicine may equally transform care for healthy individual if genomic population screening is implemented. This study examines the medical impact of opportunistic genomic screening (OGS) in a cohort of patients undergoing comprehensive genomic germline DNA testing for childhood cancer, including the impact on their relatives. Medical actionability and uptake after cascade testing in the period following disclosure of OGS results was quantified. A secondary finding was reported to 19/595 (3.2%) probands primarily in genes related to cardiovascular and lipid disorders. After a mean follow up time of 1.6 years (Interquartile range (IQR): 0.57-1.92 yrs.) only 12 (63%) of these variants were found to be medically actionable. Clinical follow up or treatment was planned in 16 relatives, and as in the probands, the prescribed treatment was primarily betablockers or cholesterol lowering therapy. No invasive procedures or implantation of medical devices were performed in probands or relatives, and no reproductive counseling was requested. After an average of 1.6 years of follow-up 2.25 relatives per family with an actionable finding had been tested. This real-world experience of OGS grants new insight into the practical implementation effects and derived health care demands of genotype-first screening. The resulting health care effect and impact on demand for genetic counseling and workup in relatives extends beyond the effect in the probands.
Similar content being viewed by others


Introduction
Extensive germline genetic sequencing is currently used in both clinical practice and research studies, generating data also covering genes not related to the primary clinical question. The American College of Medical Genetics and Genomics (ACMG) has published a list of “actionable” genes, recommending universal reporting of known or expected disease-causing variants within these genes irrespectively of the clinical indication leading to germline sequencing, resulting in opportunistic genomic screening (OGS) for susceptibility to other preventable diseases in patients [1,2,3,4]. Previous studies of several cohorts have determined the rate of secondary genetic findings to be 1.0–3.4% [5,6,7]. However, within the genetic community the European Society of Human Genetics recommends a different approach, where OGS only is performed within a research setting generating more knowledge of genotype-first genetics [8].
Established clinical practice recommends cascade testing of a pathogenic (PV) or likely pathogenic (LPV) variant within a family for many of the genes listed as actionable by the ACMG. Testing at-risk individuals can provide tailored counseling and management of potentially life-threatening and treatable diseases [9]. As such, return of a secondary finding (SF) may lead to additional genetic consultations of both probands and relatives as well as genetic testing, clinical workup, treatment, and long-term follow-up of several individuals [7, 10]. While previous studies have described OGS findings in patients from patient cohorts or population genomic screening [7, 11,12,13,14,15] the down-stream impact that such a finding has on the index patient, as well as relatives requires further attention.
The aim of this study was to quantify the medical impact of OGS in a cohort of patients undergoing comprehensive genomic DNA testing for childhood cancer as well as the medical impact on their relatives. This was performed by quantifying actionability and uptake after cascade testing in the time following disclosure of OGS results to patients and relatives.
Materials and methods
Inclusion and exclusion
Participants (probands and parents), enrolled in the “Sequencing of Tumor And Germline DNA – Implications and National Guidelines” (STAGING) study were included. The STAGING study is a Danish, prospective multicenter genomic sequencing study offering inclusion to children diagnosed with any cancer or CNS tumor before 18 years of age since January 1st 2017 [16]. Patients were excluded from the STAGING study if they or the legal guardian did not consent to the return of an actionable genetic finding. Four-generation pedigrees were obtained.
Sampling and variant interpretation
DNA sampling and sequencing of probands is described in supplementary methods. Variants were annotated and filtered using VarSeq (Golden Helix) as recently described [16]. Briefly, all variants with a minor allele frequency of >1% in any large population (gnomAD v2.1) were excluded. Only coding and splicing variants (+/− 10 bp) were included in the analysis. Moreover, only variants with a variant allele fraction > 20% were kept in the analysis. Finally, data was filtered for variants in 314 genes associated with cancer as well as the genes listed as actionable in the ACMG v.2.0 policy statement [2, 16]. This version of the ACMG policy statement was the recommended version when data was analyzed, and used throughout the study period to ensure continuity in data analysis, despite the publication of updated recommendations [4]. Only variants in ACMG genes, excluding cancer predisposing genes, were included.
Variants were assessed by a team of clinical geneticists and molecular biologists based on variant ontology (e.g., frameshift, nonsense, missense), in silico predictions of effect on protein and RNA function (e.g., Combined Annotation Dependent Depletion [CADD], PHRED quality score, ADA splice prediction score), and database searches for published literature on each variant. Health care records for the proband in some instances including an EKG or a lipid profile and a detailed pedigree was available to the interpreters. The ACMG guidelines regarding variant interpretation was not published when the study was planned, and not implemented at our institution when the first samples were analyzed [17]. During the study period the ACMG classification system was gradually implemented, and the previous local standard phased out. Only pathogenic or likely pathogenic variants were returned to the probands in accordance with the ACMG v.2.0 guidance. Challenging cases were discussed in special academic fora, see supplementary methods for details.
Genetic counseling, clinical work up and cascade testing
Probands with a SF in one of the genes not associated with cancer predisposition on the ACMG v 2.0 list (36 genes), were informed of the results by a research team member and the findings were noted in the proband’s electronic medical record. The proband was referred to the local department of Clinical Genetics for clinical genetic workup and counselling, and if relevant, the proband/family was referred to clinical management with a relevant specialist (typically pediatrician or specialized cardiologist). Any clinical evaluation or counseling that was performed as result of the return of the SF was undertaken and funded by the Danish health care system and was not a part of the research protocol. Further clinical work up entailed obtaining a medical and family history, clinical exam, and further diagnostic testing such as imaging (echocardiography, cardiac and/or vascular MRI), EKG, 48-hour holter-monitoring, lipid profile measurements, assessment of ICD implantation risk, medical treatment initiation etc, at the discretion of the treating physician. See supplementary methods for further details. If relatives were identified as eligible for genetic counseling and/or cascade testing by the physician in charge of the clinical follow up in the proband, they were referred directly to genetic counselling in the relevant clinical specialty.
Actionability and follow-up
Electronic medical records in probands and relatives were reviewed for family history, diagnostic testing results, referral to medical specialists, as well as medical treatment plans relating to the return of the SF. Actionability of a SF was defined as planned regular follow-up (e.g., lipid profile evaluation, clinical follow up in specialized cardiology or pediatric clinic, repeat cardiac imaging), prescription of medical or dietary treatment, invasive medical procedures, and risk reducing medical procedures (e.g., pacemaker or implantable cardioverter defibrillator implantations). Reproductive decisions due to the SF (prenatal diagnostic testing and preimplantation genetic testing) were also included. Uptake was defined as number of relatives tested for the variant per proband, when cascade testing was recommended for the disclosed SF variant.
Statistics
Descriptive statistics were performed in Microsoft Excel (version 2016). Ninety-five percent confidence limits for point estimates were calculated by non-parametric bootstrap resampling in SAS (version 9.4).
Ethical considerations
Ethical approval was obtained through the regional scientific ethical committee (the Ethical Scientific Committees for the Capital Region, H-15016782) and the Danish Data Protection Agency (RH-2016-219, I-Suite no: 04804). All parents/guardians and patients 18 years or older gave formal written consent to germline WGS in the proband as well as written, informed consent to the return of actionable findings as well as scientific reporting of this. Relatives beyond parents that were referred to genetic counselling or clinical workup due to a SF gave written consent to the inclusion of their data in a scientific publication.
Results
Secondary finding variants
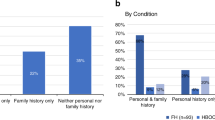
837 consecutive patients were invited to enroll in the STAGING study between January 1st 2017 and December 31st 2021, of which 665 patients consented and were included. At the time of analysis (April 2022), final data had been reported in 595 patients. Twenty-six pathogenic or likely pathogenic variants were detected in non-oncogene actionable genes in twenty-six probands, of which seven did not meet the gene-specific reporting criteria (heterozygosity of the recessive gene in question or likely pathogenic variant in a gene for which only known pathogenic should be reported), see Supplementary Table 1. Thus, a SF was returned to 19/595 (3.2% with 95% confidence limits 2.1%-4.2%) patients (Table 1). Of the 19 reported findings, seven were classified as PV and twelve as LPV using the aforementioned classification strategy, see Table 1. Variant interpretation using the ACMG guidelines is also noted in Table 1, further details on the criteria used can be found in supplementary table 2 Secondary findings were reported in 10 of the 36 SF genes, and primarily related to cardiovascular diseases (9/19 genes, Fig. 1), and in genes related to lipid disorders (8/19 genes) or connective tissue disorders (2/19).

Findings are primarily related to genes related to cardiomyopathies or lipid disorders.
Probands and genetic counseling uptake
The median age at return of the SF to the probands was 13.4 years (inter quartile range (IQR): 6.15–17.2 yrs), and median follow-up time since return of SF was 1.6 years (IQR: 0.57–1.92 yrs). One patient was referred to genetic counseling but did not attend the planned session, two cases were handled by the treating oncological pediatrician and the family did not wish further genetic consultation, but the remainder (n = 16) received genetic counselling and/or referral to relevant specialist (cardiologist or specialized pediatrician). One proband died before the return of the SF, which subsequently was reported to the parents, who were referred to genetic counseling. In nine families there was a suggestive family history of a relevant cardiovascular disease, most commonly of a lipid disorder, but without previously identified genetic or clinical diagnosis in the family.
Clinical actionability
Clinical workup of the probands or families in which a SF was reported led to ongoing changes in clinical management in eleven of the living probands, and in the one family where the proband died before genetic counseling (Fig. 2). Further details regarding actionability can be found in Table 2 No invasive procedures or implantation of medical devices were performed in probands or relatives, and no reproductive genetic counseling or testing was requested. On average, 2.25 family members underwent genetic testing per proband in the families with actionable findings (27 relatives from 12 families). Family pedigrees are found in Supplementary Fig. S1.

Actionability of disclosed variants by disease category after clinical and/or genetic workup in proband and relatives.
Lipid disorders (n = 8)
Five of the variants disclosed in which actionable therapy was planned were in LDLR and APOB, genes which are associated with lipid disorders. Statin therapy or dietary treatment was prescribed after evaluation in a specialized lipid clinic in four probands, and further follow up in adulthood has been planned in the fifth proband. In one of these probands, who harbored a pathogenic APOB variant, the prescription of statin treatment was complicated by the fact that these drugs are contraindicated during posaconazol treatment, prescribed as fungal prophylaxis as pediatric oncological care. While cascade testing is ongoing, statin therapy was initiated or add-on lipid lowering therapy was prescribed in four relatives found to be a carrier (one a sibling), as well as regular appointments in the lipid clinic. In families 13 and 10, segregation analysis and lipid profiles of the parents resulted in reclassification of the variants and no additional treatment. The probands in the two families that were handled by the treating oncology pediatrician (family 18 and 19) did not have elevated lipid levels. Lipids profile testing was offered to these parents: reevaluation was recommended in one parent due to slightly elevated LDL levels, and in one family additional testing or counselling was actively declined.
Cardiomyopathy disorders (n = 6)
Two probands (family 1 and 3) underwent clinical work up in the outpatient cardiology clinic after DSG2 and MYBPC3 variants were reported: no cardiomyopathy was detected by echocardiography, EKG and holter-monitoring were also normal, and there was no relevant family history or reported symptoms, why regular follow-up is planned in 3–5 years. Cascade testing revealed an asymptomatic carrier parent in family 3 with normal EKG and holter-monitoring. Echocardiography in this individual was of an unclear phenotype, but at diagnosis of hypertrophic cardiomyopathy made after cardiac MRI, due to indicative imaging findings. Regular follow up with cardiac evaluation is planned. A healthy sibling also underwent genetic testing in this family, after diagnosis in the parent, and found to also carry the MYBPC3 variant. Cardiac evaluation (clinical exam, echocardiography and EKG) did not reveal a hypertrophic cardiomyopathy phenotype; a follow up in the specialized cardiology clinic is planned in 5 years. In family 17, the PKP2 variant was reported to the parents, who both underwent genetic testing. Subsequently, clinical workup of the asymptomatic carrier parent entailing echocardiography, holter-monitoring, and EKG was performed. These exams and medical history were normal in the carrier parent; annual checkups in a specialized cardiology clinic are planned. Four cases of sudden death were reported in this family, in the parent’s third- and fourth-degree relatives, but it was not possible to attribute these deaths to PKP2 carrier status after testing three relatives in this family. Interestingly, three relatives in this family actively declined genetic testing. The proband in family 7 (MYBPC3), did not attend genetic counselling. Despite this, the pediatric oncologists have planned muligated acquisition (MUGA) scans every three years due to the perceived risk of dilated cardiomyopathy as a carrier of a MYBPC3 variant, after reading the results of the SF in the medical records. No further clinical testing or clinical management was necessary in the proband or family following genetic consultation in family 11 (MYL2) and family 14 (MYBPC3), due to reclassification of the variant as a variant of unknown significance (VUS), or because the variant was only clinically relevant if biallelic. In total, five at-risk-relatives were tested for SF variants in cardiomyopathy genes, of which three are carriers, and cascade testing is ongoing in two families.
Arrythmia disorders (n = 3)
In the two probands (from family 2 and 8) with KCNQ1 variants (associated with long QT syndrome), medical treatment with betablockers was prescribed by the treating cardiologist as well as annual follow up in a specialized cardiology clinic. As part of the counseling, information was given specifically to avoid hypokalemia and QT prolonging drugs. Cascade testing of ten at risk relatives identified two carriers of which data only was available from one, in whom betablocker therapy was prescribed despite normal EKG and echocardiogram. Thus, betablocker therapy was initiated in both asymptomatic patients and carrier relatives with no family history of sudden death, but as carriers of disease causing variants, as is recommended clinical practice [18, 19]. A shared decision-making strategy was planned in family 5 (SCN5A) with the clinical geneticist and cardiologist. The family was offered full clinical work up (ajmaline challenge to variant carriers and advice regarding possible triggers), requiring the family to commute to a university hospital. There was no relevant family history of arrhythmic disorders and based on the situation in their family, the parents chose only to have tests done that could be done by the local general practitioner. The family’s wishes of no further referrals was respected. They have the option of referral at a later time, should their wishes change. As such, Family 2 and 8 had a similar offer and weighed their options differently. The fact that a pharmaceutical intervention to ameliorate risk is available for patient with LQTS but not in Brugada syndrome may have influenced their decision, but this has not been explored in depth.
Connective tissue disorders (n = 2)
The diagnosis of vascular Ehlers-Danlos syndrome (COL3A1), was made in a proband and two of the six tested relatives. These individuals were referred to regular clinical follow-up in a specialized pediatrics center for rare diseases where a dedicated team manages syndrome-related issues such as cardiovascular follow up. Baseline echocardiography in the proband and carrier relatives were normal. Regular checkups at a specialized cardiovascular clinic with annual cardiovascular MRI scans for the adults and echocardiograms in the child are planned. Treatment with the specific betablocker celiprolol has been prescribed in both adult carriers. For the proband in family 6 (FBN1 LPV) testing of the parents was undertaken. The mother carried the reported variant and was extensively examined for Marfan syndrome phenotype (ophthalmological evaluation, echocardiography, cardiac MRI, clinical evaluation of Marfan systemic score) and a medical and family history were obtained. She did not fulfill the diagnostic clinical criteria, resulting in reclassification of the variant as a VUS, and no further clinical testing or treatment was warranted in the proband or relatives.
Genetic counseling issues
While cascade testing is ongoing in seven families (median time since return of SF 1.89 years, IQR 1.63-2.02 yrs), we are aware of four relatives, from two families, who actively declined genetic testing (family 17 and 19). In one family, relatives initially declined genetic testing due to fear that non-paternity unknown to the relatives’ children would be disclosed. Additional genetic consultation of the mother in family 8 was planned at the local genetics department after the initial genetics consult regarding the SF, due to a detected familial risk of breast cancer that was independent of the proband’s disease and SF. Analysis of a panel of breast and ovarian cancer genes in the mother was performed with normal results, why she was recommended annual mammographies due to the moderately increased risk of breast cancer. Similarly, five relatives were recommended additional evaluation, due to a family history of ischemic cardiovascular disease or slightly elevated lipid levels in a non-carrier parent that came to attention during the genetic counseling pertaining the unrelated SF. When these individuals are accounted for, additional follow up or treatment was planned in a total of 16 relatives because of both cascade testing and “downstream” genetic counseling initiated by the return of the SF. While most relatives were tested due to cascade testing (n = 27), eleven relatives were tested or underwent medical evaluation such as segregation analysis.
Discussion
This study elucidates the impact OGS of actionable genes has on patients and relatives in a country with free nationwide health care. The SF rate of 3.2% is higher than previously reported, e.g. by Haer-Wigman et al., in which the detection rate in 1640 healthy individuals was 1.5% in cardiovascular and connective tissue genes [20]. This may be due to the smaller sample size in the current study or differences in population frequencies of certain diseases, such as lipid disorders, which are more prevalent in northern Europeans (1/137 for familial hypercholesterolemia in Denmark) and not reported in the Haer-Wigman paper [21, 22]. A recent study from Germany reported a frequency of SF (2,6%) which is within the confidence limits in the current study, though this study used the ACMG version 3.0 SF list, and did not report lipid disorders [23]. Differences in variant interpretation may also play a role: if only pathogenic variants were reported, the rate would have been 1.2%, which is more in line with previous reports [6]. In this paper, only findings in non-cancer genes are reported, which makes the findings less generalizable, as many (23/59) of the actionable genes are cancer genes. Also, the population has low rates of consanguinity, hence the prevalence of recessively inherited disorders may be higher in other populations.
The uptake of cascade testing was 2.25 pr index patient during an average of 1.6 years of follow up. In seven families, cascade testing is ongoing, why this number will rise. Frey et al. studied uptake of cascade testing in a cohort of relatives to patients with pathogenic variants in hereditary cancer syndromes and similarly found an average of 1.9 relatives pr index case over two years [24]. This underscores that in a public health care setting with easy access to genetic counseling, the effect of disclosing an actionable variant extends beyond the effect for the index case.
In the probands, uptake to genetic counseling or clinical testing for the SF was high (84%, 16/19). We are aware of four relatives that actively declined genetic testing. It is a weakness of our design that we cannot distinguish between relatives that have actively declinedand relatives that simply have not yet been tested. In a recent review of genetic testing in relatives from families with hereditary cancer, uptake was found to be 48% (95% CI 38-58) overall [25]. In a similar meta-analysis of relatives in families with hypertrophic cardiomyopathy uptake varied from 37% to 84% [26]. It appears that uptake was high in our cohort. We propose that this is due to the direct contact with the families and the accessibility of health care in Denmark. Families that have experienced pediatric cancer may be less worried about the risk of cardiovascular disease. Conversely, the families’ behavior and responses may be atypical due a challenging situation with either on-going cancer treatment, terminal disease, or follow-up in survivors. Also, actionability of SFs may be limited by other health issues until later, such as the case of the delayed statin therapy due to concomitant antifungal therapy. Uptake could possibly be facilitated by providing letters to the families explaining the actionable finding and recommended follow-up.
Medical actionability is a key argument for implementation of OGS and the return of SFs. In this study, medical interventions such as regular follow up, dietary or pharmacological treatment were initiated in 61% of probands with a SF, as well as in 17 relatives. In five families, the patients and/or relatives had a phenotype consistent with the SFs, and typically a lipid disorder (families 4, 12, 9, 15 and 16). In one family (family 3, MYBPC3) the phenotype was not quite as clear- only findings on cardiac MRI in one adult carrier were consistent with hypertrophic cardiomyopathy. Interestingly, additional medical management was initiated in asymptomatic individuals on basis of the variant (families 1, 2, 8, 17 and 18) in five families. In the current study, all genetic results including SFs, were documented in the electronic medical record. In this setting, information is accessible to the patient and medical staff, not only researchers. In family 7, where a MYBPC3 variant was reported, but the family did not seek genetic counseling. Thus, reclassification to a benign variant based on ACMG criteria was not changed accordingly in the medical records, resulting in pediatric assessment of a need for additional MUGA scans. Conversely, knowledge of an actionable finding that isn’t acted upon may create ethical or legal issues, which may be elucidated by further research.
Several families reported a family history of cardiovascular disease, but not a previous diagnosis of hereditary disease. This finding emphasizes the fact that SFs can be expected to be disclosed to families unaware of a risk of inherited disease. Counseling in these families is complicated by paucity of penetrance estimates for many diseases in patients without relevant family history, which is highlighted by the fact that several of the carriers of a SF were asymptomatic in this study [27]. Neither probands nor relatives underwent invasive procedures or the implantation of medical devices. This could be expected to be different if cancer predisposition genes such as BRCA1 or BRCA2 were included in the return of SFs, as in a recent paper from the BabySeq project, where the return of a genomic finding in children led to risk reducing surgeries in parents [15].
The risk of burdening patients with concerns that later are unfounded is a general argument against screening [8]. In this study 3 variants were reclassified from LP to VUS as a result of the clinical work up and genetic counseling with implementation of ACMG variant interpretation. Since the development of this study the ACMG guidelines on variant interpretation have become widely accepted and implemented. This development, along with the increased access to variant databases and ongoing work from ClinGen to provide gene specific classification guidelines (e.g. for FBN1), may reduce the probability of variant misclassification in the future [28]. Variant interpretation is a common challenge, both in diagnostic testing as well as in a setting of OGS as reported by Hart et al., where 14 variants of the 76 reported variants (18%) were reclassified as a VUS after evaluation of new evidence [7]. An example of variant misclassification in this study is the reported variant APOB: c.11330 C > A, p.(Ser3777Ter), family 13. This variant was mistakenly classified as a likely pathogenic variant associated with familial hypercholesterolemia type 2 (OMIM# 144010) at the time of reporting, while truncating APOB variants are associated with hypobetalipoproteinemia (OMIM # 615558). Difficult cases were discussed in the academic fora we described, but this variant was not considered there. In hindsight this error highlights that it would have been preferable to discuss all variants in a routine fashion before reporting. Careful variant interpretation is especially important in the setting of SFs and should be undertaken according to best available evidence. Further research in the related psychological and emotional impact in these individuals will be interesting.
While the ACMG position statement provides guidance for actionability in a setting of OGS, certain genetic variants may be more or less “actionable” in different populations or patient cohorts. For example, patients with Charcot-Marie-Tooth disease due to duplication of the PMP22 gene are at higher risk of permanent neurological complications after treatment with certain antineoplastic agents [29]. Knowledge of such a predisposition could potentially be useful when planning oncological treatment. Recently, the list of medically actionable genes was expanded to 81 genes, and could potentially grow after each version, as more knowledge of genotype-phenotype relationships are understood and novel medical therapies become available [3, 4, 30]. As reported by Johnson et al., the returnable variant frequency rate increased by 22% when 73 actionable genes from the third version of the ACMG actionable list were analyzed compared to 59 genes from the second version [31]. If implemented, this can be expected to increase the work load on both variant interpretation laboratories and genetic counseling resources as well as impact more patients and their families. Likewise, it is helpful to have strategies in place, such as academic fora with expertise within different specialties (cardiovascular genetics, genetic oncology etc), to ensure that the management of such secondary findings happens in a standardized manner, while taking into account the specifics of each case such as shared decision-making strategies.
Conclusion
In conclusion, OGS of 595 pediatric cancer patients resulted in the disclosure of a SF to 19 probands (3.1%) and led to clinical workup and genetic counseling in 38 relatives and planned follow up or treatment in 27 individuals. The number of relatives from this cohort offered medical intervention is expected to rise since cascade screening is ongoing. If OGS is undertaken, the resulting health care impact and demand for genetic counseling and workup in relatives extends far beyond the effect in the probands. This real-life experience further emphasizes the need for robust set-ups to ensure expert contribution and clinical consensus from clinicians in all specialties and institutions to care for both patients and relatives. Frameworks for the return of SFs are important to ensure systematic approaches and platforms for the return of such findings [10, 32, 33]. Finally, research in the patient experience, both when correctly, and incorrectly identified as a person at risk of an inherited disorder is warranted, for example via semi-structured interviews of the relatives involved in this study. This study demonstrates the feasibility of genetic screening and illustrates that the health care system in our setting can effectively handle the resulting needs for care in these families.
Data availability
Data analyzed during this study can be found within the tables, figures, and supplementary materials.
References
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. American College of Medical Genetics and Genomics ACMG Recommendations for Reporting of Incidental Findings in Clinical Exome and Genome Sequencing. Genet Med. 2013;15:565–74.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55.
Miller DT, Lee K, Abul-Husn NS, Amendola LM, Brothers K, Chung WK, et al. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2022;24:1407–14.
Miller DT, Lee K, Chung WK, Gordon AS, Herman GE, Klein TE, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23:1381–90.
Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, et al. Actionable exomic incidental findings in 6503 participants: Challenges of variant classification. Genome Res. 2015;25:305–15.
Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013;93:631–40.
Hart MR, Biesecker BB, Blout CL, Christensen KD, Amendola LM, Bergstrom KL, et al. Secondary findings from clinical genomic sequencing: prevalence, patient perspectives, family history assessment, and health-care costs from a multisite study. Genet Med. 2019;21:1100–10.
de Wert G, Dondorp W, Clarke A, Dequeker EMC, Cordier C, Deans Z, et al. Opportunistic genomic screening. Recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2021;29:365–77.
Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20:899–909.
Katz AE, Nussbaum RL, Solomon BD, Rehm HL, Williams MS, Biesecker LG. Management of secondary genomic findings. Am J Hum Genet 2020;107:3–14.
Cannon A, McMillan O, Kelley WV, East KM, Cochran ME, Miskell EL, et al. Medical and psychosocial outcomes of state-funded population genomic screening. Clin Genet. 2023;104:434–42.
Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS, et al. Interpreting secondary cardiac disease variants in an exome cohort. Circ Cardiovasc Genet. 2013;6:337–46.
Ormondroyd E, Harper AR, Thomson KL, Mackley MP, Martin J, Penkett CJ, et al. Secondary findings in inherited heart conditions: a genotype-first feasibility study to assess phenotype, behavioural and psychosocial outcomes. Eur J Hum Genet. https://doi.org/10.1038/s41431-020-0694-9 (2020).
Sapp JC, Facio FM, Cooper D, Lewis KL, Modlin E, van der Wees P, et al. A systematic literature review of disclosure practices and reported outcomes for medically actionable genomic secondary findings. Genet Med. 2021;23:2260–9.
Green RC, Shah N, Genetti CA, Holm IA, Beggs AH, Babyseq T, et al. Actionability of unanticipated monogenic disease risks in newborn genomic screening: Findings from the BabySeq Project Authors ll Actionability of unanticipated monogenic disease risks in newborn genomic screening: Findings from the BabySeq Project. Am J Hum Genet 2023;110:1034–45.
Byrjalsen A, Hansen TVO, Stoltze UK, Mehrjouy MM, Barnkob NM, Hjalgrim LL, et al. Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high frequency of cancer prone syndromes. PLoS Genet. 2020;16:1–24.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Dansk Cardiologisk Selskab: Arvelige hjertesygdomme. 2013. https://www.cardio.dk/arvelige-hjertesygdomme2013.
Aepc CC, Gregers B, Task W, Coordinator F, Behr ER, Kingdom U, et al. ESC Guidelines for the management of V tachyarrhytmias and the prevention of sudden cardiac death death of the European Society of Cardiology (ESC). 2022;3997–4126.
Haer-Wigman L, van der Schoot V, Feenstra I, Vulto-van Silfhout AT, Gilissen C, Brunner HG, et al. 1 in 38 individuals at risk of a dominant medically actionable disease. Eur J Hum Genet. 2019;27:325–30.
Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide prevalence of familial hypercholesterolemia: meta-analyses of 11 million subjects. J Am Coll Cardiol. 2020;75:2553–66.
Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: Prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012;97:3956–64.
Wagener R, Walter C, Surowy HM, Brandes D, Soura S, Alzoubi D, et al. Noncancer-related Secondary Findings in a Cohort of 231 Children With Cancer and Their Parents. J Pediatr Hematol Oncol. 2023;45:E244–8.
Frey MK, Ahsan MD, Badiner N, Lin J, Narayan P, Nitecki R, et al. What happens in the long term: Uptake of cancer surveillance and prevention strategies among at-risk relatives with pathogenic variants detected via cascade testing. Cancer. 2022;128:4241–50.
Frey MK, Ahsan MD, Bergeron H, Lin J, Li X, Fowlkes RK, et al. Cascade testing for hereditary cancer syndromes: should we move toward direct relative contact? A systematic review and meta-analysis. J Clin Oncol. 2022;40:21–5.
Cirino AL, Harris SL, Murad AM, Hansen B, Malinowski J, Natoli JL, et al. The uptake and utility of genetic testing and genetic counseling for hypertrophic cardiomyopathy—A systematic review and meta-analysis. J Genet Couns. 2022;31:1290–305.
Turner H, Jackson L. Evidence for penetrance in patients without a family history of disease: a systematic review. Eur J Hum Genet. 2020;28:539–50.
ClinGen FBN1 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1. 2022;1. https://clinicalgenome.org/working-groups/sequence (accessed 23 Oct2023).
Ibañez-Juliá MJ, Berzero G, Reyes-Botero G, Maisonobe T, Lenglet T, Slim M, et al. Antineoplastic agents exacerbating Charcot Marie Tooth disease: red flags to avoid permanent disability. Acta Oncol (Madr). 2018;57:403–11.
Miller DT, Lee K, Abul-Husn NS, Amendola LM, Brothers K, Chung WK, et al. ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2023;25:1–6.
Johnston JJ, Brennan ML, Radenbaugh B, Yoo SJ, Hernandez SM, Lewis KL, et al. The ACMG SF v3.0 gene list increases returnable variant detection by 22% when compared with v2.0 in the ClinSeq cohort. Genet Med. 2022;24:736–43.
Willis AM, Terrill B, Pearce A, McEwen A, Ballinger ML, Young MA. My Research Results: a program to facilitate return of clinically actionable genomic research findings. Eur J Hum Genet. 2022;30:363–6.
Middleton A, Patch C, Wiggins J, Barnes K, Crawford G, Benjamin C, et al. Position statement on opportunistic genomic screening from the Association of Genetic Nurses and Counsellors (UK and Ireland). Eur J Hum Genet. 2014;22:955–6.
Acknowledgements
We would like to thank all the patients and families that participated in the study and the many colleagues we have worked with through different networks that have contributed to discussions regarding patientcare.
Funding
This study was financially supported by the Danish Childhood Cancer Foundation, The Danish Cancer Society, The European Union’s Interregional Øresund–Kattegat–Skagerrak grant. No funding sources played a role in study design, data collection, analysis, decision to publish, or preparation of the manuscript. Open access funding provided by National Hospital.
Author information
Authors and Affiliations
Contributions
SHH, US, EB, KS, KW and BRD designed the study. SHH, US, EB, TVH, AB, ATH, KJ, KS, JTH, HB, KW and BRD acquired data and played an important role in interpreting the results. SHH, KW and BRD drafted the manuscript. All authors contributed to revising the manuscript, approved the final version, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Ethical approval was obtained through the regional scientific ethical committee (the Ethical Scientific Committees for the Capital Region, H-15016782) and the Danish Data Protection Agency (RH-2016-219, I-Suite no: 04804). All parents/guardians and patients 18 years or older gave formal written consent to germline WGS in the proband as well as written, informed consent to the return of actionable findings as well as scientific reporting of this. Relatives beyond parents that were referred to genetic counselling or clinical workup due to a SF gave written consent to the inclusion of their data in a scientific publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hammer-Hansen, S., Stoltze, U., Bartels, E. et al. Actionability and familial uptake following opportunistic genomic screening in a pediatric cancer cohort. Eur J Hum Genet (2024). https://doi.org/10.1038/s41431-024-01618-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41431-024-01618-7
