
Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) infections are a significant global health challenge due to the emergence of strains exhibiting resistance to nearly all classes of antibiotics. This necessitates the rapid development of novel antimicrobials to circumvent this critical problem. Screening of compounds based on phenotypes is one of the major strategies for finding new antibiotics at present. Hence, we here performed a phenotypic screening against MRSA and identified NPS-2143 exhibiting activity against MRSA with an MIC value of 16 μg ml−1. In order to discover more potent anti-MRSA agents, a series of derivatives of NPS-2143 were designed and synthesized. The most promising compounds 48 and 49 exhibited favorable antimicrobial activity with an MIC value of 2 μg ml−1.
Similar content being viewed by others
Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) remains one of the most important multidrug-resistant organisms causing healthcare infections. Studies indicate that the incidence of MRSA in the past few years has extensively increased worldwide. Infection due to MRSA imposes a high and increasing burden on healthcare resources, as well as increasing morbidity and mortality. In a recently published report, it is estimated that by 2050, 10 million lives a year will be at risk due to the rise of drug-resistant infections [1,2,3]. Indeed, clinical isolates of both community-associated MRSA (CA-MRSA) [2] and healthcare-associated MRSA (HA-MRSA) have been documented that exhibit resistance to an array of different antibiotic classes including the β-lactams [4, 5], macrolides [6], quinolones [7, 8], tetracyclines [9, 10], and lincosamides [9]. Further exacerbating the problem, strains have emerged which exhibit resistance to first-line antibiotics (such as mupirocin [9, 11] for the treatment of MRSA skin infections) and agents of last resort (such as vancomycin [12, 13] and linezolid [10, 14, 15]). Therefore, there is an urgent need for the development of novel therapeutic agents and treatment strategies to circumvent this significant global health issue.
The discovery of new antibacterial agents which relies on new structures and new action modes plays a significant role in achieving the goal of overcoming bacterial resistance [16, 17]. Several strategies have been employed in the process of traditional antimicrobials discovery, i.e., high-throughput screening based on biological targets, structural modification of the known antimicrobial agents, and phenotypic compounds screening. Although many hit compounds [17, 18] were found by target-based high-throughput screening method, most of these hit compounds exhibit low phenotypic antibacterial activities, difficulty in crossing cell membrane or not absorbed because of the intrinsic efflux pump mechanism of bacteria [17]. Some leading pharmaceutical companies have carried out a large number of high-throughput screening based on molecular targets, but few hit compounds advanced to the clinical research [19]. In contrast, phenotypic screening is highly productive in the history of antimicrobial agents discovery campaign, and a brand new action mechanism may be disclosed with new agents were found in this way [20]. Notably, most antibiotics used clinically are directly obtained through phenotypic screening [17]. Furthermore, novel antibiotic discovery studies, with high originality and new skeletons, are often derived from phenotypic screening. For example, the listed antimicrobial agents bedaquiline [21], retapamulin [22], and GSK299423 [23] currently in clinical research stage in recent years, were all found by phenotypic screening strategy.
Hence, we here performed a phenotypic screening of around 2600 compounds from MCE library against MRSA and identified NPS-2143 (Fig. 1) as a potential hit, which exhibiting activity against MRSA with minimal inhibitory concentration (MIC) value of 16 μg ml−1. The hit molecule, named NPS-2143, is a calcilytic drug which acts as an antagonist at the calcium-sensing receptor (CaSR). The chemical structure of NPS-2143 [24] consists of three parts, i.e., a linear linker with heteroatom incorporated in and substituted, the two aromatic groups in left-hand and right-hand side. In order to find more potent antibacterial candidates and explore the structure-activity relationships (SAR), a series of NPS-2143 derivatives with modifications to these three parts (Fig. 1b) were designed and synthesized.

(a) The chemical structure of NPS-2143; (b) Scheme of the three parts to modify
Result and discussion
Chemistry
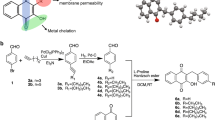
In order to examine the SAR of the right-hand side part of NPS-2143 (Fig. 1b), compounds 3–18 were synthesized following the routes detailed in Fig. 2a. The (R)-2-chloro-6-(2-(oxiran-2-yl)ethoxy)benzonitrile (2a) was synthesized by a regioselective nucleophilic substitution reaction of 2-chloro-6-hydroxybenzonitrile (1a) with commercially available (R)-glycidyl nosylate [25,26,27]. It was previously shown by Sharpless that such nucleophilic displacement proceeded with no racemization [28, 29]. Then, solutions of different amines in ethanol reacted with 2a providing a range of substituted analogs by changing its right-hand side part (3–18) (Table 1).

Synthetic routes of target compounds. Reagents and conditions: (I) (R)-glycidyl nosylate and NaOH, DMF, rt, 5 h; (II) (S)-epichlorhydrin, K2CO3, KI, DMF, 60 °C,4 h; (III) HNR1R2, Et3N, EtOH, reflux, 7 h; (IV) (S)-glycidyl nosylate NaOH, DMF, rt, 5 h; (V) (R)-epichlorhydrin, K2CO3, KI, DMF, 60 °C,4 h; (VI) CDI, Et3N, DCM, r.t. 30 min; (VII) CH3I, NaOH, MeOH, r.t. 1 h; (VIII) Boc2O, Et3N, DCM, r.t. 40 min. CH3I, NaH, DMF, r.t. 1 h. HCl, EtOH, r.t. 20 min; (IX) NaS2O3:5H2O, EtOH, 0–5 °C, 1 h; (X) (S)-epichlorhydrin, K2CO3, acetone, reflux, 8 h; (XI) (R)-epichlorhydrin, K2CO3, acetone, reflux, 8 h; (XII) (R)-epichlorhydrin, AcOH, 75 °C, 8 h; (XIII) (S)-epichlorhydrin, AcOH, 75 °C, 8 h; (XIV) KF, MeCN, reflux, 5 h
Analogs 19–30 and 38–40 were designed to explore the SAR of left-hand side part of NPS-2143. As outlined in Fig. 2a, b), the synthesis started from nucleophilic substitution reaction of various phenols 1b-m [27] or thiophenols 36a-c [30] with (R)-glycidyl nosylate and (S)-epichlorhydrin. Treatment of phenols 1c-i with NaOH, followed by (R)-glycidyl nosylate provided epoxides 2c–2i. By heating substituted phenols 1b,1j-m with excess (S)-epichlorhydrin, K2CO3 and KI, we obtained the corresponding epoxides 2b,2j-m. Thiophenols 36a-c were captured with (S)-epichlorhydrin to yield epoxides 37a-c. Then, solutions of 2-methyl-1-(naphthalen-2-yl)propan-2-amine in ethanol reacted with epoxides 2b-m, 37a-c providing a range of substituted analogs in the left-hand side part (19–30, 38–40). Enantiomers 31 and 32 were obtained in the same ways as NPS-2143 and 30, replacing (R)-glycidyl nosylate and (S)-epichlorhydrin with (S)-glycidyl nosylate and (R)-epichlorhydrin. The chemical structures of analogs 19–30, 38–40 were recorded in Table 2.
Synthesis of analogs 43, 44, 48, and 49 was outlined in Fig. 2c, d. Treatment of 7-bromo-6-chloro-4-(3 H)quinazolinone (41) with (S)-epichlorhydrin and (R)-epichlorhydrin, respectively, in the presence of K2CO3 afforded epoxides 42a and 42b. 3,5-Bis(trifluoromethyl)aniline (45) was reacted with (R)-epichlorhydrin or (S)-epichlorhydrin to yield substituted propanolamines 46a and 46b which afforded epoxides 47a and 47b by treating with KF. Then, solutions of commercially available 2-methyl-1-(naphthalen-2-yl)propan-2-amine in ethanol reacted with corresponding epoxides provide target analogs 43, 44, 48 and 49.
As showed in Fig. 2a, d), analog 33 was produced by treatment of 31 with 1,1′-carbonyldiimidazole (CDI) and triethylamine. Methylation of secondary amine of 31 and analog 48 by using iodomethane afforded target compounds 34 and 50. Methylation of hydroxyl group of 32, whose secondary amine was protected by using di-tert-butyl dicarbonate (Boc2O), produced analog 35 after removal of the protective group.
Anti-MRSA activity
Phenotypic screening results in the identification of the moderately potent hit compound NPS-2143 against MRSA (MIC = 16 μg ml−1). To ascertain the SAR of the lead compound more thoroughly, our initial efforts focused on the SAR of the right-hand side moiety as detailed in Table 1. Replacement of the right-hand side aromatic moiety with alkyl groups provided analogs 3 and 4, which were inactive to MRSA versus NPS-2143 and warranted no further investigation. Thereby, we designed and synthesized a series of analogs 5–16 which contained a benzene ring or pyridine ring in hopes of identifying analogs with greater potency. Unfortunately, each of analogs 5–16 did not exhibit any activities against MRSA at all. Analogs 17 and 18 which contained a naphthalene ring without geminal dimethyl group were also inactive. These results strongly suggest that the geminal dimethyl moiety contained within this amino alcohol template plays a critical role in anti-MRSA activity.
The structure-activity studies to explore the role of the left-hand side part began with holding the right-hand side part beta-naphtyl amine group with the geminal dimethyl moiety as in lead compound NPS-2143 invariant, focusing on variations of substitution on the benzene ring as detailed in Table 2. The 5-hydroxy-1,3-benzodioxole (compound 22, MIC = 64 μg ml−1), 5-bromo-2-methoxyphenyl (compound 25, MIC = 32 μg ml−), 3-methoxyphenyl (compound 28, MIC = 32 μg ml−1) analogs exhibited a 2 or 4-fold decrease in activity versus the lead compound NPS-2143, which clearly demonstrates that electron donating groups on the left-hand side part is detrimental to increasing antibacterial activity. A series of analogs 19, 20, 21, 23, 24, 26 and 27 which contained electron withdrawing groups only afforded an approximately equipotent anti-MRSA activity versus the lead compound NPS-2143 except the analog 30 (MIC = 8 μg ml−1). Replacement of left-hand side moiety with phenyl-sulfhydryl (compounds 38–40) did not give an improvement of anti-MRSA activity neither. However, analog 43 (MIC = 4 μg ml−1) exhibited a 4-fold increase in anti-MRSA activity. As above, we obtained analog 48 which gave a gain in activity anti-methicillin-susceptible Staphylococcus aureus which is not resistant to methicillin (MSSA, MIC = 2 μg ml−1).
Assessment of the role of the stereochemistry of the C-2 secondary hydroxyl group in influencing potency was performed utilizing analogs 30, 43, 49, and the lead compound NPS-2143. As we can see from the Table 3, the C2 S enantiomers 31, 44, and 48 were 2-fold more active than the C2 R enantiomers NPS-2143, 30, 43, and 49. The structure-activity studies focused on the central propanolamine linker were initiated around analogs 31, 32, and 48 (Table 3). Either methylation of the hydroxyl group or cyclization between the hydroxyl group and the secondary amine as in analogs 35 and 33 resulted in a significant loss in activity, suggesting that hydrogens on oxygen atom is critical for anti-MRSA activity. Methylation of the secondary amine as in analogs 34 and 50 did not provoke an obvious increase in activity.
After confirming that analogs 48 and 49 possessed strong antimicrobial activity against MRSA ATCC 33591 and MSSA ATCC 25923, we next assessed their activity against two other MSSA strains which are not resistant to methicillin, MRSA ATCC 43300 and Escherichia coli (E. coli) (Table 4). We found analogs 48 and 49 also have strong activity against other MRSA and MSSA strains. However, they do not have obvious antimicrobial activity against E. coli.
Conclusion
We present herein a novel series of NPS-2143 derivatives exhibiting potent activity against MRSA. A rigorous analysis of the SAR of these analogs reveals the geminal dimethyl groups contained within this amino alcohol moiety and unsubstituted hydroxyl group are critical for the compound’s anti-MRSA activity. Furthermore, substitution with aniline contained electron withdrawing groups results in a compound that exhibits favorable anti-MRSA activity with an MIC value of 2 μg ml−1. But the most potent compounds 48 and 49 did not possess obvious antimicrobial activity against Gram-negative bacteria E. coli. Consequently, it is the first time that naphthylamine compounds were reported to display such low MIC on MRSA strains, and the results present critical information necessary for further analysis and development of these compounds containing an amino alcohol linker and geminal dimethyl moiety as novel antimicrobial agents for treatment of infections caused by MRSA.
Experimental
Chemistry
Chemical reagents and solvents used were purchased from commercial sources (mainly Sigma-Aldrich. Acros and Fisher Scientific).1H-NMR and 13C-NMR spectra were recorded in a DMSO-d6 or CDCl3 solution on a Bruker 400 spectrometer (400 and 100 MHz) using tetramethylsilane as internal standard. Monitoring the reactions and checking the purity of the final products were carried out by thin layer chromatography using visualization with ultraviolet light (UV) at 365 and 254 nm.
General procedure for the synthesis of compounds 3–18
To a solution of 2-chloro-6-hydroxybenzonitrile 1a (1.315 g, 10.6 mmol) in DMF (8 ml) was added NaOH (625 mg, 14.5 mmol). After stirring for 30 min, a solution of (R)-glycidyl nosylate (2.5 g, 9.65 mmol) in DMF (8 ml) was added. The mixture was stirred at room temperature for 5 h (TLC-monitoring). Then the reaction was quenched by adding water (100 ml) and the resulting mixture was extracted with ethyl acetate (3 × 100 ml). The organic phase was washed with brine (100 ml), dried over sodium sulfate, filtrated, and evaporated. Thereafter, column chromatography was performed over silica gel (petroleum ether/CH2Cl2 = 2:1), 1.3 g (68%) of (R)-2-chloro-6-(oxiran-2-yl)methoxybenzonitrile (2a) was obtained as a white solid.
A mixture of the appropriate amine (1.2 mmol) and compound 2a (1.0 mmol) in 20 ml of ethanol was refluxed for 7 h. After cooling, the solvent was removed in vacuo. The crude product was purified on a silica gel column to give pure compounds 3–18.
General procedure for the synthesis of compounds 19–32 and 38–40
To a solution of the phenol 1c-i (1.315 g, 10.6 mmol) in DMF (8 ml) was added NaOH (625 mg, 14.5 mmol). After stirring for 30 min, a solution of (R)-glycidyl nosylate (2.5 g, 9.65 mmol) in DMF (8 ml) was added. The mixture was stirred at room temperature for 5 h (TLC-monitoring). Then the reaction was quenched by adding water (100 ml) and the resulting mixture was extracted with ethyl acetate (3 × 100 ml). The organic phase was washed with brine (100 ml), dried over sodium sulfate, filtrated, evaporated, and purified by silica gel column chromatography (EtOAc/petroleum benzin = 1:8) to give the desired compounds 2c-i.
To a solution of the phenol 1b, 1j-m (1.315 g, 10.6 mmol) in DMF (8 ml) was added K2CO3 (625 mg, 14.5 mmol) and KI. (S)-epichlorhydrin (3.2 ml, 40.8 mmol) was added dropwise and reaction stirred at 60 °C for 6 h (TLC-monitoring). Then the reaction mixture was cooled and quenched by adding water (100 ml) and the resulting mixture was extracted with ethyl acetate (3 × 100 ml). The organic phase was washed with brine (100 ml), dried over sodium sulfate, filtrated, evaporated, and purified by silica gel column chromatography (EtOAc/petroleum benzin = 1:8) to furnish the desired compounds 2b and 2j-m. Enantiomers 2n and 2o were obtained in the same ways as 2a and 2b, replacing (R)-glycidyl nosylate and (S)-epichlorhydrin with (S)-glycidyl nosylate and (R)-epichlorhydrin.
A solution of the thiophenol 36a-c (7.46 g, 41.6 mmol) and Na2S2O3 (13.15 g, 83.2 mmol) in aqueous EtOH (5 ml, 1:1) was cooled to 0 °C. (S)-Epichlorhydrin (3.2 ml, 40.8 mmol) was added dropwise and reaction stirred at 0 °C for 1 h before warming to room temperature for 5 h (TLC-monitoring). The EtOH was removed in vacuo, and the residual aqueous phase was saturated with NaCl and extracted with EtOAc (3 × 10 ml). The combined extract was washed with brine (1 × 10 ml), dried over sodium sulfate, filtrated, evaporated, and purified by silica gel column chromatography (EtOAc/petroleum benzin = 1:8) to give the desired compounds 37a-c.
A mixture of epoxides 2b-o, 37a-c (1.0 mmol), 2-methyl-1-(naphthalen-2-yl)propan-2-amine hydrochloride (1.2 mmol) and trimethylamine (1.2 mmol) in 20 ml of ethanol was refluxed for 7 h (TLC-monitoring). After cooling, the solvent was removed in vacuo. The crude product was purified on a silica gel column to give pure compounds 19–32 and 38–40.
General procedure for the synthesis of compounds 43 and 44
To a solution of 7-bromo-6-chloro-4-(3 H)quinazolinone 41 (2.0 mmol) in acetone (40 ml) was added K2CO3 (2.4 mmol) and (S)-epichlorohydrin or (R)-epichlorhydrin (2.3 mmol), respectively. After refluxing for 12 h, the resulting mixture was concentrated under vacuum and the product was extracted with AcOEt (3 × 100 ml). The AcOEt extract was dried (Na2SO4), filtered and evaporated in vacuo. The crude product was purified on silica gel to afford compounds 42a or 42b.
A mixture of epoxide 42a (0.47 mmol) or 42b (0.47 mmol) and 2-methyl-1-(naphthalen-2-yl)propan-2-amine hydrochloride (0.47 mmol) in 20 ml of ethanol was refluxed for 7 h (TLC-monitoring). After cooling, the solvent was removed in vacuo. The crude product was purified on a silica gel column to give pure compounds 43 or 44.
General procedure for the synthesis of compounds 48 and 49
A mixture of 3,5-bis(trifluoromethyl)aniline (1.31 mmol) (45) and (R)-epichlorhydrin (6.55 mmol) or (S)-epichlorhydrin (6.55 mmol) in acetic acid (10 ml) was heated to 75 °C. After 2 h, the reaction mixture was cooled to room temperature. The resulting mixture was concentrated under vacuum and the product was extracted with AcOEt (3 × 100 ml). The AcOEt extract was dried (Na2SO4), filtered and evaporated in vacuo. The crude was purified on the silica gel to afford compound 46a or 46b. The solution of compound 46a (0.83 mmol) or 46b (0.83 mmol) and KF (2.49 mmol) in acetonitrile (10 ml) was refluxed for 3 h. Then the resulting mixture was filtered and the filtrate was concentrated under vacuum, the product was purified on the silica gel to afford epoxides 47a or 47b.
A mixture of epoxide 47a (0.29 mmol) or 47b (0.29 mmol), 2-methyl-1-(naphthalen-2-yl)propan-2-amine hydrochloride (0.43 mmol), and triethylamine (0.43 mmol) in 15 ml of ethanol was refluxed for 7 h (TLC-monitoring). After cooling, the solvent was removed in vacuo. The crude product was purified on a silica gel column to give pure compounds 48 or 49.
General procedure for the synthesis of compounds 33–35, 50
A mixture of 31 (0.073 mmol), 1,1′-carbonyldiimidazole (CDI) (0.183 mmol) and trimethylamine (0.073 mmol) in 10 ml of dichloromethane was stirred at room temperature for 5 h (TLC-monitoring). Then the solvent was removed in vacuo. The crude product was purified on a silica gel column to give pure compound 33.
A mixture of 31 (0.145 mmol) or compound 48 (0.145 mmol), iodomethane (0.290 mmol), and NaOH in 10 ml of methanol was stirred at room temperature for 3 h (TLC-monitoring). Then the solvent was removed in vacuo. The crude product was purified on a silica gel column to give pure compounds 34 and 50.
To a solution of compound 32 (0.30 mmol) and NaOH (0.90 mmol) in H2O (4 ml) and 1,4-dioxane (4 ml) was added di-tert-butyl dicarbonate (Boc2O) (0.90 mmol). The mixture was stirred at room temperature for 1 h (TLC-monitoring). Then the resulting mixture was extracted with AcOEt (3 × 10 ml). The AcOEt extract was dried (Na2SO4), filtered and evaporated in vacuo. The crude was purified on the silica gel to afford N-Boc intermediate. The intermediate was treated with NaH (2.40 mmol) and iodomethane (3.40 mmol) in DMF (3 ml), after stirring for 3 h (TLC-monitoring) at room temperature. Then the resulting mixture was extracted with AcOEt (3 × 10 ml). The AcOEt extract was dried with Na2SO4, filtered and evaporated in vacuo. The crude was purified on the silica gel to afford another N-protected methoxy intermediate. Then this intermediate was treated with hydrochloric acid in ethanol (10 ml). After stirring for 1 h at room temperature, the resulting mixture was concentrated under vacuum and the product was extracted with AcOEt (3 × 10 ml). The AcOEt extract was dried (Na2SO4), filtered and evaporated in vacuo. The crude product was purified on the silica gel to afford compound 35.
Microbiology
In vitro anti-MRSA activity
Bacteria strains MRSA ATCC 33591, MRSA ATCC 43300, MSSA ATCC 25923, MSSA ATCC 6538, MSSA ATCC 25913, and E. coli ATCC 25922 were purchased from the American Type Culture Collection. All synthesized compounds were tested for their in vitro antibacterial activity against MRSA, MSSA, and E. coli by performing a microdilution (MIC). Methicillin-resistant Staphylococcus aureus (MRSA and MSSA) was cultured in Mueller–Hinton broth (MH, Oxoid, Basingstoke, England) at 37 °C under aerobic conditions. MICs of NPS-2143 analogs used against MRSA were determined in triplicate using flat-bottom 96-well microtiter plates (TPP, Trasadingen, Switzerland).
References
O'neill J. Tackling drug-resistant infections globally: final report and recommendations (2016). http://amr-review.org/sites/default/files/160525_Final%20paper_with%20cover.pdf. March 22, 2019.
Rodvold KA, McConeghy KW. Methicillin-resistant Staphylococcus aureus therapy: past, present, and future. Clin Infect Dis. 2014;58(Suppl 1):S20–27.
Lee AS, de Lencastre H, Garau J, Kluytmans J, Malhotra-Kumar S, Peschel A, et al. Methicillin-resistant Staphylococcus aureus. Nat Rev Dis Prim. 2018;4:18033.
Henry F, Chambers MD. Community-associated MRSA—resistance and virulence converge. N Engl J Med. 2005;352:1485–7.
Mun SH, Kim SB, Kong R, Choi JG, Kim YC, Shin DW, et al. Curcumin reverse methicillin resistance in Staphylococcus aureus. Molecules. 2014;19:18283–95.
Moran GJ, Krishnadasan A, Gorwitz RJ. Methicillin-Resistant S. aureus Infections among Patients in the Emergency Departmen. N Engl J Med. 2006;355:666–74.
Frazee BW, Lynn J, Charlebois ED, Lambert L, Lowery D, Perdreau-Remington F. High prevalence of methicillin-resistant Staphylococcus aureus in emergency department skin and soft tissue infections. Ann Emerg Med. 2005;45:311–20.
Fridkin ScottK, Hageman JeffreyC, Morrison Melissa. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352:1436–44.
Han LL, McDougal LK, Gorwitz RJ, Mayer KH, Patel JB, Sennott JM, et al. High frequencies of clindamycin and tetracycline resistance in methicillin-resistant Staphylococcus aureus pulsed-field type USA300 isolates collected at a Boston ambulatory health center. J Clin Microbiol. 2007;45:1350–2.
Khan A, Wilson B, Gould IM. Current and future treatment options for community-associated MRSA infection. Expert Opin Pharmacother. 2018;19:457–70.
Patel JB, Gorwitz RJ, Jernigan JA. Mupirocin resistance. Clin Infect Dis. 2009;49:935–41.
Hiramatsu K. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect Dis. 2001;1:147–55.
Elsebaei MM, Mohammad H, Abouf M, Abutaleb NS, Hegazy YA, Ghiaty A, et al. Alkynyl-containing phenylthiazoles: systemically active antibacterial agents effective against methicillin-resistant Staphylococcus aureus (MRSA). Eur J Med Chem. 2018;148:195–209.
Locke JB, Morales G, Hilgers M, G CK, Rahawi S, Jose Picazo J, et al. Elevated linezolid resistance in clinical cfr-positive Staphylococcus aureus isolates is associated with co-occurring mutations in ribosomal protein L3. Antimicrob Agents Chemother. 2010;54:5352–5.
Wilson P. Linezolid resistance in clinical isolates of Staphylococcus aureus. J Antimicrob Chemother. 2002;51:186–8.
Conlon BP. Staphylococcus aureus chronic and relapsing infections: Evidence of a role for persister cells: an investigation of persister cells, their formation and their role in S. aureus disease. Bioessays. 2014;36:991–6.
Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov. 2015;14:529–42.
de Jonge BL, Walkup GK, Lahiri SD, Huynh H, Neckermann G, Utley L, et al. Discovery of inhibitors of 4′-phosphopantetheine adenylyltransferase (PPAT) to validate PPAT as a target for antibacterial therapy. Antimicrob Agents Chemother. 2013;57:6005–15.
Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40.
Gavrish E, Sit CS, Cao S, Kandror O, Spoering A, Peoples A, et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol. 2014;21:509–18.
Cholo MC, Mothiba MT, Fourie B, Anderson R. Mechanisms of action and therapeutic efficacies of the lipophilic antimycobacterial agents clofazimine and bedaquiline. J Antimicrob Chemother. 2017;72:338–53.
Septimus EJ, Schweizer ML. Decolonization in prevention of health care-associated infections. Clin Microbiol Rev. 2016;29:201–22.
Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature. 2010;466:935–40.
Marquis RobertW, Lago AmparoM, Callahan JamesF, Lee Trout RobertE, Gowen Maxine, DelMar. EricG, et al. Antagonists of the calcium receptor I. Amino alcohol-based parathyroid hormone secretagogues. J Med Chem. 2009;52:3982–93.
Yang W, Wang Y, Roberge JY, Ma Z, Liu Y, Michael Lawrence R, et al. Discovery and structure-activity relationships of 2-benzylpyrrolidine-substituted aryloxypropanols as calcium-sensing receptor antagonists. Bioorg Med Chem Lett. 2005;15:1225–8.
Martinez A, Dutasta J-P. Hemicryptophane–oxidovanadium(V) complexes: Lead of a new class of efficient supramolecular catalysts. J Catal. 2009;267:188–92.
Hansen HD, Lacivita E, Di Pilato P, Herth MM, Lehel S, Ettrup A, et al. Synthesis, radiolabeling and in vivo evaluation of [(11)C](R)-1-[4-[2-(4-methoxyphenyl)phenyl]piperazin-1-yl]-3-(2-pyrazinyloxy)-2-p ropanol, a potential PET radioligand for the 5-HT(7) receptor. Eur J Med Chem. 2014;79:152–63.
Teixeira F, Mosquera R, Melo A, Freire C, Cordeiro MN. Driving forces in the sharpless epoxidation reaction: a coupled AIMD/QTAIM study. Inorg Chem. 2017;56:2124–34.
Corey EJ. On the origin of enantioselectivity in the katsuki-sharpless epoxidation procedure. J Org Chem. 1990;55:1693–4.
Apparano S, Schmidt RR. Synthesis of functionally substituted α,β-unsaturated carbonyl compounds. Synthesis. 1987;10:896–9.
Acknowledgements
The authors acknowledge National Natural Science Foundation of China (No.81473253) for financial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, Y., Ju, Y., Li, C. et al. Design, synthesis, and antibacterial evaluation of novel derivatives of NPS-2143 for the treatment of methicillin-resistant S. aureus (MRSA) infection. J Antibiot 72, 545–554 (2019). https://doi.org/10.1038/s41429-019-0177-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0177-9
