
Abstract
Tunicamycins are nucleoside natural products and show antibacterial, antiviral and antitumor activities, which are attributed to their inhibition of enzymatic reactions between polyisoprenyl phosphate and UDP-GlcNAc or UDP-MurNAc-pentapeptide. Because of their various intriguing biological activities, tunicamycins have potential as therapeutic agents for infectious diseases or cancers. Structurally, tunicamycins have a unique structure composed of an undecodialdose skeleton, a lipid chain and a GlcNAc fragment linked by a 1,1-β,α-trehalose-type glycosidic bond. In this mini review, we summarize the total chemical syntheses and biosynthetic studies of tunicamycins.
Similar content being viewed by others

Introduction
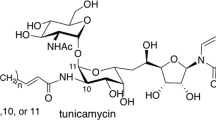
Tunicamycins (Fig. 1) [1,2,3,4,5,6,7] are nucleoside natural products that were first isolated from the fermentation broths of Streptomyces lysosuperficus and later Streptomyces chartreusis. Tunicamycins inhibit enzymatic reactions catalyzed by bacterial phospho-N-acetylmuramyl-pentapeptide transferase (MraY), which is responsible for the biosynthesis of peptidoglycan, and undecaprenyl-phosphate α-N-acetylglucosaminyl 1-phosphate transferase (WecA), which is responsible for the synthesis of lipopolysaccharide and enterobacterial common antigen [8,9,10,11,12,13,14,15,16]. These mechanisms are modes of antibacterial activity of tunicamycins. MraY is an essential enzyme for bacterial survival and a good target for antibacterial drug discovery [17,18,19]. Tunicamycins also strongly inhibit human UDP-N-acetylglucosamine-dolichyl phosphate N-acetylglucosamine-phosphotransferase (GPT), which is responsible for the first N-acetylglucosamination of N-linked glycopeptides in the endothelial reticulum [20]. Tunicamycins are broadly used as biological tools in studies of the N-glycosylation of proteins.

Structures of tunicamycins. The structure shown in a box with broken line is called to tunicamine (2), which is composed of a D-ribose and D-galactosamine. These antibiotic congeners bear a various length of fatty acyl side chains
Tunicamycins have a unique structure comprising an undecodialdose core, referred to as tunicamine (2), decorated with uracil, D-N-acetylglucosamine (GlcNAc) which attached to the core by a 1,1-β,α-trehalose-type glycosidic bond, and an amide-linked fatty acid. Streptovirudins [21], corynetoxins [22], MM19290 [23], mycospocidin [24] and antibiotic 24010 [25] share the same pseudotrisaccharide core, and quinovosamycins [26] are a deoxy congener. These unique structural features and various biological activities have attracted the attention of many researchers to attempt their challenging chemical synthesis or revealed their biosynthetic pathways. Here, we review previous total chemical syntheses and biosynthetic analyses of tunicamycins. The total synthesis of tunicamycin V (1) has been accomplished by Suami’s [27,28,29], Myers’ [30], Yu’s [31] and our groups [32], and many other synthetic studies have been conducted [33,34,35,36,37,38]. Of all the synthetic studies, this review focuses only on the total syntheses.
Total synthesis
The construction of the tunicamine (2) core by a C5′–C6′ bond formation and a 1,1-β,α-trehalose-type glycosylation, which could result in four stereoisomers, are challenging tasks in total synthesis of tunicamycin. Suami and co-workers completed the first total synthesis of 1 in 1985 (Scheme 1) [27,28,29]. They achieved the C5′–C6′ bond formation to afford product 7 via the Henry reaction between 5-deoxy-5-nitro-D-ribose derivative 4 and an aldehyde 6, which was prepared from N-acetylgalactosamine derivative 5. Coupling product 7 was converted to a suitably protected tunicamine derivative 9 via 8, and subsequent protecting group manipulation provided hexaacetate 10. Then, a uracil moiety was introduced to 10 with bis(trimethylsilyloxy)pyrimidine in the presence SnCl4, and the substitution of the 11′-OMe group with Cl via a two-step reaction provided glycosyl chloride 11.

Total synthesis of tunicamycin V by Suami and co-workers. Ac acetyl, Cbz benzyloxycarbonyl, DCC dicyclohexylcarbodiimide, MS4A molecular sieves 4A, TMS trimethylsilyl
The challenging trehalose-type glycosylation using glycosyl chloride 11 and acceptor 12 was conducted in the presence of two silver salts and provide desired (11′-β,1′′-α)-product 13, but undesired (11′-β,1′′-β)-product 14 was also obtained in addition to 13 and no selectivity was observed (~1:1) despite the α-configuration of the anomeric hydroxyl group of the GlcNAc moiety. Glycoside 13 was then converted into tunicamycin V (1) via four additional transformations, including the installation of the fatty acyl side chain.
The second total synthesis was achieved by Myers and co-workers [30]. In their synthetic strategy, the trehalose-type linkage was constructed first, and a C5′–C6′ bond was formed in the last stage of the synthesis. In this type of glycosylation, the choice of glycosyl donor and acceptor played a very important role in the selectivity. The glycosylation of glycosyl acceptor 16 and donor 17 in the presence of TMSOTf as a promoter in CH2Cl2 provided desired glycoside (β,α)-18 and undesired glycoside (β,β)-19 (Scheme 2, eq. 1). The β-selectivity at the anomeric position on the galactosamine side was controlled by neighboring group participation of the phthaloyl (Phth) protecting group on donor 17. However, no selectivity at the anomeric position of the GlcNAc moiety was observed due to the α-dominance of acceptor 16 (α/β = 2/1). On the other hand, the glycosylation of donor 20 and Phth-protected acceptor 21 preferentially provided desired glycoside (β,α)-22 (22/23 ratio = 7/1; eq. 2). In this case, an anomeric hydroxyl group of acceptor 21 was β-dominant (α/β = 1/10) due to the steric effect of the Phth group. The high α-selectivity at the anomeric position of 2-azidoglucose putatively resulted from the restricted transition state conformation of donor 20 in or an SN2-like mechanism in the glycosylation using an imidate donor [39, 40].

Studies of 1,1-trehalose-type glycosylation in total synthesis by Myers and co-workers. Bn benzyl, Bz benzoyl, BOM benzyloxymethyl, Phth phthaloyl, TBS tert-butyldimethylsilyl, Tf trifluoromethanesulfonyl
To achieve the C5′–C6′ bond formation between the disaccharide unit and uridine, they studied intramolecular radical cyclization (Scheme 3). Glycoside 22 was converted to allyl alcohol 24 over several steps, and the formation of a silylene bridge with uridine 5′-aldehyde derivative 25 provided radical cyclization precursor 26 (eq. 1). The intramolecular radical cyclization of 26 proceeded cleanly in the presence of Et3B/Bu3SnH, but undesired (5′S)-product 27 was obtained (eq. 2). On the other hand, precursor 28 without a TBS groups on the uridine moiety preferentially provided desired (5′R)-product 29, which is consistent with the stereochemistry of tunicamycins (eq. 3). After introducing the fatty acyl chain and global deprotection, the total synthesis of tunicamycin V (1) was accomplished.

Total synthesis of tunicamycin V via radical cyclization by Myers and co-workers. Boc tert-butyloxycarbonyl
The total synthesis of tunicamycin V (1) by Yu and co-workers [31] was reported almost two decades after the Myers’ report. Their strategy included an aldol reaction to realize the C5′–C6′ bond formation and two in-house-developed Au-catalyzed glycosylation to introduce the uracil and GlcNAc units (Scheme 4). Linear enol silyl ether 30, used as the C6′–C11′ unit in tunicamycin, was prepared from D-galactosamine in 14 steps. Mukaiyama aldol reaction of 30 and 31 was conducted in the presence of SnCl4, leading to (5′R)-product 32 with high diastereoselectivity (d.r. = 13/1). The resulting (5′R)-hydroxyketone 32 was converted to Phth-protected glycosyl acceptor 35 over several steps, including a 1,3-trans-selective ketone reduction of 32 and formation of lactone 34. Next, they studied a stereoselective trehalose-type glycosylation to introduce the GlcNAc moiety. Glycosylation between 2-azidoglucosyl alkynylbenzoate 36 and acceptor 35 proceeded in the presence of a catalytic amount of Ph3PAuNTf2 in toluene as a solvent at room temperature to give desired pseudotrisaccharide 37 with moderate selectivity in addition to the other three isomers. After the conversion of 37 to alkynylbenzoate 38 over four steps, the second Au-catalyzed glycosylation provided uracil-bearing product 39. Finally, the conversion of the azide group to the corresponding acetamide and the introduction of the fatty acyl chain provided tunicamycin V (1).

Total synthesis of tunicamycin V by Yu and co-workers. AW MS acid washed molecular sieves, BAIB bis-(acetoxy) iodobenzene, MMTr monomethoxytrytyl, PMP p-methoxyphenyl, TBDPS tert-butyldiphenylsilyl, TEMPO 2,2,6,6-tetramethyl-1-piperidinyloxyl, TFA trifluoroacetic acid
Recently, we accomplished the total synthesis of tunicamycin V (1) with a de novo sugar synthesis strategy (Scheme 5) [32]. The Mukaiyama aldol reaction of enol silyl ether 40 and aldehyde 41 in the presence of BF3·OEt2 as a promotor provided desired (5′R)-C5′–C6′ coupling product 42 with very high diastereoselectivity (d.r.>98/2). On the other hand, the use of SnCl4 as a promotor gave the undesired (5′S)-product (d.r. = 96/4). Surprisingly, this stereoselectivity was in contrast to that observed in Yu’s synthesis (Scheme 4, 32). This diastereoselectivity may reflect the difference in the conformation of the ribose ring or in the substituent at the 1′-position [41, 42]. A multistep transformation involving an oxidative furan ring rearrangement of 43 and a stereoselective reduction of enone 44 afforded allyl carbamate 45 and set the stage for the allylcyanate rearrangement to introduce the 10′-amino functional group. Dehydration of the carbamate group provided cyanate 46, which smoothly underwent a [3,3] sigmatropic rearrangement to give 47. The sequential treatment of 47 with TAS-F afforded cyclic carbamate 48. The facial selectivity of the dihydroxylation of olefin in 48 was controlled by the cis-fused structure. After the transformation of diol 49 to Phth-protected glycosyl acceptor 50, the trehalose-type glycosylation between 50 and 2-azidoglucosyl imidate 51 in the presence of a catalytic amount of TfOH in Et2O was conducted according to Myers’ reports [30]. As a result, desired β,α-glycoside 53 was provided in a higher selectivity than was observed in previous reports (β,α-53/β,β-52 = 14/1). Finally, introducing the fatty acyl chain and removing the protection group accomplished the total synthesis of tunicamycin V (1).

Total synthesis of tunicamycin V by our group. mCPBA m-chloroperbenzoic acid, MOM methoxymethyl, NMO N-methylmorpholine-N-oxide, TAS-F tris(dimethylamino)sulfonium difluorotrimethylsilicate, TBAI tetrabutylammonium iodide
Biosynthesis
The Tunicamycin family has a unique and curious structure that contains a C5′–C6′-linked disaccharide, which is unparalleled in nature in terms of biosynthesized compounds. Although numerous studies on the chemical synthesis of tunicamycins have been reported since shortly after its isolation in 1971, biosynthetic studies were not reported until the 2000’s. The biosynthetic study of tunicamycins by Price and co-workers reported a new tunicamycin-producing microorganism (S. chartreusis), and the metabolic origin of tunicamine was revealed [43]. LC-ESI-CD-MS analyses of the stable isotope-labeled tunicamycin indicated that both the D-GlcNAc and D-galactosamine moieties originated from D-glucose (or UDP-GlcNAc) via the sugar metabolic pathway (Fig. 2), which suggested the presence of UDP-GlcNAc 4′-epimerase to produce the galactosamine moiety.

Labeling site by tunicamycin production experiment using labelled D-glucose
In addition, a minimal gene cluster in tunicamycin biosynthesis was identified by Davis and co-workers in 2010 (Fig. 3) [44]. Because tunicamycins are not polyketides or peptide natural products biosynthesized by polyketide synthase or non-ribosomal peptide synthase (NRPS) with highly conserved sequences, they utilized bioinformatics tools and searched genes involving 1 → 1-linking glycosyltransferases, N-acetylhexosamine N-deacetylases, lipid-processing proteins and NDP-hexose epimerase/dehydrase, which were expected to be involved in the construction of the unique substructure of tunicamycins. Through these analyses, tun gene clusters were identified from several microorganisms containing S. chartreusis, which was shown to be a tunicamycin-producing microorganism in a previous report [43]. Considering the putative functions of tun gene products, it was suggested that a TunB/M-catalyzed, enol-aldehyde reductive C5′-C6′ bond formation in the radical process formed the tunicamine scaffold (56 + 58 → 59, Scheme 6), and this was a previously unknown enzymatic reaction, and TunD formed the 11′-β,1′′-α trehalose-type glycosidic bond (59 + UDP-GlcNAc → 60, Scheme 6). Additionally, the diversity of fatty acyl chains in the tunicamycin family is attributed to the use of lipids present in the cells because of the lack of a fatty acid synthase in the tun gene cluster. These insights were consistent with a previous report [44], in which D-glucose (or UDP-GlcNAc) was used as a biosynthetic precursor.

Genetic organization of the tunicamycin biosynthetic gene cluster. The figure was modified from Chem. Sci. 2010 [22]

Proposed biosynthetic pathway by Davis and co-workers in 2010
Although tunicamycin-producing gene clusters were identified and the biosynthetic pathway was proposed, the details of the C–C radical coupling mechanism remained unclear. Davis and co-workers elucidated the true precursor of the C–C radical coupling in 2012 (Fig. 4) [45]. A detailed analysis of the enzymatic reactions revealed that TunF catalyzes the epimerization of the equatorial-hydroxyl groups at the 4-positions in UDP-GlcNAc (57) and UDP-6-deoxy-5,6-ene-GlcNAc (63) to give UDP-GalNAc (62) and UDP-6-deoxy-5,6-ene-GalNAc (64), respectively. In addition, TunA catalyzes the 5,6-dehydration of 57 but not 62. These results indicated that 57 was dehydrated by TunA to give 5,6-ene-product 63, which was subsequently epimerized by TunF to give 64 in the biosynthesis of tunicamycin. Therefore, as shown in the revised biosynthetic pathway (Scheme 7), the C–C radical coupling is predicted to occur between uridine 5′-carbon radical 56 and allyl alcohol 64, instead of unsaturated ketone (58, Scheme 6), which directly gives tunicamine scaffold 59. Interestingly, the total synthesis by Myers’ group [30] mimicked this revised biosynthetic pathway, especially in the construction of the tunicamine scaffold, 18 years before Davis’ study.

The enzymatic experiments of TunA, F

Revised biosynthetic pathway by Davis and co-workers in 2012
In recent studies [46], mutant experiments of a tun gene cluster suggested that six genes (tunABCDEH) are essential for tunicamycin production, but the functions of the five gene products (TunFGKLN) could be replaced by other primary metabolic enzymes. It has also been shown that the deletion of tunMIJ genes induced an increase in tunicamycin sensitivity in the producing bacterium, meaning that the tunMIJ gene products are involved in self-resistance mediated by deactivation via methylation (tunM) and the expulsion (tunIJ) of tunicamycins.
Conclusion
Tunicamycins have various biological activities, including antimicrobial, antivirus and antitumor activities, which result from the inhibition of MraY in prokaryotes and GPT in eukaryotes. Tunicamycins also have a unique C5′-C6′-linked undecodialdose core, which is rare in nature. This has attracted the attention of many researchers, and many chemical synthetic and biosynthetic studies have been conducted. In the total syntheses of tunicamycin V (1), the C5′–C6′ bond formation has been accomplished with a nitroaldol reaction, an intramolecular radical cyclization and a Mukaiyama aldol reaction. On the other hand, biosynthetic studies indicated that the tunicamine core was produced via a radical coupling between uridine and UDP-6-deoxy-5,6-ene-GalNAc, which is a new type of enzymatic reaction in biosynthesis.
The usefulness of tunicamycins as antibacterial agents is limited by their off-target inhibition of human-GPT. To decrease this off-target effect, structural modifications of tunicamycins to achieve target selectivity is required. Recently, the structures of tunicamycin complexed with MraY and GPT were revealed [47,48,49]. According to the co-crystal structure of MraY and GPT, the recognition of GlcNAc moiety in tunicamycin is quite different. Therefore, a GlcNAc-modified analogue was designed, and this analogue inhibited only MraY but not GPT (IC50 = 640 nM vs 15 μM) [48]. As just described, it is expected that the various synthetic methods of tunicamycins in conjunction with the structural information provided by the ligand-protein complexes will facilitate rational drug design to improve selectivity among enzymes, biological activities, metabolic stabilities and so on to improve the applicability of these compounds as antibacterial or anticancer agents.
References
Takatsuki A, Arima K, Tamura G. Tunicamycin, a new antibiotic. I. J Antibiot. 1971;24:215–23.
Takatsuki A, Tamura G. Tunicamycin, a new antibiotic. II. J Antibiot. 1971;24:224–31.
Takatsuki A, Tamura G. Tunicamycin, a new antibiotic. III. J Antibiot. 1971;24:232–8.
Takatsuki A, Tamura G. Effect of tunicamycin on the synthesis of macromolecules in cultures of chick embryo fibroblasts infected with new castle disease virus. J Antibiot. 1971;24:785–95.
Ito T, et al. The structure of tunicaminyl uracil, a degradation product of tunicamycin. Agric Biol Chem. 1977;41:2303–5.
Takatsuki A, et al. The structure of tunicamycin. Agric Biol Chem. 1977;41:2307–9.
Ito T, Takatsuki A, Kawamura K, Sato K, Tamura G. Isolation and structures of components of tunicamycin. Agric Biol Chem. 1980;44:695–8.
Anderson JS, Matsuhashi M, Haskin MA, Strominger JL. Lipid-phosphoacetylmuramyl-pentapeptide and lipid-phosphodisaccharide-pentapeptide: presumed membrane transport intermediates in cell wall synthesis. Proc Natl Acad Sci USA. 1965;53:881–9.
Struve WG, Neuhaus FC. Evidence for an initial acceptor of UDP-NAc-muramyl-pentapeptide in the synthesis of bacterial mucopeptide. Biochem Biophys Res Commun. 1965;18:6–12.
Lehrman MA. A family of UDP-GlcNAc/MurNAc: polyisoprenol-P GlcNAc/MurNAc-1-P transferases. Glycobiology. 1994;4:768–71.
BrandishP. E, et al. Modes of action of tunicamycin, liposidomycin B, and mureidomycin A: inhibition of phosphor-N-acetylmuramyl-pentapeptide translocase from Escherichia coli. Antimicrob Agents Chemother. 1996;40:1640–4.
Boyle DS, Donachie WD. mraY is an essential gene for cell growth in Escherichia coli. J Bactetiol. 1998;180:6429–32.
Schmidt G, Myer H, Mäkelä PH. Presence of rfe genes in Escherichia coli: their participation in biosynthesis of O antigen and enterobacterial common antigen. J Bacteriol. 1976;127:755–62.
Anderson MS, Eveland SS, Price NPJ. Conserved cytoplasmic motifs that distinguish sub-groups of the polyprenol phosphate:N-acetylhexosamine-1-phosphate transferase family. FEMS Microbiol Lett. 2000;191:169–75.
Lehrer J, Vigeant KA, Tatar LD, Valvano MA. Functional characterization and membrane topology of Escherichia coli WecA, a sugar-phosphate transferase initiating the biosynthesis of enterobacterial common antigen and O-antigen lipopolysaccharide. J Bacteriol. 2007;189:2618–28.
Dabbagh BA, Lecreulx DM, Bouhss A. Purification and characterization of the bacterial UDP-GlcNAc:undecaprenyl-phosphate GlcNAc-1-phosphate transferase WecA. J Bacteriol. 2008;190:7141–6.
Kimura K, Bugg TDH. Recent advances in antimicrobial nucleoside antibiotics targeting cell wall biosynthesis. Nat Prod Rep. 2003;20:252–73.
Bugg TDH, Lloyd AJ, Roger DI. Phospho-MurNAc-pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect Dis Drug Targets. 2006;6:85–106.
Winn M, Goss RJM, Kimura K, Bugg TDH. Antimicrobial nucleoside antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat Prod Rep. 2010;27:279–304.
Lehrman MA. Biosynthesis of N-acetylglucosamine-P-P-dolichol, the committed step of asparagine-linked oligosaccharide assembly. Glycobiology. 1991;1:553–62.
Thrum H, et al. Streptovirundins, new antibiotics with antibacterial and antiviral activity. I. Culture taxonomy, fermentation and production of streptovirudin complex. J Antibiot. 1975;28:514–21.
Vogel P, et al. Isolation of a group of glycolipid toxins from seedheads of annual ryegrass (Lolium rigidum Gaud.) infected by Corynebacterium rathayi. Aust J Exp Biol Med Sci. 1981;59:455–67.
Kenig M, Reading C. Holomycin and an antibiotic (MM 19290) related to tunicamycin, metabolites of Streptomyces clavuligerus. J Antibiot. 1979;32:549–54.
Nakamura S, Arai M, Karasawa K, Yonehara H. An antibiotic, mycospocidin. J Antibiot. 1957;10:248–53.
Mizuno M, Shimojima Y, Sugawara T, Takeda I. An antibiotic 24010. J Antibiot. 1971;24:896–9.
Price NPJ, et al. Quinovosamycins: new tunicamycin-type antibiotics in which the α,β-1′′,11′-linked N-acetylglucosamine residue is replaced by N-acetylquinovosamine. J Antibiot. 2016;69:637–46.
Suami T, Sasai H, Matsuno K. Synthesis of methyl hexaacetyl-tunicaminyl uracil. Chem Lett. 1983;12:819–22.
Suami T, et al. Synthetic approach toward antibiotic tunicamycins–VI Total synthesis of tunicamycins. Tetrahedron Lett. 1984;25:4533–6.
Suami T, Sasai H, Matsuno K, Suzuki N. Total synthesis of tunicamycin. Carbohydr Res. 1985;143:85–96.
Myers AG, Gin DY, Rogers DH. Synthetic studies of the tunicamycin antibiotics. Preparation of (+)-tunicaminyluracil, (+)-tunicamycin-V, and 5′-epi-tunicamycin-V. J Am Chem Soc. 1994;116:4697–718.
Li J, Yu B. A modular approach to the total synthesis of tunicamycins. Angew Chem Int Ed. 2015;54:6618–21.
Yamamoto K, Yakushiji F, Matsumaru T, Ichikawa S. Total synthesis of tunicamycin V. Org Lett. 2018;20:256–9.
Danishefsky SJ, DeNinno SL, Chen SH, Boisvert L, Barbachyn M. Fully synthetic stereoselective routes to the differentially protected subunits of the tunicamycins. J Am Chem Soc. 1989;111:5810–8.
Ramza J, Zamojski A. New convenient synthesis of tunicamine. Tetrahedron. 1992;48:6123–34.
Karpiesiuk W, Banaszek A. Highly stereoselective synthesis of α,β-linked, nonreducing disaccharides related to tunicamycin. Carbohydr Res. 1994;261:243–53.
Karpiesiuk W, Banaszek A. Stereoselective syntheses of the O, N-protected subunits of the tunicamycins. Carbohydr Res. 1997;299:245–52.
Sarabia F, Martin-Ortiz L, López-Herrera FJ. Synthetic studies towards the tunicamycins and analogues based on diazo chemistry. Total synthesis of tunicaminyl uracil. Org Biomol Chem. 2003;1:3716–25.
Ichikawa S, Matsuda A. Synthesis of tunicaminyluracil derivatives. Nucleosides, Nucleotides Nucleic Acids. 2004;23:239–53.
Schmidt RR. New methods for the synthesis of glycosides and oligosaccharides – Are there alternatives to the Koenigs-Knorr method? Angew Chem Int Ed. 1986;25:212–35.
Zhu X, Schmidt RR. New principles for glycoside-bond formation. Angew. Chem. Int. Ed. 48, 1900–34 (2009).
Danishefsky SJ, DeNinno MP, Phillips GB, Zelle RE, Lartey PA. On the communication of chirality from furanose and pyranose rings to monosaccharide side chains: anomalous results in the glucose series. Tetrahedron. 1986;42:2809–19.
Chen X, Wiemer DF. α-Phosphono lactone analogues of cytidine and cytosine arabinoside diphosphates: synthesis via ring closing metathesis. J Org Chem. 2003;68:6597–604.
Tsvetanova BC, Kiemle DJ, Price NPJ. Biosynthesis of tunicamycin and metabolic origin of the 11-carbon dialdose sugar, tunicamine. J Biol Chem. 2002;277:35289–96.
Wyszynski FJ, Hesketh AR, Bibb MJ, Davis BG. Dissecting tunicamycin biosynthesis by genome mining: cloning and heterologous expression of a minimal gene cluster. Chem Sci. 2010;1:581–9.
Wyszynski FJ, et al. Biosynthesis of the tunicamycin antibiotics proceeds via unique exo-glycal intermediates. Nat Chem. 2012;4:539–46.
Widdick D, et al. Analysis of the tunicamycin biosynthetic gene cluster of Streptomyces chartreusis reveals new insights into tunicamycin production and immunity. Antimicrob Agents Chemother. 2018;62:e00130–18.
Hakulinen JK, et al. MraY-antibiotic complex reveals details of tunicamycin mode of action. Nat Chem Biol. 2017;13:265–7.
Yoo J, et al. GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat Struct Mol Biol. 2018;25:217–24.
Dong YY, et al. Structures of DPAGT1 explain glycosylation disease mechanisms and advance TB antibiotic design. Cell. 2018;175:1045–58.
Acknowledgements
This research was supported in part by JSPS Grant-in-Aid for Scientific Research (B) (Grant Number 16H05097 to S.I.), Grant-in Aid for Scientific Research on Innovative Areas “Frontier Research on Chemical Communications” (No. 18H04599 to S.I.), Takeda foundation, Astellas Foundation for Research on Metabolic Disorders, The Tokyo Biomedical Research Foundation and was partly supported by Hokkaido University, Global Facility Center (GFC), Pharma Science Open Unit (PSOU), funded by MEXT under “Support Program for Implementation of New Equipment Sharing System”, the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research; BINDS) from the Japan Agency for Medical Research and Development (AMED).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This manuscript is dedicated to Dr Kiyoshi Isono’s 88 years’ anniversary and his long-standing contribution to the study of antibiotics, especially nucleoside antibiotics, polyoxins.
Rights and permissions
About this article

Cite this article
Yamamoto, K., Ichikawa, S. Tunicamycin: chemical synthesis and biosynthesis. J Antibiot 72, 924–933 (2019). https://doi.org/10.1038/s41429-019-0200-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0200-1
This article is cited by
-
Capturing time-dependent activation of genes and stress-response pathways using transcriptomics in iPSC-derived renal proximal tubule cells
Cell Biology and Toxicology (2023)
-
Involvement of Sec71 and Ubp2 in tunicamycin-induced ER stress response in the fission yeast
Molecular Biology Reports (2022)
-
CCAAT/Enhancer-Binding Protein Homologous Protein (CHOP) Deficiency Attenuates Heatstroke-Induced Intestinal Injury
Inflammation (2022)
-
ALG3 contributes to stemness and radioresistance through regulating glycosylation of TGF-β receptor II in breast cancer
Journal of Experimental & Clinical Cancer Research (2021)
-
Liposidomycin, the first reported nucleoside antibiotic inhibitor of peptidoglycan biosynthesis translocase I: The discovery of liposidomycin and related compounds with a perspective on their application to new antibiotics
The Journal of Antibiotics (2019)
