
Abstract
Three cyclic oxoester-thioester hybrid monomers, 1 (3-methyl-1,4-oxathiane-2,5-dione), 2 (6-methyl-1,4-oxathiane-2,5-dione), and 3 (3,6-dimethyl-1,4-oxathiane-2,5-dione), were studied for anionic and cationic ring-opening polymerizations. These monomers are six-membered cyclic cross-dimers corresponding to combinations of glycolic and lactic acids with their thiol analogs. Anionic polymerizations using thiol as the initiator and 2,6-lutidine as the base catalyst were successful for the chemoselective cleavage of the thioester with the thiol propagating end. The polymerizability increased in the order of 3 < 1 < 2, which was in good agreement with the increasing ring strain order evaluated by Density Functional Theory calculations. The living character, to some extent, was suggested by the postpolymerization reactions, which involved a two-stage feed of the monomers and a thiol-ene terminal coupling reaction to form a block copolymer with PEG. Additionally, it was found that the polymerization took place in 2,6-lutidine without a thiol initiator and produced macrocyclic polymers. The cationic polymerizations took place with the aid of CF3SO3H and benzyl alcohol but involved side reactions with low chemoselective ring cleavage. The thioester unit caused the polymers to exhibit a lower Tg with greater thermal and photo degradability.
Similar content being viewed by others

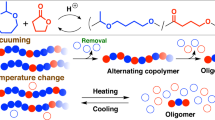

Introduction
Compared with the usual carboxylic ester, thioester has a more electrophilic carbonyl carbon (Character 1) and forms a more stable carbonyl α-anion (Character 2) due to the weaker π-n conjugation between the carbonyl group and the sulfur atom. These characteristics play essential roles in the bioreaction of trans-acylation and C-C bond formation, representatively, through metabolic pathways related to CoA. However, thioesters have rarely been studied for polymer synthesis, while there are a huge number of studies on polyesters and polyamides.
We have engaged in the elucidation of thioester chemistry in the field of polymerization. Characters 1 and 2 of thioester prompted two research projects, i.e., ring-opening polymerization (ROP) of thiolactone and anionic polymerization of (meth)acrylic thioester, respectively. We have already reported that highly strained β-thiolactone undergoes thiol-initiated ROP without the use of a catalyst [1]. Less-strained rac-thiolactide was found to require amine catalysts for equilibrium polymerization [2]. It has been revealed that an acrylic thioester undergoes anionic polymerization via a twitter ionic mechanism initiated by amines, demonstrating higher anionic polymerizability than that of acrylate [3].
Recently, related studies based on Character 1 have increased in number. They include not only studies on ROPs but also those focusing on polycondensation, polyaddition, polymer reactions at the side and main chains, and ligations at the polymer terminal groups [4,5,6,7,8,9,10,11,12]. Trends in sustainable chemistry prompted the study of reversible ROPs by using the dynamic property of thiol-thioester exchange reactions [7, 9,10,11,12]. Tao et al. reported that six-membered dithiolactones, including thiolactide, are monomers that perform well [11]. His group very recently extended a similar work for monothiodilactones consisting of a glycolic thioester component [12]. Independently, we have also extended our previous studies mentioned above for the ROP of six-membered cyclic hybrid dimers composed of an oxoester and thioester (monomers 1, 2, and 3, Scheme 1) since 2019. We undertook this project to examine the selective ring cleavage of the thioester and oxoester parts. The anionic condition is expected to result in thioester cleavage due to its higher electrophilicity. Monomer 1 has been employed also by Tao et al. and revealed to undergo such ROP [12]. On the other hand, it is possible that the oxoester is more reactive under acidic conditions, which was investigated in this article. How the methyl substituent on the monomer ring affects the ROP has also been investigated. The chemo- and regioselective ROPs are known for other six-membered cyclic hybrid dimers consisting of amide-oxoester, amide-thioester, and glycolic-lactic oxoesters [13, 14]. Therein, ROPs are generated at the oxoester and thioester due to the much lower reactivity of the amide group [13], and methylglycolide undergoes ROP via lactyl cleavage [14]. These works produce polymers consisting of alternating units; amide-oxoester, amide-thioester, and glycolic-lactic oxoester polymers are obtained in a controlled fashion. The hybrid monomers studied herein are expected to yield polymers composed of alternating oxoester-thioester units.
Experimental section
General methods and materials
The instruments, measurement conditions, and material information are described in the Supplementary Information.
Synthesis of monomer 1 (3-methyl-1,4-oxathiane-2,5-dione)
Thiolactic acid (8.7 mL, 0.1 mol) was slowly added via a syringe to bromoacetyl bromide (8.8 mL, 0.1 mol) in a two-neck flask under N2. The mixture was stirred at r.t. overnight, while the evolved HBr gas was introduced over NaOH aq. through an outlet natural rubber tube with an N2 stream. Water (200 mL) was added to the reaction mixture, which was subsequently extracted with CH2Cl2 (2×200 mL). The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The product was used for cyclization without further purification.
The residue (16.46 g, 72.4 mmol) was dissolved in dry MeCN (200 mL) under N2 and cooled to 0 °C. Dry (iPr)2NEt (12 mL, 70.6 mmol) dissolved in dry MeCN (17 mL) was added via a syringe. The mixture was then allowed to warm to room temperature and stirred overnight. Diethyl ether was added, and (iPr)2NEt hydrobromide was removed by filtration. The filtrate was evaporated under reduced pressure, and the crude mixture was purified by silica gel column chromatography (CH2Cl2, Rf = 0.47). The product was distilled (131 °C/270 Pa), yielding 1 as a colorless liquid (2.16 g, 20% yield), which was crystallized in a refrigerator (m.p. 41.4–42.5 °C). 1H NMR (CDCl3, Fig. S1): δ 1.62 (d, J = 6.9 Hz, 3H), 4.21 (q, J = 6.9 Hz, 1H), 4.75 (d, J = 15.3 Hz, 1H), 4.93 (d, J = 15.3 Hz, 1H). 13C NMR (CDCl3, Fig. S2): δ 14.45, 36.00, 74.07, 168.20, 194.88. IR (KBr): Fig. S3. MS (EI, m/z): Found 146.0027, Calc. 146.0038 (M+).
Synthesis of monomer 2 (6-methyl-1,4-oxathiane-2,5-dione)
Monomer 2 was prepared by the same procedure as 1. Starting from thioglycolic acid (6.14 mL, 0.093 mol) and 2-bromopropionyl bromide (9.75 mL, 0.093 mol), 2 was produced via the crude precursor (17.6 g, 77.5 mmol). Monomer 2 was purified by silica gel column chromatography (CH2Cl2, Rf = 0.41) and subsequent vapor-diffusion recrystallization (EtOAc with CCl4) in 37% yield (4.14 g, m.p. 101.3–102.4 °C). 1H NMR (CDCl3, Fig. S4): δ 1.58 (d, J = 6.5 Hz, 3H), 3.70 (d, J = 15.2 Hz, 1H), 4.13 (d, J = 15.2 Hz, 1H), 4.98 (q, J = 6.5 Hz, 1H). 13C NMR (CDCl3, Fig. S5): δ 14.91, 30.06, 79.84, 165.29, 196.13. IR (KBr): Fig. S6. MS (EI, m/z): Found 146.0039, Calc. 146.0038 (M+).
Synthesis of monomer 3 (3,6-dimethyl-1,4-oxathiane-2,5-dione)
Monomer 3 was prepared by the same procedure as 1. Starting from thiolactic acid (4.42 mL, 0.05 mol) and 2-bromopropionyl bromide (5.27 mL, 0.093 mol), 3 was produced via the crude precursor (8.77 g, 36.4 mmol). Monomer 3 was purified twice by silica gel column chromatography (hexane/EtOAc = 5/3, Rf = 0.46, and subsequently CH2Cl2, Rf = 0.55) in 42% yield (2.44 g, RR and SS isomers/RS and SR isomers = 95/5, m.p. 34.3–35.5 °C). 1H NMR (CDCl3, Fig. S7) of the RR and SS isomers: δ 1.57 (d, J = 6.5 Hz, 3H), 1.60 (d, J = 6.8 Hz, 3H), 4.28 (q, J = 6.8 Hz, 1H), 5.02 (q, J = 6.5 Hz, 1H); the RS and SR isomers: δ 1.67 (d, J = 7.1 Hz, 3H), 1.71 (d, J = 7.2 Hz, 3H), 4.19 (q, J = 7.2 Hz, 1H), 5.03 (q, J = 7.1 Hz, 1H). 13C NMR (CDCl3, Fig. S8) of the RR and SS isomers: δ 14.18; 14.68; 37.03; 79.90; 168.70; 196.76. IR (KBr): Fig. S9. MS (EI, m/z): Found 160.0219, Calc. 160.0194 (M+).
A representative procedure for anionic polymerization
Monomer 1 (100 mg, 0.684 mmol) was dissolved in dry CH2Cl2 (0.46 mL) in a test tube under N2. A solution of dry 2-ethylhexyl thioglycolate (140 µL, 136 mg, 0.666 mmol) in dry CH2Cl2 (2.0 mL, 2.730 g) was prepared in advance, and a portion (147 mg, 0.0342 mmol of 2-ethylhexyl thioglycolate) was added to the test tube. Another solution of dry 2,6-lutidine (163 µL, 150 mg, 1.40 mmol) in dry CH2Cl2 (2.0 mL, 2.688 g) was also prepared in advance, and a portion (138 mg, 0.068 mmol of 2,6-lutidine) was subsequently added to the test tube. The mixture was stirred under a closed system at r.t. for 24 h. Amberlyst 15E (15 mg, H+ = 0.071 mmol) was added to the reaction mixture to exclude 2,6-lutidine. After 30 min of stirring, the supernatant collected by filtration was dried under vacuum and analyzed by 1H NMR spectroscopy and GPC. The polymer was isolated by reprecipitation into diethyl ether. The 1H and 13C NMR and IR spectra of Poly1–3 are available in the Supplementary Information (Figs. S11–S19).
A representative procedure for cationic polymerization
Monomer 1 (100 mg, 0.684 mmol) was dissolved in dry CH2Cl2 (0.65 mL) in a test tube under N2. A solution of dry benzyl alcohol (35.5 µL, 37.0 mg, 0.342 mmol) in dry CH2Cl2 (0.3 mL, 0.396 g) was prepared in advance, and a portion (43.6 mg, 0.0342 mmol of benzyl alcohol) was added to the test tube. Subsequently, trifluoromethanesulfonic acid (5.2 mg, 0.034 mmol) was also added. The mixture was stirred under a closed system at r.t. for 24 h. Amberlyst 21 (8 mg, base = 0.034 mmol) was added to the reaction mixture to remove the acid. After 30 min of stirring, the supernatant collected by filtration was dried under vacuum and analyzed by 1H NMR spectroscopy and GPC.
Synthesis of the block copolymer by the thiol-ene coupling reaction
The dried reaction mixture (64.9 mg) of poly1, which was prepared in the presence of 2-ethylhexyl thioglycolate (6.93 mol% for monomer 1, 0.023 mmol) with 2,6-lutidine, was transferred into a Pyrex® test tube. Norbornene-terminated PEG 2000 (73 mg, 0.034 mmol) and DMPA (2,2-dimethoxy-2-phenylacetophenone) (2.9 mg, 0.009 mmol) were added, and then CH2Cl2 (0.4 mL) was added under N2. The solution was degassed by three consecutive freeze‒pump–thaw cycles. This mixture was subjected to irradiation from a high-pressure Hg lamp for 4 h. The resultant solution was dried in vacuo and analyzed by GPC.
A typical procedure for UV-photolysis of the polymer
A CH2Cl2 (0.3 mL) solution of poly1 (3 mg, [C( = O)S] = 68.5 mM) in a quartz test tube was exposed to a low-pressure Hg lamp under N2 at r.t. After 3 h, a small aliquot from the CH2Cl2 solution was subjected to GPC analysis.
Calculation methods
The density functional theory (DFT) calculation methods are described in our previous study [2], and the detailed results are available in the Supplementary Information.
Results and discussion
Monomer synthesis
Monomer 1 was prepared from bromoacetyl bromide and thiolactic acid (Scheme 1) by a modified procedure from a previous report [15]. This procedure was also applied in the preparation of monomers 2 and 3. The 1H NMR spectrum (Fig. S7) suggested that the obtained monomer 3 was a diastereomeric mixture (95/5). The major diastereomers were identified by the NOESY spectrum as the RR and SS isomers (Fig. S10). A cross peak between the two methine protons of the major diastereomer was observed, suggesting that it has RR and SS stereochemistry.

Monomer syntheses
Anionic polymerization by combining a thiol initiator with 2,6-lutidine
In a previous report from our group, pyridine was found to effectively catalyze the bulk polymerization of thiolactide [2]. Thus, the ring-opening polymerizations of monomers 1, 2, and 3 were investigated by using a combination of 2-ethylhexyl thioglycolate as the initiator with pyridine as the catalyst in CH2Cl2 at r.t. (Table 1). However, the polymerizations were so slow in producing the oligomer that the stronger base 2,6-lutidine (pKa = 6.60 in H2O) (https://pubchem.ncbi.nlm.nih.gov/) was employed in place of pyridine (pKa = 5.23 in H2O) (https://pubchem.ncbi.nlm.nih.gov/). As expected, the polymerizations were more efficient for monomers 1 and 2 (Table 2). Increasing the amount of 2,6-lutidine from 5 mol% to 10 mol% apparently accelerated the polymerization. Figure 1 shows the living characteristics of the polymerizations of 1 and 2 at the stages at which the low-molecular-weight polymers were produced. However, decreasing the amount of initiator produced a higher molecular weight polymer, which was not precisely controllable. For example (Entry 9), using 1 mol% initiator afforded a polymer with Mn = 7100, which was much smaller than the theoretical value of 11500 (monomer conv. = 79%); this is probably due to the backbiting reaction (vide infra), the relative rate of which increases in comparison with that of propagation at a low concentration of the initiator, causing a larger PDI.

Conversion dependence of Mn and Mw/Mn of polymers from 1 (a) and 2 (b) in the presence of 2-ethylhexyl thioglycolate (5 mol%) and 2.6-lutidine (10 mol%) in CH2Cl2 ([Monomer] = 1 M) at r.t
In agreement with our expectation, as suggested by the 1H NMR spectra of the obtained polymers, ring opening selectively occurs at the C(=O)-S bond and the propagating chain end is the thiol group. Figure 2 shows signals due to the terminal protons, i.e., the methine and thiol, produced from monomer 1. Their peak assignment was assisted by the spectrum of the stoichiometric reaction mixture of monomer 1 with the initiator (Fig. S20). The signal due to the thiol proton disappeared upon the addition of a drop of D2O. Similarly, selective ring cleavage at the thioester was also confirmed for monomers 2 and 3 (Figs. S21–S24).

1H NMR spectrum (CDCl3) of the reaction mixture after 3 days of Entry 1 (Table 2)
Table 2 suggests that monomers 3, 1, and 2 are in order of increasing polymerizability. The time-conversion curves of monomers 1 and 2 also suggested that 2 was more reactive (Fig. 3A). As shown in Fig. 3B, the propagating end of poly1 is the secondary thiol, which attacks the α-unsubstituted thioester carbonyl. On the other hand, in the propagation of poly2, the primary thiol reacts with the α-methyl thioester carbonyl; the experimental results suggest that the latter is faster, which is consistent with the literature about regioselective ring opening in the polymerization of methylglycolide, initiated by alcohol with a base [14]. The propagating end is the primary alcohol, which selectively attacks the α-methyl ester carbonyl. Because of the steric hindrance between the two methyl groups located at the propagating end and at the monomer, the polymerization rate of monomer 3 is the lowest.

A Time-conversion curves of the polymerizations of monomers 1 and 2 in the presence of 2-ethylhexyl thioglycolate (5 mol%) and 2.6-lutidine (10 mol%) in CH2Cl2 ([Monomer] = 1 M) at r.t. B Propagations in the polymerizations of monomers 1 and 2
Thermodynamic consideration of the ring strain of monomers 1–3 provides additional insight into their polymerizability. DFT calculations (ωB97X-D/6-311 + G(d,p) with PCM toluene) [2] were used to evaluate the ring strain energy by using homodesmotic reactions (Eq. (1)).
The ring strain energy values (ΔH) are 8.96, 9.82, and 8.47 kcal/mol for monomers 1, 2, and 3 (the RR and SS isomers), respectively. Thus, the thermodynamic aspect also suggests that the order of increasing polymerizability is 3 < 1 < 2, which is consistent with the experimental results. In a previous study by our group [2], the same DFT calculations for rac-thiolactide and rac-lactide revealed that their ring strain energies were 5.45 and 11.27 kcal/mol, respectively. Monomer 3 is a hybrid compound and reasonably has a medium ring strain. The longer bond length of the sulfur atom reduces the strain of the six-membered bislactone ring.
Figure 4 shows the MALDI-TOF-MS spectrum of the polymerization mixture of monomer 1 (Entry 2, Table 2). Two types of regular peak series were observed for the polymers. One is due to the linear polymer, whose terminals have an initiator fragment and a thiol proton, and the other is ascribable to the cyclic polymer, which is produced by the backbiting reaction. Poly2 was also revealed to contain both linear and cyclic structures (Fig. S25). However, their relative contents are not informed by the MS spectra, so chain extension experiments were performed. Either monomer 1 or monomer 2 was added to the polymerization mixture, and the products were analyzed by GPC before and after polymerization (Fig. 5). The GPC profiles clearly shift to the higher molecular weight region, suggesting that chain extension occurred from the thiol terminals of the linear polymers and that the relative contents of the cyclic polymers were very small. Another chain extension experiment involves a thiol-ene reaction to form a block copolymer [1]. The terminal thiol groups of the linear polymers from 1 and 2 were subjected to photoinitiated radical addition to the norbornene group at the PEG (MW2000) terminal (Scheme 2). Compared with those of the starting polymers, the GPC profiles of the reaction mixtures shifted to the higher molecular weight region (Fig. 6 and S26). When 1.5 eq of PEG was added, the RI detector showed a bimodal GPC profile due to the block copolymer with unreacted PEG. On the other hand, the UV detector shows no peak due to the presence of PEG, which absorbs no UV. UV-detected GPC profile is monomodal, however, with two shoulders in the higher and lower molecular weight regions; the former is probably due to disulfide-forming coupling at the polymer terminals, and the latter is ascribable to the cyclic polymer slightly involved in the starting polymer.

MALDI-TOF-MS spectrum of the polymerization mixture of monomer 1 (Entry 2, Table 2)

Postpolymerization of monomers 1 and 2 in the presence of 2-ethylhexyl thioglycolate (6 mol%) and 2.6-lutidine (10 mol%)

Thiol-ene reaction to produce the block copolymer

GPC profiles of the thiol-ene reaction mixture from poly2, detected by RI (upper) and UV (lower)
Anionic polymerization in 2,6-lutidine as the solvent
The polymerization of monomer 3 using a catalytic amount of 2,6-lutidine (Entry 10, Table 2) was so slow that 2,6-lutidine was used as the polymerization solvent. As shown in Table 3, the polymerization was accelerated; however, a decrease in the amount of initiator did not increase the molecular weight of the polymer. This finding prompted us to investigate whether the polymerization can occur even in the absence of the thiol initiator. As a result, monomer 1 was found to undergo polymerization in 2,6-lutidine without a thiol initiator (Entries 1–3, Table 4); however, multimodal GPC profiles were obtained (Fig. 7). The MALDI-TOF-MS spectrum of the polymerization mixture revealed that a cyclic polymer was exclusively produced (Fig. 8). The polymerization is reasonably initiated via nucleophilic attack of 2,6-lutidine on the thioester carbonyl group of 1, and the generated zwitterion propagates at the thiolate anion site (Scheme 3).

GPC profiles of the polymerization mixtures of monomer 1 in 2,6-litidine without a thiol initiator. [1] = 0.5 M (left), and 0.3 M (right)

MALDI-TOF-MS spectrum of the polymerization mixture (after 1 day) of monomer 1 in 2,6-lutidine without a thiol initiator (Entry 1, Table 4)

Polymerization initiated with 2,6-litidine and the formation of cyclic polymers
The backbiting reaction to the initiating end cation produces a high-molecular-weight cyclic polymer (route A in Scheme 3), and that to the polymer main chain results in the formation of smaller rings (route B in Scheme 3). It is reasonable that a lower monomer concentration decreases the molecular weight of the product polymer (Entries 1, 2, and 3, Table 4) by increasing the relative rate of the backbiting reaction (first-order for the concentration) in comparison with the propagation rate (second-order for the concentration). Figure 7 shows that the molecular weights of the product polymers decreased with increasing reaction time. The cyclic polymers undergo nucleophilic ring-opening by a large amount of 2,6-lutidine at the thioester carbonyl group, and the thiolate anion site of the generated zwitterion undergoes backbiting to form smaller rings. Monomers 2 and 3 are also polymerizable without the thiol initiator, producing cyclic polymers, as suggested by the MALDI-TOF-MS spectra (Figs. S27 and S28). Since monomer 2 is insoluble in 2,6-lutidine, a mixed solvent of 2,6-lutidine and CH2Cl2 (1/1 v/v) was used. The relative polymerization rates of the three monomers are 2 > 1 > 3 (Entries 4–6, Table 4), which are in the same order as those in the presence of the thiol initiator in CH2Cl2 (Table 1, vide supra). Monomer 2 gave a higher molecular weight polymer because of the larger ratio of the propagation rate to the backbiting reaction rate. The polymer from 3 showed an unimodal GPC profile in the high-molecular weight region; this is probably due to steric hindrance, which favors the backbiting reaction to the cationic initiation terminal instead of to the polymer mid-chain.
As mentioned above, the direct attack of a base on the monomer hinders the control of the polymerization. Thus, a nonnucleophilic base, i.e., 2,6-di-tert-butylpyridine (DBP), was examined as the catalyst. As expected, no polymerizations occurred without a thiol initiator in DBP or DBP/CH2Cl2 (1:1) as the solvent for the three monomers. The addition of the thiol initiator induced polymerization, however, which was very slow at low initiator concentrations (Table S1). The steric bulkiness of the two tert-butyl groups of DPB hinders the activation of the thiol at the propagating end. In addition, a backbiting reaction to the polymer mid-chain occurred, as suggested by the ESI-MS spectra (Fig. S29).
Cationic polymerization
The combination of PhCH2OH as an initiator with CF3SO3H as an acid catalyst was examined for the cationic polymerization of the three monomers since it is an efficient system for the controlled polymerization of lactide via an activated monomer mechanism [16]. Monomers 1, 2, and 3 were found to undergo polymerization (Table 5). Decreasing the initiator amount increased the molecular weights of the product polymers with decreasing polymerization rates. The 1H NMR spectrum (Fig. S30) of the Et2O-insoluble polymer from 1 showed a signal (5.17 ppm) due to the methylene protons of the benzyl ester group, supporting the initiation of the reaction. The MALDI-TOF-MS spectrum (Fig. S31) revealed that PhCH2OH was incorporated into the polymer terminals, but other unidentified peak series were observed. The polymerization mixtures from 2 and 3 showed no 1H NMR signals due to the benzyl ester group and provided no support according to their mass spectra (Figs. S32–S35).
The equimolar reactions between the monomers and PhCH2OH in the presence of the CF3SO3H catalyst (5 mol%) were performed to obtain information about the chemoselectivity of the ring-opening reactions. The 1H NMR spectrum of the reaction mixture from 1 (Fig. S36) showed signals due to the thiol and methine protons of the -CH(Me)SH structure, which is formed by ring opening at the thioester site. However, the relative integral intensities of these signals (0.24 and 0.27 for 1H and 1H, respectively) are smaller than the theoretical values based on the signal intensities of the phenyl and benzylic protons (5.00 and 1.94 for 5H and 2H, respectively) of the benzyl ester group. This finding suggests that ring opening occurs not only at the thioester site but also at the oxoester site. The latter reaction produces the -CH2OH structure, whose methylene protons labeled as i are detected as overlapping signals, whose relative integral intensity is 1.82 for 3H in the 1H NMR spectrum (Fig. S36). Thus, these relative signal intensities allow the selectivity ratio of the ring-opening site to be evaluated as thioester/oxoester = 24/76 ~ 39/61. The stoichiometric reactions of monomers 2 and 3 with PhCH2OH were also investigated by 1H NMR spectroscopy (Figs. S37 and S38), which indicated that the oxoester ester was mainly cleaved; however, many small signals attributed to side reactions were observed.
The chemoselectivity of ring cleavage is affected by the basicity of the carbonyl oxygen, which is protonated, and by steric hindrance around the carbonyl carbon. The latter factor can be excluded for the reaction of monomer 3, which has α-methyl groups at each of the 3- and 6-positions of the ring. Accordingly, the above findings suggest that the oxoester has more basic carbonyl oxygen than does the thioester. The 6-methyl group of monomer 2 exerts steric hindrance on the thioester carbonyl, consequently maintaining the ring-opening at the oxoester site, whereas the 3-methyl group of monomer 1 disfavors the reaction at the oxoester. This finding is consistent with the abovementioned experimental results. However, these chemoselective ring-opening reactions cannot be directly applied to ROP. As mentioned above, the benzyl ester group was not detected by 1H NMR or mass spectrometry for the polymerization mixtures of monomers 2 and 3. In the additional experiment, when the amount of CF3SO3H was increased to 10 mol%, the polymer from monomer 1 also showed no benzyl ester group in the 1H NMR spectrum. These inconsistencies are probably due to the low concentration of benzyl alcohol in the polymerization system, leading to ROP via the cationic chain-end mechanism instead of the activated monomer mechanism. To prevent side reactions and achieve a controllable ROP, CH3CN, a more polar solvent, and (CF3SO2)2NH, a less acidic catalyst [17], were tested. Consequently, CH3CN with CF3SO3H (5 mol%) accelerated the reaction but promoted frequent side reactions, producing an oligomer. (CF3SO2)2NH (10 mol%) also produced an oligomer in CH2Cl2 and no reaction in CH3CN.
Thermal properties of the polymers obtained by the anionic polymerization
Since the Tg and Td10 (10%-weight-loss temperature) of low-molecular-weight polymers have some degree of dependence on the molecular weight, discussion about these parameters is limited herein (Table 6 and Figs. S39 and S40). However, it is obvious that the thioester group reduces the Tg compared with the oxoester. Thermal degradation is most likely initiated by the backbiting reaction of the thiol terminal; thus, poly3 shows greater thermal stability than poly1 and poly2 because of steric hindrance due to the presence of two methyl groups. The backbiting reaction can lead to depolymerization of the monomer so that a more thermodynamically stable monomer with a smaller ring strain can be produced at a lower temperature. The lowest Td10 of poly(rac-thiolactide) is ascribed to the smallest ring strain of rac-thiolactide, as shown by the abovementioned DFT calculations.
Photodegradation of the polymers obtained by the anionic polymerization
Thioester absorbs UV at approximately 235 and 280 nm due to the π-π* and n-π* transitions, respectively. Our group previously demonstrated the photodegradation of poly(rac-thiolactide) [2]. Thus, polymers prepared from 1, 2, and 3 by the anionic polymerization were examined for UV degradation. The CH2Cl2 solutions of the polymers in the quartz tubes were irradiated for 3 hr with a low-pressure Hg lamp, which has an emission band of 254 nm. The resultant polymers showed shifts in their GPC profiles to the lower molecular weight region (Fig. 9; S41 and S42). There was no large difference in the degradability among the three polymers. In contrast, poly(rac-lactide) showed little photodegradation under the same conditions (Fig. 9) since oxoester has almost no absorption at 254 nm.

GPC profiles of poly3 (upper) and poly(rac-lactide) (lower) before and after UV irradiation in CH2Cl2 ([C( = O)S] = 68.2 mM and [C( = O)O] = 139 mM) for 3 h
Conclusion
Monomers 1 and 2, which are cyclic cross-dimers composed of oxoester-thioester units from glycolic and thiolactic acids and from lactic and thioglycolic acids, respectively, underwent anionic ROP in a controlled manner to some extent by combining the thiol as the initiator with 2,6-lutidine as the catalyst in CH2Cl2 ant r.t. However, small amounts of cyclic polymers were involved. Monomer 3, which is a cyclic cross-dimer from lactic and thiolactic acids, requires 2,6-lutidine as the solvent for ROP. The absence of the thiol initiator in 2,6-lutidine exclusively produced macrocyclic polymers by backbiting reactions. In these polymerizations, the ring-opening occurred selectively at the thioester site, producing a polymer consisting of alternating oxoester-thioester units. The polymerization rates increased in the order of 3 < 1 < 2, which was consistent with the increasing ring strain order evaluated by DFT calculations. The thioester unit caused the polymers to exhibit a lower Tg with greater thermal and photo degradability. The cationic ROPs of three monomers, 1, 2, and 3, occurred in the presence of benzyl alcohol as the initiator with CF3SO3H as the catalyst in CH2Cl2 at r.t., but the selectivity of the ring-opening site was low, and frequent side reactions occurred to produce low-molecular-weight polymers.
References
Suzuki M, Makimura K, Matsuoka S. Thiol-mediated controlled ring-opening polymerization of cysteine derived β‑thiolactone and unique features of product polythioester. Biomacromolecules. 2016;17:1135–41.
Suzuki M, Watanabe A, Kawai R, Sato R, Matsuoka S, Kawauchi S. Ring-opening polymerization of thiolactide by using thiol-amine combination. Polymer 2021;215:123386. Corrigendum. 2022;242:124617
Suzuki M, Kaneko T, Ishikawa Y, Matsuoka S. Anionic polymerization of acrylic thioester by using organic base. Polym Chem. 2020;11:1145–50.
Wang C, Mavila S, Worrell BT, Xi W, Goldman TM, Bowman CN. Productive exchange of thiols and thioesters to form dynamic polythioester-based polymers. ACS Macro Lett. 2018;7:1312–6.
Aksakal S, Aksakal R, Becer CR. Thioester functional polymers. Polym Chem. 2018;9:4507–16.
Worrell BT, Mavila S, Wang C, Kontour TM, Lim C-H, McBride MK, et al. A User’s guide to the thiol-thioester exchange in organic media: scope, limitations, and applications in material science. Polym Chem. 2018;9:4523–34.
Purohit VB, Pięta M, Pietrasik J, Plummer CM. Recent advances in the ring-opening polymerization of sulfur-containing monomers. Polym Chem. 2022;13:4858–78.
Illy N, Mongkhoun E. Thiolactone chemistry, a versatile platform for macromolecular engineering. Polym Chem. 2022;13:4592–614.
Li H, Guillaume SM, Carpentier J-F. Polythioesters prepared by ring-opening polymerization of cyclic thioesters and related monomers. Chem Asian J. 2022;17:e202200641.
Narmon AS, van Slagmaat CAMR, De Wildeman SMA, Dusselier M. Sustainable polythioesters via thio(no)lactones: monomer synthesis, ring-opening polymerization, end-of-life considerations, and industrial perspectives. ChemSusChem. 2023;16:e202202276.
Wang Y, Li M, Chen J, Tao Y, Wang X. O-to-S substitution enables dovetailing conflicting cyclizability, polymerizability, and recyclability: dithiolactone vs. Dilactone. Angew Chem Int Ed. 2021;60:22547–53.
Wang Y, Zhu Y, Lv W, Wang X, Tao Y. Tough while recyclable plastics enabled by monothiodilactone monomers. J Am Chem Soc. 2023;145:1877–85.
Ura Y, Al-Sayah M, Montenegro J, Beierle JM, Leman LJ, Ghadiri MR. Dynamic polythioesters via ring-opening polymerization of 1,4-thiazine-2,5-diones. Org Biomol Chem. 2009;7:2878–84. and references therein.
Takojima K, Makino H, Saito T, Yamamoto T, Tajima K, Isono T, et al. An organocatalytic ring-opening polymerization approach to highly alternating copolymers of lactic acid and glycolic acid. Polym Chem. 2020;11:6365–73.
Lozhkin BA, Shlyakhtin V, Bagrov VV, Ivchenko PV, Nifant’ev IE. Effective stereoselective approach to substituted 1,4-dioxane-2,5-diones as prospective substrates for ring-opening polymerization. Mendeleev Commun. 2018;28:61–3.
Bourissou D, Martin-Vaca B, Dumitrescu A, Graullier M, Lacombe F. Controlled cationic polymerization of lactide. Macromolecules. 2005;38:9993–8.
Makiguchi K, Kikuchi S, Satoh T, Kakuchi T. Synthesis of block and end-functionalized polyesters by triflimide-catalyzed ring-opening polymerization of e-caprolactone, 1,5-dioxepan-2-one, and rac-lactide. J Polym Sci Polym Chem. 2013;51:2455–63.
Sivalingam G, Madras G. Thermal degradation of binary physical mixtures and copolymers of poly(ε-caprolactone), poly(D, L-lactide), poly(glycolide). Polym Degrad Stab. 2004;84:393–8.
Acknowledgements
This work was supported by JSPS KAKENHI (grant number JP 17K05879). The numerical calculations were carried out on the TSUBAME3.0 supercomputer at the Tokyo Institute of Technology, Tokyo, Japan, supported by the MEXT Project of the Tokyo Tech Academy for Convergence of Materials and Informatics (TAC-MI).
Funding
Open Access funding provided by Nagoya Institute of Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hirata, M., Yoshimatsu, T., Matsuoka, Si. et al. Ring-opening polymerization of six-membered cyclic hybrid dimers composed of an oxoester and thioester. Polym J (2024). https://doi.org/10.1038/s41428-024-00915-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41428-024-00915-8
