
Abstract
Over the past two decades, there have been sizable efforts to realize condensed phase optical cooling. To date, however, there have been no verifiable demonstrations of semiconductor-based laser cooling. Recently, advances in the synthesis of semiconductor nanostructures have led to the availability of high-quality semiconductor nanocrystals, which possess superior optical properties relative to their bulk counterparts. In this review, we describe how these nanostructures can be used to demonstrate condensed phase laser cooling. We begin with a description of charge carrier dynamics in semiconductor nanocrystals and nanostructures under both above gap and below-gap excitation. Two critical parameters for realizing laser cooling are identified: emission quantum yield and upconversion efficiency. We report the literature values of these two parameters for different nanocrystal/nanostructure systems as well as the measurement approaches used to estimate them. We identify CsPbBr3 nanocrystals as a potential system by which to demonstrate verifiable laser cooling given their ease of synthesis, near-unity emission quantum yields and sizable upconversion efficiencies. Feasibility is further demonstrated through numerical simulations of CsPbBr3 nanocrystals embedded in an aerogel matrix. Our survey generally reveals that optimized semiconductor nanocrystals and nanostructures are poised to demonstrate condensed phase laser cooling in the near future.
Similar content being viewed by others

Introduction
Light/matter interactions are fundamental to modern physical sciences. They open the door to manipulating the optical, chemical, and physical behavior of materials. One of the better-known optical responses of matter involves heating, wherein absorbed radiation is converted to thermal excitation of the system. This is captured by Stokes’ empirical observation that the fluorescence from molecules generally occurs at lower energies than that of the excitation, the observed energy difference being transformed into molecular motion1.
Less recognized, but equally relevant, is the ability to cool matter with light. Early work by Hansch and Schawlow2 as well as by Wineland and Dehmelt3 illustrated how light could reduce the translational motion of gases. This ultimately led to the creation of optical molasses4,5,6, followed ~10 years later by the creation of a new quantum state of matter, a Bose–Einstein condensate, by Wieman, Cornell, and Ketterle7,8.
Today, among the remaining light/matter grand challenges is the optical cooling of condensed phases. Despite conceptual beginnings9 nearly 50 years prior to the advent of gas-phase laser cooling, condensed phase cooling remains only partially realized. This is because of significant technical hurdles that have been encountered, nearly all of which are related to material quality.
Solid/liquid state optical cooling is premised on removing thermal energy from a material through its photoluminescence. This occurs through emission at higher energies than that of the excitation and is referred to as anti-Stokes photoluminescence (ASPL). Rather than inducing translational or vibrational motion, ASPL removes thermal energy from the system, in turn cooling it. Although conceptually simple, realizing condensed phase laser cooling, in practice, represents a monumental challenge due to the delicate balance that exists between competing heating and cooling processes in a material. This will be discussed shortly.
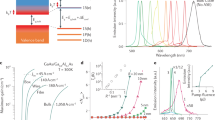
Proof-of-concept condensed phase laser cooling was first demonstrated by Epstein and co-workers in 1995 using the atomic transitions of Yb3+ dopants in a fluorozirconate glass (ZBLANP:Yb3+)10. Since then, meticulous improvements in material quality and the development of Yb3+-doped yttrium lithium fluoride (YLF) crystals have enabled rare-earth-doped hosts to be cooled to successively lower temperatures11,12,13,14,15, ultimately reaching cryogenic temperatures in 201315,16. These successes have been aided by improved cooling efficiencies, which stem from the narrow absorption linewidths and large absorption coefficients of Yb3+-doped YLF compared with those of ZBLANP:Yb3+ 16. Very recently, thermal payload cooling has been demonstrated with Yb3+-doped YLF, realizing a key end goal of the field17. A 2015 study by Roder et al. 18 also demonstrated local cooling of liquids using optically trapped Yb3+-doped YLF nanocrystals (NCs). Figure 1 summarizes key milestones achieved in cooling rare-earth-doped solids. The dashed red line denotes the NIST (National Institute of Standards and Technology)-defined cryogenic temperature threshold19. The corresponding blue line indicates the suggested minimum achievable temperature of Yb3+-doped YLF.

It is apparent from Fig. 1 that rare-earth-doped glasses/crystals only partially address the challenge of cooling solids. This is because rare-earth materials are limited in their lowest achievable temperatures due to the eventual thermal depopulation of atomic ground states. Accessing temperatures below the boiling point of liquid nitrogen thus requires laser cooling other condensed phase systems without this intrinsic limitation.
Semiconductors possess populated valence bands, even at very low temperatures, due to their Fermi–Dirac statistics. This ensures that cooling transitions are never depleted, making possible temperature floors on the order of 10 K20. Relatively large absorption coefficients also make overall cooling efficiencies larger than those of rare-earth-doped glasses/crystals. Direct integration of semiconductor optical cryocoolers into electronics is also possible given established semiconductor processing technologies. For these and other reasons, there is now significant interest in demonstrating laser cooling with semiconductors. Since 2000, much work has been done in this area on GaAs by Epstein and Sheik-Bahae, wherein near-unity external quantum efficiencies (EQEs) have been achieved through elaborate surface passivation and light management schemes21,22,23. Unfortunately, as with early attempts to cool rare-earth-doped glasses/crystals, sizable material quality issues have prevented any verifiable demonstration of laser cooling to date.
This review discusses progress made towards demonstrating semiconductor-based laser cooling. Of particular interest are semiconductor NCs and nanostructures due to the high likelihood of eventually achieving direct cooling with these materials. This stems from the nearly three decades of research invested in their synthesis and optical characterization24,25, which today has resulted in an unprecedented level of control over material quality and corresponding material properties.
In the following sections, we review the optical response of semiconductor NCs and nanostructures to both above-gap (Stokes) and below-gap (anti-Stokes) excitation. We then describe potential mechanisms leading to ASPL and discuss the critical NC/nanostructure parameters required to realize laser cooling. The accompanying tables provide literature-compiled estimates of these parameters. We end with cooling simulations of CsPbBr3 NCs, which we have identified as a promising system by which to demonstrate laser cooling. For those interested, comprehensive reviews of general condensed phase laser cooling can be found in the following refs. 23,26,27,28,29 and monographs30,31.
A delicate balance
What makes cooling a semiconductor so difficult? Realizing condensed phase laser cooling rests on achieving a delicate balance between competing cooling and heating processes in a material. This can be understood qualitatively since cooling is premised on removing a few quanta of thermal energy from a system during each cycle of excitation and subsequent emission. To put this into context, phonon energies in semiconductors range from 25 to 44 meV32,33,34. By contrast, competing nonradiative recombination processes introduce what is essentially the material’s entire band gap energy as heat into the lattice. Typical band gap energies range from 1 to 4 eV32,35. This highlights the uncomfortable reality that, given the close to two orders of magnitude difference between cooling and heating energies, cooling occurs only when radiative recombination is the near-exclusive carrier recombination process in a material.
A semiconductor’s EQE thus dictates whether optical cooling can be achieved. In semiconductor nanostructures, EQE is synonymous with the emission quantum yield (QY), as photon trapping and reabsorption processes are negligible due to the small dimensions of the samples. Near-unity QYs are therefore primarily restricted by competing nonradiative processes. This places stringent restrictions on material quality and makes much of semiconductor laser cooling a matter of materials optimization, as neither the basic principles of laser cooling nor its possibility is disputed36.
Early work on semiconductors
Early work to demonstrate semiconductor optical cooling focused on GaAs, a mature system in terms of its growth and subsequent processing. Preventing the realization of near-unity EQEs in GaAs, however, were two key problems. The first stemmed from low overall internal QYs due to the presence of surface recombination. This was ultimately addressed by developing various growth37 and surface passivation schemes22 that effectively removed undesired carrier recombination channels.
The second involved light trapping and subsequent emission reabsorption (also referred to as photon recycling). This scenario arises from the large refractive index difference that exists at the interface between a semiconductor and its surrounding medium (e.g., air). Light management schemes are therefore needed to achieve near-unity EQEs even if internal QYs are high. In practice, this has entailed introducing index matching, light extraction elements to the semiconductor surface21,38.
Only two studies have attempted the optical cooling of GaAs or any other bulk semiconductor. The first by Gauck et al. 38 involved a GaAs/GaInP double heterostructure with an impressive 96% EQE. To achieve this, GaInP layers were first used to passivate GaAs surface states. The passivation layer additionally aided in index matching the GaAs active layer to a hemispherical ZnSe light extraction dome.
Despite these optimization efforts, only net heating was observed when the excitation laser was tuned to the red of the mean emission wavelength. It was suggested that if the laser could be further detuned while maintaining an optimal excitation intensity, then cooling could be realized given the absence of any apparent parasitic absorption in the system.
The second study by Bender et al. 21 likewise focused on GaAs/GaInP double heterostructures but now with a record 99.5% EQE. This was achieved by more carefully optimizing one of two GaAs/GaInP interfaces using a stress-relieving GaP intermediary layer. As with Gauck, a (ZnS) light extraction hemisphere was added to maximize emission out-coupling. Yet, despite having a record EQE, no net cooling was observed. It was therefore suggested that cooling had been prevented by parasitic sub-band gap absorption of the incident light, leading to heating. Although the origin of this parasitic absorption was not identified, speculation centered on either GaAs/GaInP interface states or native point defects (vacancies, interstitials) within the GaInP passivation layer.
Towards nanostructures
While bulk semiconductors can display near-unity EQEs, it is evident that complex processing is required. Furthermore, achieving these near-unity values is far from routine. For these reasons, increasing attention has focused on semiconductor nanostructures, which possess notable advantages over their bulk counterparts. This includes the absence of light trapping/reabsorption effects due to their small physical dimensions as well as higher (as made) QYs, the latter occurring despite their large surface-to-volume ratios.
To illustrate these points, consider CdSe, the prototypical colloidal NC system39, which takes typical diameters between 2–12 nm40. These NCs effectively behave as dipole emitters41. Colloidal NCs also possess sizable QYs, as demonstrated by systems such as PbSe (QY ~ 85%)42 and, more recently, by CsPbBr3 (QY ~ 50−90%)43. Additional surface passivation schemes, which produce core/shell NCs44,45,46, now frequently yield near-unity QYs. When coupled to their facile chemistries as well as tunable band gaps, colloidal NCs and associated nanostructures represent obvious systems by which to demonstrate condensed phase laser cooling.
Perhaps, the most compelling reason why semiconductor nanostructures have attracted interest today is the recent report by Xiong and co-workers47,48,49, suggesting the cooling of individual CdS nanobelts (NBs). Although CdS is not the first system to come to mind when contemplating condensed phase laser cooling, the possibility of high purity/low background parasitic absorption, low carrier mobilities, low associated surface recombination velocities, and smaller Auger coefficients make it an intriguing system to investigate. Adding to this, Xiong and co-workers50 have suggested the cooling of individual hybrid perovskite nanoplatelets. An additional report by Fontenot et al. 51,52 has likewise suggested the optical cooling of commercial, overcoated CdSe NCs. Because of these tantalizing hints that semiconductor nanostructures can be cooled, the remainder of this review focuses on explaining emission upconversion in colloidal semiconductor NCs and associated nanostructures, as well as illustrating the underlying hinderances to achieving laser-induced cooling.
Semiconductor NC/nanostructure upconversion
Two common processes exist for upconverting light in isolated semiconductor nanostructures. They involve the one- or two-photon absorption of light. The former are relevant to condensed phase laser cooling, while the latter are not. This is because one-photon processes, which use sub-band gap photons, necessarily involve semiconductor lattice phonons to upconvert the initial excitation. In turn, thermal energy is removed from the system and can lead to net cooling. By contrast, two-photon processes bypass phonon involvement since the energy of two subgap photons readily spans the semiconductor band gap.
In general, within one- or two-photon processes, distinctions arise due to the identity of the intermediate state, whether real or virtual. Real states are often defect-related and have lifetimes on the order of nanoseconds. Virtual states, by contrast, possess significantly shorter lifetimes, dictated by time/energy uncertainty. This makes upconversion involving the former much more likely under conditions where excitation intensities are low.
Most existing nanostructure upconversion studies suggest the involvement of real, defect-related intermediate states that are relatively “dark” (i.e., low oscillator strength) in absorption. This includes work on TiO253,54, CdS55,56,57, CdSe58,59,60,61,62, CdTe59,63,64,65,66, InP55,58, PbS67, Ag2S68, and CsPbBrI2 NCs69. A few suggest the involvement of virtual intermediate states, especially when two-photon upconversion has been claimed (e.g., in CdS70 and CdTe71 NCs). Others (e.g., PbS72 and CsPbBr373) leave open the possibility of another upconversion mechanism, as of yet undefined.
Figure 2 schematically illustrates one-photon processes involving virtual (Fig. 2a) and real (Fig. 2b) intermediate states. For either, transitions between NC valence and conduction band states occur with G′, the subgap excitation rate, associated with an incident (subgap or anti-Stokes) excitation intensity Iexc. Relevant conduction and valence band state carrier densities are denoted in the figure by n and p, respectively. Upconversion occurs through interaction with lattice phonons (illustrated with an associated rate constant, kph) and ultimately leads to higher energy ASPL with an intensity IASPL (illustrated using an associated rate constant, kr). For simplicity, Fig. 2b assumes that the ASPL adopts the same energy as the normal band edge emission of the material and originates from the same emitting state.

a real state- and b virtual state-mediated
In the case where a real intermediate state is involved, carrier retrapping becomes possible. This is denoted in Fig. 2b using the associated rate constant kt. Similarly, radiative or nonradiative relaxation from the intermediate state back to the system’s ground state is also possible and is denoted with the associated rate constant kp. The former results in an energetically distinct, defect-related contribution to the material’s overall emission.
Figure 3 illustrates associated two-photon upconversion processes involving virtual or real intermediate states. These processes occur under significantly higher excitation intensities and are characterized by a quadratic ASPL Iexc dependency due to their two-photon nature (i.e., \(I_{\mathrm{ASPL}} \propto I_{\mathrm{exc}}^2\)). When a real state is involved, the process is sometimes referred to as a two-step, two-photon upconversion due to the fact that the intermediate state possesses a finite lifetime. Of note is the absence of any phonon involvement, making two-photon processes nominally irrelevant to achieving condensed phase laser cooling.

a real state- and b virtual state-mediated
An additional emission upconversion mechanism exists in nanostructures and stems from the Auger-induced recombination of photogenerated carriers. Auger recombination generally occurs at very large excitation intensities and involves the resonant excitation of multiple electron–hole pairs74,75. Following thermalization, energy transfer between recombining electron–hole pairs and other resident electron–hole pairs leads to carrier promotion into higher energy states, either within or associated with the nanostructure. Assuming that radiative recombination is possible from these states, upconverted emission results.
There are several points worth noting: (a) As with two-photon processes, Auger-induced emission upconversion lacks phonon involvement. (b) Under anti-Stokes excitation conditions, intermediate states must be real and must have sizable absorption cross-sections if multiple electron–hole pairs are to be generated. These states are not necessarily dark. (c) Given that involved higher energy states are generally extrinsic to the system being studied, they will be absent in isolated NCs/nanostructures. To illustrate, Si NC networks exhibit Auger-induced upconversion where interparticle energy “ladder climbing” occurs due to Auger-induced charge transfer into neighboring NCs with larger band gaps75. These Auger-induced upconversion processes will be absent if individual NCs are studied.
NC/nanostructure photophysics under above gap excitation
Given their relevance to laser cooling, we now focus on one-photon/phonon upconversion processes in NCs and nanostructures. Before discussing critical parameters required to achieve cooling, we review nanostructure photophysics under above gap (i.e., Stokes) excitation. Of particular importance is the emission QY of the material, as this is key to whether cooling can be achieved.
In NCs and nanostructures, QYs are primarily dictated by the existence of competing nonradiative recombination processes. Consequently, for laser cooling, it is important to measure QYs as well as understand their dependence on Iexc, as this contains information about competing carrier recombination processes. In NCs/nanostructures, the dependency can be assessed by examining the behavior of their emission intensity (Iem) as a function of Iexc since competing recombination mechanisms cause predictable changes to the expected power-law growth of Iem.
Specifically, it can be shown that \(I_{\mathrm{em}} \propto I_{\mathrm{exc}}^b\) with an observed power law growth exponent, b, that varies, depending on the dominant carrier recombination process occurring in a NC/nanostructure at a given excitation intensity76,77. Of specific interest are carrier trapping events, which ultimately suppress QYs.
The following equations model Iem under above gap excitation:
In Eqs. 1–3, G is the carrier generation rate, n (p) is the photogenerated electron (hole) carrier density, kr is a bimolecular radiative recombination rate constant, kt is a bimolecular trapping rate constant (the existence of electron traps is assumed for convenience), \(k_{\mathrm{ph}} = k_{\mathrm{ph}}^{\mathrm{o}}e^{ - \frac{{\Delta E}}{{kT}}}\) is a trap state depopulation rate constant, with \(k_{\mathrm{ph}}^{\mathrm{o}}\) being a detrapping attempt frequency and ΔE being the trap depth, kp is a trap recombination rate constant, and kAuger is the Auger rate constant. Nt is the native trap density of the material, with nt being the occupied density. Implicit in the model is that the material is intrinsic. Figure 4 summarizes the kinetic model, leaving out Auger processes for simplicity.

Kinetic model for above gap (Stokes) excitation
At very low excitation intensities where trapping dominates and where Nt ≫ nt, an approximate analytical solution to the model yields the following emission intensity:
Equation 4 shows that Iem grows in a power-law fashion with a slope between 2 and 1, depending on the rate constants involved. At higher intensities, prior to the onset of significant Auger recombination and where traps saturate, it can similarly be shown that Iem ∝ Iexc (i.e., b ~ 1).
Figure 5 shows exact numerical solutions to Eqs. 1–3 using rate constants and parameters established for CdS NBs57: kr = 9.55 × 10−12 cm3 s−1, kt = 6.31 × 10−11 cm3 s−1, kp = 10−11 cm3 s−1, kAuger = 10−30 cm6 s−1, \(k_{\mathrm{ph}}^{\mathrm{o}} = 10^4\) s−1, and T = 300 K. Figure 5a shows the expected power law growth of Iem and illustrates its Iexc dependency for different Nt. Figure 5b highlights specific variations of the power-law growth exponent for Nt = 1016 cm−3. Notably, at low Iexc, b adopts a value between 1 and 2, following the qualitative prediction of Eq. 4. At higher intensities, where bimolecular radiative recombination, as opposed to carrier trapping, dominates, b approaches 1. This again agrees with the approximate analytical solution to Eqs. 1–3 under conditions where bimolecular radiative recombination dominates all other processes. The condition is additionally associated with a maximum in the emission QY. At even higher intensities, Auger recombination causes b <1. Figure 5c shows the associated QYs for all Nt values and reveals peaked functions, which first grow with Iexc, but then decrease due to the Auger-induced quenching of excitations.

a Numerical simulation results for Iem as a function of Iexc for Nt = 1016−1019 cm−3. b Iexc dependency of Iem for Nt = 1016 cm−3. c Corresponding QYs for Nt from 1016 to 1019 cm−3
To illustrate that these trends are borne out experimentally, Fig. 6 shows results acquired from a single methylammonium lead iodide (MAPbI3) crystal. Figure 6a reveals that Iem grows in a power-law fashion with the b values depending on Iexc. As seen earlier and as predicted by Eq. 4, b lies between 2 and 1. Increasing Iexc causes b to decrease towards 1. Beyond this, b grows sublinearly due to the onset of Auger recombination. These trends all match the results shown in Fig. 5.

a Iem versus Iexc for a single crystal of MAPbI3. Local power-law exponents, b, indicated with dashed lines showing linear fits to the data. b Corresponding normalized Iem versus Iexc. The dashed line is a fit to the data using a kinetic model to extract the peak QY of the material
Figure 6b plots the Iexc dependence of the normalized emission intensity \(\frac{{I_{\mathrm{em}}}}{{I_{\mathrm{exc}}}}\), which is proportional to QY. \(\frac{{I_{\mathrm{em}}}}{{I_{\mathrm{exc}}}}\) peaks much like the QYs in Fig. 5c. As will be demonstrated shortly when discussing a QY estimation technique called power-dependent photoluminescence (PDPL), \(\frac{{I_{\mathrm{em}}}}{{I_{\mathrm{exc}}}}\) can be fit with the results of a kinetic model to extract an absolute optimal QY. For the MAPbI3 sample shown here, a maximum QY of 79% is found.
A critical QY exists to achieve laser cooling
A critical NC/nanostructure QY (QYcrit), required to realize laser cooling, emerges from the energy balance between competing cooling and heating processes in the NC/nanostructure. To explicitly demonstrate this, the thermal energy removed via ASPL can be expressed as Qcool = MηASPLQYΔE, where M is the number of induced sub-band gap excitations, ΔE is the energetic detuning of the laser into the semiconductor gap, and ηASPL is the fraction of those excitations promoted to the NC/nanostructure band edge.
The corresponding thermal energy introduced into the system due to nonradiative relaxation is Qheat = MηASPL(1 − QY)(Eg − ΔE) + M(1 − ηASPL)f(Eg − ΔE), where the first term reflects the fraction of upconverted excitations that recombine nonradiatively and where the second reflects the fraction (f) of excitations not upconverted, which also recombine nonradiatively. The upconversion efficiency, ηASPL, is therefore an important metric for laser cooling since it quantifies the balance between cooling and heating in a material together with QY. Optimal laser cooling conditions require large ηASPL values.
A critical QY to achieve cooling emerges when Qcool = Qheat. What results is
To put QYcrit into context, Table 1 lists QYcrit values calculated for various semiconductors, including CdS, when ΔE = 25 and 100 meV. For comparison purposes, the bulk longitudinal optical phonon energy (ℏωLO) of each semiconductor has been provided. In all cases, an upconversion efficiency of ηASPL = 1 is assumed to illustrate the minimum limiting value for QYcrit.
Table 1 reveals general trends regarding the likelihood of achieving optical cooling with a given NC/nanostructure system. Namely, whereas quantum-confinement-induced band gap shifts are important for many NC/nanostructure applications, they do not represent an advantage here since larger band gaps simply increase QYcrit. This reduces the margin of error for realizing optical cooling. By the same token, NC systems with smaller starting bulk band gaps possess lower overall QYcrit values. This means that, all things being equal, CdTe should be easier to cool than CdS. In practice, however, the ease and robustness of achieving near-unity QYs in a material will ultimately dictate its application to laser cooling. Finally, larger ΔE values lower QYcrit, which suggests that ΔE should be made as large as possible. However, upconversion efficiencies scale inversely with ΔE and, in principle, follow the Boltzmann distribution, \(e^{ - \frac{{\Delta E}}{{kT}}}\). Consequently, a limit exists to how large ΔE can be before ηASPL is adversely affected.
One last consideration exists regarding QYcrit. Table 1 lists conservative values based on the apparent band gaps of NCs/nanostructures. The existence of a Stokes shift (ΔEStokes) between the absorption and emission78, however, means that critical QYs will be slightly reduced in practice. Stokes shifts generally arise from weak oscillator strength, band edge emitting states intrinsic to NCs/nanostructures. This is evidenced by size-dependent ΔEStokes values, which range from ~10 to ~100 meV78,79.
Since the upconverted emission originates from the NC/nanostructure emitting state, Qheat can be re-expressed as Qheat = MηASPL(1 − QY)(Eem − ΔE) + M(1 − ηASPL)f(Eem − ΔE), where Eem = Eg − ΔEStokes. A QYcrit that accounts for NC/nanostructure Stokes shifts is therefore
with ΔE redefined as the energy difference between excitation and emission. Using Eq. 6, entries for the CsPbBr3 NCs in Table 1 decrease slightly to 99.0 and 96.2–95.8 for ΔE = 25 and 100 meV, respectively.
How are NC/nanostructure QYs estimated?
We now review approaches by which NC/nanostructure QYs are quantified in practice. This is motivated by the need to obtain accurate estimates as QY values approach unity. As will be seen, experimental QY measurements generally involve relative, absolute, and calorimetric approaches.
Relative QYs
Perhaps, the most popular way to measure NC/nanostructure QYs is to compare their integrated emission intensities to that of a known QY reference (usually an organic dye) under identical measurement conditions. The QY can then be estimated using
where QYref is the reference QY, Is (Iref) is the integrated emission intensity of the sample (reference), and nsolvent (nref) is the corresponding refractive index of the solvent in which the NC/nanostructure (reference) is dissolved. The fraction of light absorbed by the sample and reference at their respective excitation wavelengths, As and Aref, is determined using A = 1 − 10−Abs, where Abs is the sample/reference absorbance at the excitation wavelength used to induce emission.
Relative QY measurements can be conducted using conventional absorption and emission spectrometers. This simplicity, however, belies a number of uncertainties, which complicate accurate QY measurements. Specifically, errors stemming from the instrument response or associated with both the reference and sample must be accounted for if accurate results are to be obtained.
Regarding instrument-related issues, NC/nanostructure and reference emission spectra must be corrected for the wavelength-dependent spectral responsivity of absorption/emission spectrometers. Furthermore, if different sample and reference excitation wavelengths are used, the frequency-dependent response of the excitation source must be accounted for to eliminate any differences in the incident photon flux80,81. In principle, these instrument-related issues can be mitigated by using a suitable reference that absorbs and emits in the same wavelength region as that of the NC/nanostructure82.
It is evident that the choice of reference critically dictates the accuracy of relative QYs. Reference selection can be particularly challenging in the ultraviolet (<400 nm) and near-infrared (>750 nm) regimes83. This is because the reported QYs for dyes in these spectral windows vary greatly. To illustrate, the reported QY for blue-emitting Coumarin C153 ranges from 26 to 58%80. Red-emitting Cresyl violet has a reported QY that ranges from 51 to 67%83. Even well-characterized dyes such as Rhodamine 6G do not have consistent QYs in the literature. For Rhodamine 6G, reported QYs range from 88 to 95%82. Moreover, the use of a reference having a disparate QY relative to the sample can introduce estimation errors, especially if the sample pushes the instrument’s lower limit of detection. Obtaining accurate relative QYs therefore restricts reference dyes to those having near-identical absorption and emission wavelengths as well as QYs to the NC/nanostructure being probed80.
Beyond the choice of reference, the employed dye concentration can introduce measurement error. Too high a concentration results in emission reabsorption (also called the inner filter effect), which decreases QYs. Conversely, low concentrations become problematic for many colloidal NC/nanostructure samples due to the irreversible loss of surface passivating agents under dilute conditions. This results in both decreased emission QYs and sample instabilities that lead to eventual precipitation84,85,86. Furthermore, polarization sensitivities associated with anisotropic nanostructures as well as potential light scattering effects complicate measurements87,88. Finally, solvent light scattering and (possibly) fluorescence contribute to measurement errors85,89. It is thus evident that relative QY measurements, while simple, do not represent the most robust approach by which to estimate absolute NC/nanostructure QYs.
Absolute QY measurements
Integrating sphere
Absolute QY estimates are generally made by directly measuring a sample’s ratio of emitted to absorbed photons. As no dye reference is involved, the above-highlighted reference issues are obviated. Intensity losses due to scattering and sample anisotropies are also eliminated. In practice, absolute QY measurements entail using an integrating sphere in which QYs are estimated using
In Eq. 8, Iem is the integrated emission intensity of the sample under direct illumination, while Iexc,s(Iexc,ref) is the integrated excitation intensity of the incident beam illuminating the sample (an empty sphere). The denominator of Eq. 8 represents the total number of photons absorbed by the sample.
Two sources of uncertainty exist in Eq. 8. The first involves the spectral responsivity of the integrating sphere/detection system. The second stems from potential sample reabsorption effects resulting from the large effective optical path length introduced by the integrating sphere. The former can be addressed by correcting the overall spectral responsivity of the system to the known spectral radiance of a reference light source. For the latter, emission reabsorption can be accounted for by correcting measured QYs using90
where r is the reabsorption probability, estimated from the ratio of integrated emission intensities of concentrated and dilute NC/nanostructure solutions82.
Power-dependent photoluminescence
An alternative approach to measuring absolute QYs is called PDPL. PDPL involves measuring Iem as a function of Iexc. The resulting Iem versus Iexc dependency exhibits a peaked structure that can be fit with a kinetic model to yield absolute QYs. These optimal, excitation intensity-dependent QYs are crucial parameters for establishing the likelihood of laser cooling. Peaked QY Iexc dependencies have previously been illustrated in Fig. 5c and Fig. 6b. Whereas both pulsed57,91,92 and continuous wave (CW)93 laser sources have been employed for PDPL, the following model description assumes the use of a CW laser. Specifics regarding the pulsed case modeling can be found in refs. 57,91,92.
A generic model for carrier recombination in a semiconductor is first constructed by consolidating Eqs. 1 and 2 into
under the assumption of equal photogenerated electron and hole carrier densities. In Eq. 10, N is the free charge carrier density, νexc is the excitation frequency, α(υexc) is the NC/nanostructure absorption coefficient at νexc, ηe is a photon extraction efficiency, and A, B, and C are the semiconductor’s rate constants, associated with first-order carrier trapping, second-order radiative recombination, and third-order Auger nonradiative recombination.
At steady state, \(\frac{{{\mathrm{d}}N\left( t \right)}}{{{\mathrm{d}}t}} = 0\); thus, Iexc can alternatively be expressed in terms of the steady-state carrier density, N, as
A corresponding emission intensity is
where κ is a proportionality constant with units of length that depends upon instrument geometry, sample thickness, and detector collection efficiency. The ratio of Iem to Iexc then gives
where
In practice, QY(N) exhibits a peaked functional form with Iexc due to the various competing carrier recombination processes that underlie it. This has previously been illustrated in Figs. 5c and 6b, where, at low excitation intensities, the QY grows with Iexc, being limited by nonradiative carrier trapping processes. At higher intensities, the QY peaks due to trap saturation. At even higher intensities, the QY decreases with increasing Iexc due to the onset of nonradiative Auger recombination.
By taking the derivative of Eq. 14 with respect to N, the carrier density associated with the maximum observable QY is
and is linked with a corresponding optimal excitation intensity of
Iopt is found from Eqs. 13 and 14 when BN2 ≫ AN + CN3. The sample’s peak QY, in terms of Nopt, is then
In the ideal case, the rate constants A, B, and C are known. The photon extraction efficiency, ηe, is furthermore measurable94. Consequently, Eq. 17 immediately yields the maximum absolute QY of a sample.
In practice, however, NC/nanostructure rate constants are often not well characterized and, for NCs, can be size-dependent95,96. The practical employment of PDPL thus requires additional steps to be taken. To proceed, a dimensionless excitation intensity, \(\overline I _{\mathrm{exc}}\), is therefore defined by normalizing Iexc to Iopt. What results is
Assuming the limit of near-unity QYs and replacing N in Eq. 14 with \(N = N_{\mathrm{opt}}\sqrt {\overline I _{\mathrm{exc}}}\) (Eq. 18) yields the following QY expression:
Then, using Nopt from Eq. 15 gives
In terms of QYopt (Eq. 17), this is
Finally, introducing \({\mathrm{QY}}_{\mathrm{opt}}\left( {\overline I _{\mathrm{exc}}} \right)\) into the ratio \(\frac{{I_{\mathrm{em}}}}{{I_{\mathrm{exc}}}}\) (Eq. 13) gives
Experimental \(\frac{{I_{\mathrm{em}}}}{{I_{\mathrm{exc}}}}\) versus Iexc data can therefore be fit with Eq. 22 to find a NC’s/nanostructure’s QYopt. This fitting procedure has previously been illustrated in Fig. 6b for a MAPbI3 single crystal. In general, the approach is relatively fast and does not require any special corrections for detector responsivity. PDPL, however, yields accurate QYs only when they are near-unity in value given approximations made in the modeling. Additional details of the technique can be found in ref. 93.
Calorimetric approaches
A third class of QY measurements entails calorimetric approaches. These measurements estimate NC/nanostructure QYs based on local heating that arises from nonradiative relaxation in the sample following excitation. Of this class of techniques, popular examples include thermal lensing and photoacoustic spectroscopy97,98,99. Neither, however, is commonly used to estimate NC/nanostructure QYs due to difficulties in achieving accurate values. This stems, in part, from the fact that these techniques work best on lower-QY samples, where a sizable fraction of the excitation causes local heating.
All optical scanning laser calorimetry
An alternative approach that circumvents these issues is called all optical scanning laser calorimetry (ASLC), which measures QYs through a sample’s temperature-dependent emission spectrum91,100. To illustrate how ASLC links a sample’s peak emission frequency to its QY, we first describe its heating power, Pheat, due to nonradiative recombination, following excitation at a frequency, νexc. Ignoring parasitic/background absorption, Pheat can be expressed as the difference between absorbed and emitted powers:
where νem is the mean emission frequency and Pem = hνemηeBN2 is the corresponding emission power. If Pheat proportionally induces a temperature change, ΔT, Eq. 23 can be rewritten as
In practice, a specimen’s temperature-dependent emission spectrum is acquired to generate a ΔT calibration curve. Then, a separate measurement varies νexc while keeping Pem constant. At each frequency, ΔT is estimated and plotted against νexc. Equation 24 shows that a linear relationship exists between the two parameters such that the acquired experimental data can be extrapolated to the intercept where ΔT = 0. This is illustrated conceptually in Fig. 7. The sample’s emission QY is then found using the ratio

Schematic representation of temperature-dependent data obtained by ASLC
This illustrates the convenience of ASLC and demonstrates its suitability to samples not readily amenable to relative or absolute integrating sphere approaches. Of note, ASLC is applicable to conducting single NC/nanostructure QY measurements50.
In all cases, accurate ASLC QYs require accounting for the spectral responsivity of the detection system as well as maintaining a high degree of sample temperature stability. More importantly, specimens should exhibit clear temperature-dependent changes to νem. In this regard, systems such as CsPbBr3 NCs exhibit small ΔT variations of their emission spectra101, making ASLC measurements problematic. Finally, Figs. 5c and 6b show that QYs depend upon Iexc. Consequently, ASLC should be conducted at Iopt to establish a sample’s maximum QY. In practice, Iopt is found using PDPL, carried out in conjunction with ASLC.
Table 2 summarizes the experimentally reported maximum QYs of popular semiconductor NC/nanostructure systems established using the above-mentioned relative, absolute, and calorimetric approaches.
Estimating upconversion efficiencies
Beyond QY, Eq. 5 shows that equally important to achieving condensed phase laser cooling are experimental NC/nanostructure ηASPL values. Notably, although NC/nanostructure emission upconversion has been reported for many years53,59,60,61,63,64,65,72,102, few studies have actually estimated ηASPL. Those that have report values between 3.34 × 10−4 and 2.7 × 10−2.
In practice, experimental ηASPL values can be established using
where Iexc,Stokes and Iexc,ASPL are the Stokes and anti-Stokes excitation intensities required to achieve identical Stokes/ASPL emission intensities (i.e., Iem = IASPL). AStokes and Aanti-Stokes are the corresponding Stokes and anti-Stokes absorptance values found using A = 1 − 10−Abs, where Abs is the absorbance of the NC/nanostructure at Iexc,Stokes or Iexc,ASPL. Equation 26 implicitly assumes the involvement of real intermediate states in the upconversion process.
Using Eq. 26, we find that CsPbBr3 and CdSe/CdS NCs possess sizable ηASPL values. Specifically, for CsPbBr3 NCs, ηASPL = 0.75 (ηASPL = 0.32) for ΔE = 23 (ΔE = 102) meV103. For CdSe/CdS core/shell NCs, ηASPL = 0.55 (ηASPL = 0.03) for ΔE = 56.6 (ΔE = 107). These and all other reported ηASPL values are tabulated below in Table 3.
Experimental NC/nanostructure QYs and η ASPL values
Table 3 now summarizes all experimental NC/nanostructure QY and corresponding ηASPL values we are aware of in the literature. Upon inspection, it is evident that many NC/nanostructure systems exhibit ASPL. It is also clear that much of the older literature addresses samples with suboptimal QYs. Newer systems such as CdS, CdSe, and hybrid/all inorganic lead halide perovskites [e.g., MAPbI3 and cesium lead bromide (CsPbBr3)] show much more promising QY and ηASPL values. These systems should therefore be focused on in future NC-based laser cooling studies. Other NCs/nanostructures, which today show high QYs, will likely upconvert and should also be investigated.
Towards a mechanistic understanding of NC/nanostructure I ASPL
We now summarize what is understood about NC/nanostructure ASPL using the above-compiled literature observations. To begin, it generally involves real intermediate states related to defects. This is because excitation intensities in many experiments are often low and are below the values expected for two-photon processes. Consequently, only real intermediate states with finite lifetimes are expected to yield sizable ASPL intensities.
Next, NC/nanostructure upconversion is most often a one-photon process relevant to laser cooling. This conclusion is supported by the observation of linear IASPL and Iexc dependencies in NC systems, such as TiO253, CdS56, CdSe58,59,60,61,62,104, CdTe59,63,64,65,105, PbS67, CsPbBr373,103, and CsPbBrI269,106. Other nanostructures exhibiting linear IASPL versus Iexc behavior include CdS NBs47,57,107, ZnTe nanoribbons108, bulk MAPbI350, and PhEPbI3 nanoplatelets50. Although occasional quadratic/near-quadratic growth has been reported68,70,71,109,110,111,112,113, the bulk of the available literature points to the dominant role played by one-photon and, by corollary, defect-mediated processes in NC/nanostructure ASPL.
Phonon involvement is confirmed by reported ASPL temperature (T) dependencies. Namely, IASPL increases (decreases) with increasing (decreasing) temperature. This has been reported for CdS57, CdSe61,62, CdTe59,63,64,65, and CsPbBr3 NCs73, as well as for ZnTe nanoribbons108. These positive dependencies, in turn, are consistent with the Boltzmann population of phonon states. As a point of contrast, NC/nanostructure Iem values generally scale inversely with temperature.
Less frequently reported but equally relevant is the observation that IASPL increases (decreases) exponentially with decreasing (increasing) detuning (i.e., ΔE) of the excitation laser into the semiconductor gap. This has been reported for CdS57 and CsPbBrI2 NCs69, as well as for free-standing GaN films114. These dependencies again point to phonon involvement given the exponential ΔE dependency of the Boltzmann distribution.
The qualitative picture that emerges is ASPL from defect-mediated, one-photon/phonon upconversion to the semiconductor band edge. A schematic of the process is illustrated in Fig. 2b. This conclusion is further supported by associated kinetic modeling using
where G' is the absorbed subgap excitation rate (G' ∝ Iexc,ASPL).
At low excitation intensities, IASPL takes the limiting form
and grows linearly with Iexc,ASPL. This is corroborated by numerical solutions to Eqs. 27–29 using parameters previously used to simulate Eqs. 1–3. The only difference is the use of a larger kph value (i.e., \(k_{\mathrm{ph}}^{\mathrm{o}} = 10^8\) s−1) to model efficient upconversion. Figure 8 shows plots of IASPL for different Nt values, wherein IASPL follows the predicted linear dependence at low excitation intensities.

The inset shows the linear dependence of IASPL on \(\frac{{G^\prime }}{{N_{\mathrm{t}}}}\) under conditions relevant to Eq. 30. Here, Nt is the native trap density
Equation 30 additionally reveals that IASPL increases (decreases) with increasing temperature (increasing ΔE). Specifically, \(\ln \left( {I_{\mathrm{ASPL}}} \right) \propto - \left( {\frac{{\Delta E}}{k}} \right)\frac{1}{T} = - \left( {\frac{1}{{kT}}} \right)\Delta E\). These predictions agree with the above-mentioned experimental observations.
Notably, Eq. 30 predicts that IASPL possesses an inverse Nt dependence. Lowering NC/nanostructure trap densities should therefore increase IASPL. This last prediction is important because it has been observed that IASPL increases with increasing QYs in both CdSe60,61 and CsPbBr3 NCs73. We also observe this behavior in CdSe/CdS NCs studied here.
This positive IASPL/QY correlation was initially interpreted as a sign that ASPL may not be defect-mediated, as Nt is directly associated with NC/nanostructure QYs. Minimizing defect-state densities might therefore be expected to reduce upconversion activity. The kinetic model, however, rationalizes this observation and reveals that increased QYs are, in fact, consistent with enhanced IASPL values.
This observation is explicitly illustrated in the inset of Fig. 8, where IASPL has been plotted as a function of \(\frac{{G^\prime }}{{N_{\mathrm{t}}}}\) using numerical solutions to Eqs. 27–29. A clear linear dependence is seen, as first predicted by Eq. 30. The linearity spans the range of trap densities Nt = 1020−1021 cm−3, where the condition Nt ≫ nt holds.
Finally, at high excitation intensities and low trap state densities (e.g., Nt = 3.24 × 1016 cm−3), trap saturation occurs such that nt ≈ Nt. Under these conditions, it can be shown analytically that
IASPL saturates and is corroborated by the numerical results in Fig. 8. Similar saturation behavior is predicted at other Nt values.
There is one final observation to explain. This entails observations of redshifted ASPL spectra relative to the normal NC/nanostructure band edge emission. These redshifts have been observed in CdS1 − xSex110, CdSe59,62,109, CdTe59,63,65,66,71,105,112,113, PbS NC-doped glasses67,115, CsPbBr373,103, and CsPbBrI2 NCs69. Figure 9a provides an example, showing the apparent ~8 meV redshift between the emission and ASPL spectra of a CsPbBr3 NC ensemble. These redshifts have, in turn, been used to suggest that ASPL originates from different NC/nanostructure (defect) states, unrelated to the normal, above gap-excited, NC/nanostructure emitting state62,65,67.

a Ensemble absorption, emission, and ASPL spectra of CsPbBr3 NCs. b Single CsPbBr3 NC emission (solid line) and ASPL (open circles) spectra superimposed. c Single CdS NB emission (solid line) and ASPL (open circles) spectra superimposed. More information can be found in ref. 103
We suggest that these redshifts are artifacts related to NC/nanostructure ensemble residual size distributions. This is because apparent redshifts have not been observed in the ASPL spectra of individual CsPbBr3 NCs103 and CdS NBs57. Figure 9b, c provide illustrations, revealing that single CsPbBr3 NC103 and CdS NB57 emission and ASPL spectra exhibit near-coincident energies.
Additionally, an ASPL redshift arises naturally when accounting for an ensemble’s residual size distribution. In particular, convoluting a single NC’s thermally broadened emission spectrum with a modified Gaussian function that accounts for the ensemble size distribution, the Boltzmann upconversion probability in Eq. 30, and the exponential decrease in absorption into the gap [Abs(x)]
yields an IASPL spectrum naturally redshifted from the normal emission spectrum. In Eq. 32, σx reflects the ensemble’s size distribution, and \(\overline x\) is the mean NC/nanostructure size. σE accounts for the thermal broadening of a single NC’s/nanostructure’s emission linewidth, with E′(x) representing the mean emission energy for a given size and Eexc denoting the excitation energy.
By plotting IASPL(E), it is evident that a redshift of the ASPL spectrum appears naturally. This is illustrated in Fig. 10, where the ASPL redshift due to a CsPbBr3 NC ensemble’s size distribution has been simulated. Employed parameters include \(\overline x = 9.2\) nm, σx = 0.5−1.5 nm, and σE = 0.04 eV. Abs(x) is found by fitting the red edge of the experimental NC absorption spectrum, while E′(x) is found using a sizing curve from the literature, linking emission maxima to NC size 79.

Simulated ASPL redshift of a CsPbBr3 NC ensemble relative to its normal above gap-excited emission
CsPbBr3 NC cooling simulation
A global survey of Tables 1, 2, and 3 identifies CsPbBr3 NCs as a particularly promising material for laser cooling demonstrations. This stems from critical QYs as low as QYcrit = 96%, experimental near-unity QYs, and sizable ηASPL values. To investigate the potential cooling performance of this material, we numerically simulated its cooling when dispersed within a thermally isolating aerogel matrix. The simulations consider photogenerated charge carrier recombination, include Auger recombination and model heat diffusion across the NC composite.
Figure 11a illustrates the NC solid, where CsPbBr3 NCs have been uniformly dispersed within the inner portion of a 20-mm-diameter aerogel disk. The inner diameter is 2 mm, and the overall aerogel thickness is 0.1 mm. An associated NC density in the inner disk is 3.1 × 1016 cm−3. The aerogel is thermally clamped to a To = 300 K reservoir around its perimeter, and NCs are excited below-gap using a 532 nm laser with an intensity resulting in an effective carrier generation rate of G′ = 1.22 × 1022 cm3 s−1. Due to the relatively small absorption cross-section for below-gap excitation, the incident excitation is assumed to occur homogeneously throughout the inner disk containing NCs.

a Schematic illustration of an aerogel disk with CsPbBr3 NCs embedded in its center. The entire aerogel diameter is 20 mm. The diameter of the inner region containing NCs is 2 mm. b Steady-state temperature distribution along the aerogel radius. The shaded green region denotes the inner disk containing NCs. Inset: transient temperature at the disk center. c Color map of the resulting aerogel steady-state temperature distribution
The kinetic expressions for charge generation and recombination are analogous to those shown in Eqs. 27–29 with one difference. Given reports of unity QYs (Table 2), nt ≪ n. What results is
with kr ~ 10−10 cm3 s−1 116 and kAuger = 3 × 10−28 cm6 s−1 117. Steady-state n and p values are then found for a given Iexc,ASPL.
To account for heat diffusion across the solid, the heat equation is simultaneously solved. Its one-dimensional form is used, given the rotational and translational (depth) symmetry of the system
In Eq. 34, Da = 10−4 cm2 s−1 118 is the aerogel heat diffusivity, ρa = 0.1 g cm−3 118 is the aerogel density, and Ca = 2 J g−1 K−1 118 is its specific heat.
Qnet is given by
where the first term reflects the Auger-induced heating of the NC and the second captures its cooling through emission upconversion. Blackbody contributions from the surrounding environment are excluded for simplicity. Equation 34 is solved using a forward-time center-space finite difference approach, where time steps are determined using the Von Neumann stability criterion119. An identical approach involving the simultaneous solution of both charge generation/recombination and heat diffusion equations in a solid has previously been used to model the optical cooling of bulk GaAs120.
Figure 11b shows results of the simulation, where the final steady-state temperature at various points along the disk radius have been plotted. At the disk center, a ΔT = 37 K cooling, starting from room temperature, is predicted, with a final steady-state temperature of T = 263 K. The inset of Fig. 11b shows that the final disk center temperature is reached on a 0.5 s timescale. Towards the disk edges, temperatures progressively rise to 300 K. Figure 11c shows a false color map of the aerogel’s resulting steady-state temperature profile.
These results clearly indicate the feasibility of cooling CsPbBr3 NCs. Improvements that can be considered in future modeling and experimental studies include the use of better thermal isolation schemes to improve the predicted/achieved cooling floor. They also include optimizing NC concentrations to maximize the absorption of subgap photons while maintaining the thermal properties of the aerogel. Of additional note is that the simulation approach employed here is not exclusive to CsPbBr3 and can be applied to study the potential cooling of other promising nanostructures.
Prior suggestions of nanostructure laser cooling
Finally, before concluding, we discuss prior suggestions of NC/nanostructure laser cooling. In this regard, we are aware of only four reports suggesting to have successfully cooled a semiconductor. These are ref. 47 (CdS NBs), ref. 50 (hybrid perovskite nanoplatelets), and refs. 51,52 (core/shell CdSe NCs). Concerns, however, exist regarding the validity of these studies, as outlined in ref. 121 and below.
In the first two reports by Xiong and co-workers, a major discrepancy centers on the timescale over which individual CdS NBs or hybrid perovskite platelets reportedly cool/heat. Specifically, Fig. 12a, b show extracted cooling and heating data from these studies and reveal that cooling occurs on a timescale of thousands of seconds. This is highlighted by the data in both figures, where the subgap excitation laser has been kept on. When Iexc,ASPL = 0, both figures likewise reveal that nanostructure heating ensues over the course of ~103 s.

a CdS NB (λexc = 532 nm) b MAPbI3 crystal (λexc = 785 nm)
However, numerical simulations, which consider charge generation/recombination and heat diffusion, predict that individual CdS NBs should cool (or heat) on a timescale of ~10−4 s. In particular, an irradiated rectangular CdS cantilever (length of 20 µm, width of 5 µm, and thickness of 110 nm), modeled in ref. 107, cools (heats) over tens of microseconds. This is true for both 514 and 532 nm (below-gap) excitation. This cooling timescale easily differs by 7 orders of magnitude from what has been reported by Xiong and co-workers in Fig. 12. This discrepancy becomes even larger when a doubly clamped CdS cantilever is modeled.
An upper bound to the expected cooling timescale can be estimated for the case of a thermally isolated NB. Namely, an energy balance expression for its temperature change is
where the first term on the right-hand side of the equation stems from upconversion-induced NB cooling. The second term reflects NB heating due to blackbody radiation from the surroundings. In Eq. 36, V is the NB volume, Cp is its volumetric heat capacity, and α(νexc) is the sub-band gap absorption coefficient. The accompanying blackbody heating load can be expressed as
where A is the total NB surface area and σ is the Stefan–Boltzmann constant.
Solving Eq. 36 then yields
and reveals a characteristic cooling/heating time constant of
Upon introducing the following parameters for a CdS NB (Cp = 1.62 × 106 J m−3 K−1 122, A = 2.5 × 10−6 cm2, and V = 5 × 10−11 cm3 47) one finds that τ = 29 ms. A perfectly isolated NB thus cools/heats on a timescale ~4 orders of magnitude faster than what has been reported in Fig. 12. A similar analysis can be conducted for hybrid perovskite nanoplatelets50 to show that a large discrepancy in reported cooling/heating timescales also exists with this material. To illustrate, using the following parameters for MAPbI3 (a molar heat capacity of Cp = 9.7 J K−1 mol−1 123, a corresponding density of ρa = 4.286 g cm−3 124, and \(\frac{A}{V} = \frac{1}{d} = 5 \times 10^4\) cm−1, where the platelet thickness d = 200 nm50), Eq. 39 yields τ = 2.2 ms. This time constant is ~6 orders of magnitude smaller than the cooling/heating timescales in Fig. 12b.
In principle, slow cooling/heating timescales can stem from the presence of an additional thermal load in the system. For the NBs and nanoplatelets in question, this could arise from their respective Si or mica substrates. In fact, this scenario has been explored by Xiong and co-workers in ref. 49, where the NB-induced cooling of a SiO2/Si substrate has been modeled. However, what has not been addressed in this study and in refs. 47,48,50 is substrate heating due to the absorption of the incident light. A simple calculation for the NB case shows that ~79% of the incident power will be absorbed by the Si substrate. Given its low emission quantum efficiency, subsequent nonradiative relaxation will heat both the substrate and the CdS NB. In this case, the estimated heating power is 50-fold greater than the reported NB cooling power. This suggests that cooling should not be observed. Together with the cooling/heating timescale discrepancies outlined above, this casts doubts on Xiong’s suggestions of successful laser cooling.
Finally, Fontenot et al. 51,52 has suggested that a suspension of commercial overcoated CdSe NCs can be macroscopically cooled. While intriguing, the QY of 80% reported by the manufacturer is below QYcrit for CdSe in Table 1 and again implies that cooling should not be observed. Beyond this, other questions arise, as there are no details regarding the ASPL spectrum of the NCs or their T and ΔE dependencies. There are also no estimates of ηASPL, which we have shown is critical to achieving condensed phase laser cooling. Last, there are concerns over the long-term temperature stability of the measurements despite the use of control specimens. Overall, it is our opinion that it is premature to suggest that condensed phase laser cooling has been achieved in any NC/nanostructure system to date.
Outlook
Six years ago, a purported breakthrough was reported, describing the optical cooling of individual CdS NBs47. Although a debate now exists on whether cooling was actually achieved, the possibility that semiconductor nanostructures can be cooled has intrigued the community. This would capitalize on nearly three decades of research on their synthesis and optical characterization. Today, reports of unity or near-unity QYs in colloidal NCs coupled with the ability to upconvert light suggest the possibility of verifiably demonstrating condensed phase laser cooling. Although there remains much to be understood about NC/nanostructure upconversion, for example, the chemical identity of participating intermediate states as well as the apparent ubiquity of NC/nanostructure ASPL, it is clear that the field is within reach of realizing the century-old concept of optically cooling matter via anti-Stokes emission.
References
Stokes, G. G. On the change of refrangibility of light. Philos. Trans. R. Soc. 142, 463–562 (1852).
Hansch, T. W. & Schawlow, A. L. Cooling of gases by laser radiation. Opt. Commun. 13, 68–69 (1975).
Wineland, D. & Dehmelt, H. Proposed 1014 δν<ν laser fluorescence spectroscopy on TI+ mono-ion oscillator II (spontaneous quantum jumps). Bull. Am. Phys. Soc. 20, 637 (1975).
Phillips, W. D. & Metcalf, H. Laser deceleration of an atomic beam. Phys. Rev. Lett. 48, 596–599 (1982).
Chu, S., Hollberg, L., Bjorkholm, J. E., Cable, A. & Ashkin, A. Three-dimensional viscous confinement and cooling of atoms by resonance radiation pressure. Phys. Rev. Lett. 55, 48–51 (1985).
Aspect, A., Arimondo, E., Kaiser, R., Vansteenkiste, N. & Cohen-Tannoudji, C. Laser cooling below the one-photon recoil energy by velocity-selective coherent population trapping. Phys. Rev. Lett. 61, 826–829 (1988).
Anderson, M. H., Ensher, J. R., Matthews, M. R., Wieman, C. E. & Cornell, E. A. Observation of Bose–Einstein condensation in a dilute atomic vapor. Science 269, 198–201 (1995).
Davis, K. B. et al. Bose–Einstein condensation in a gas of sodium atoms. Phys. Rev. Lett. 75, 3969–3973 (1995).
Pringsheim, P. Zwei bemerkungen über den unterschied von lumineszenzund temperaturstrahlung. Z. Phys. 57, 739–746 (1929).
Epstein, R. I., Buchwald, M. I., Edwards, B. C., Gosnell, T. R. & Mungan, C. E. Observation of laser-induced fluorescent cooling of a solid. Nature 377, 500–503 (1995).
Mungan, C. E., Buchwald, M. I., Edwards, B. C., Epstein, R. I. & Gosnell, T. R. Laser cooling of a solid by 16 K starting from room temperature. Phys. Rev. Lett. 78, 1030–1033 (1997).
Luo, X., Eisaman, M. D. & Gosnell, T. R. Laser cooling of a solid by 21 K starting from room temperature. Opt. Lett. 23, 639–641 (1998).
Gosnell, T. R. Laser cooling of a solid by 65 K starting from room temperature. Opt. Lett. 24, 1041–1043 (1999).
Thiede, J., Distel, J., Greenfield, S. R. & Epstein, R. I. Cooling to 208 K by optical refrigeration. Appl. Phys. Lett. 86, 154107 (2005).
Melgaard, S. D., Seletskiy, D. V., Di Lieto, A., Tonelli, M. & Sheik-Bahae, M. Optical refrigeration to 119 K, below National Institute of Standards and Technology cryogenic temperature. Opt. Lett. 38, 1588–1590 (2013).
Melgaard, S. D., Albrecht, A. R., Hehlen, M. P. & Sheik-Bahae, M. Solid-state optical refrigeration to sub-100 Kelvin regime. Sci. Rep. 6, 20380 (2016).
Hehlen, M. P. et al. First demonstration of an all-solid-state optical cryocooler. Light Sci. Appl. 7, 15 (2018).
Roder, P. B., Smith, B. E., Zhou, X., Crane, M. J. & Pauzauskie, P. J. Laser refrigeration of hydrothermal nanocrystals in physiological media. Proc. Natl. Acad. Sci. USA 112, 15024–15029 (2015).
Seletskiy, D. V. et al. Laser cooling of solids to cryogenic temperatures. Nat. Photonics 4, 161–164 (2010).
Rupper, G., Kwong, N. H. & Binder, R. Large excitonic enhancement of optical refrigeration in semiconductors. Phys. Rev. Lett. 97, 117401 (2006).
Bender, D. A., Cederberg, J. G., Wang, C. & Sheik-Bahae, M. Development of high quantum efficiency GaAs/GaInP double heterostructures for laser cooling. Appl. Phys. Lett. 102, 252102 (2013).
Imangholi, B., Hasselbeck, M. P., Sheik-Bahae, M., Epstein, R. I. & Kurtz, S. Effects of epitaxial lift-off on interface recombination and laser cooling in GaInP/GaAs heterostructures. Appl. Phys. Lett. 86, 081104 (2005).
Seletskiy, D. V., Epstein, R. I. & Sheik-Bahae, M. Laser cooling in solids: advances and prospects. Rep. Prog. Phys. 79, 096401 (2016).
Gaponenko, S. V. Optical Properties of Semiconductor Nanocrystals 1st edn (Cambridge University Press, Cambridge, UK, 1998).
Yoffe, A. D. Semiconductor quantum dots and related systems: electronic, optical, luminescence and related properties of low dimensional systems. Adv. Phys. 50, 1–208 (2001).
Mungan, C. E. & Gosnell, T. R. Laser cooling of solids. Adv. At. Mol. Opt. Phys. 40, 161–228 (1999).
Rayner, A., Heckenberg, N. R. & Rubinsztein-Dunlop, H. Condensed-phase optical refrigeration. J. Opt. Soc. Am. B 20, 1037–1053 (2003).
Sheik-Bahae, M. & Epstein, R. I. Optical refrigeration. Nat. Photonics 1, 693–699 (2007).
Sheik-Bahae, M. & Epstein, R. I. Laser cooling of solids. Laser Photon. Rev. 3, 67–84 (2009).
Epstein, R. I. & Sheik-Bahae, M. Optical Refrigeration: Science and Appli-cations of Laser cooling of Solids (WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009).
Petrushkin, S. & Samartsev, V. Laser Cooling of Solids (Woodhead Publishing, Cambridge, UK, 2009).
Amirtharaj, P. M. & Seiler, D. G. in Handbook of Optics, Vol. IV (ed. Bass, M.) (McGraw-Hill Inc., New York, 1995).
Tell, B., Damen, T. C. & Porto, S. P. S. Raman effect in cadmium sulfide. Phys. Rev. 144, 771–774 (1966).
Lin, C., Kelley, D. F., Rico, M. & Kelley, A. M. The “surface optical” phonon in CdSe nanocrystals. ACS Nano 8, 3928–3938 (2014).
Ninomiya, S. & Adachi, S. Optical properties of cubic and hexagonal CdSe. J. Appl. Phys. 78, 4681–4689 (1995).
Sheik-Bahae, M. & Epstein, R. I. Can laser light cool semiconductors? Phys. Rev. Lett. 92, 247403 (2004).
Wang, J. B., Ding, D., Johnson, S. R., Yu, S. Q. & Zhang, Y. H. Determination and improvement of spontaneous emission quantum efficiency in GaAs/AlGaAs heterostructures grown by molecular beam epitaxy. Phys. Stat. Sol. B 244, 2740–2751 (2007).
Gauck, H., Gfroerer, T. H., Renn, M. J., Cornell, E. A. & Bertness, K. A. External radiative quantum efficiency of 96% from a GaAs/GaInP heterostructure. Appl. Phys. A 64, 143–147 (1997).
Kuno, M. Colloidal quantum dots: a model nanoscience system. J. Phys. Chem. Lett. 4, 680–680 (2013).
Murray, C. B., Norris, D. J. & Bawendi, M. G. Synthesis and characterization of nearly monodisperse CdE (E = sulfur, selenium, tellurium) semiconductor nanocrystallites. J. Am. Chem. Soc. 115, 8706–8715 (1993).
Empedocles, S. A., Neuhauser, R. & Bawendi, M. G. Three-dimensional orientation measurements of symmetric single chromophores using polarization microscopy. Nature 399, 126–130 (1999).
Wehrenberg, B. L., Wang, C. & Guyot-Sionnest, P. Interband and intraband optical studies of PbSe colloidal quantum dots. J. Phys. Chem. B 106, 10634–10640 (2002).
Protesescu, L. et al. Nanocrystals of cesium lead halide perovskites (CsPbX3, X=Cl, Br, and I): novel optoelectronic materials showing bright emission with wide color gamut. Nano Lett. 15, 3692–3696 (2015).
Hines, M. A. & Guyot-Sionnest, P. Synthesis and characterization of strongly luminescing ZnS-capped CdSe nanocrystals. J. Phys. Chem. 100, 468–471 (1996).
Dabbousi, B. O. et al. (CdSe)ZnS core-shell quantum dots: synthesis and characterization of a size series of highly luminescent nanocrystallites. J. Phys. Chem. B 101, 9463–9475 (1997).
Peng, X., Schlamp, M. C., Kadavanich, A. V. & Alivisatos, A. P. Epitaxial growth of highly luminescent CdSe/CdS core/shell nanocrystals with photostability and electronic accessibility. J. Am. Chem. Soc. 119, 7019–7029 (1997).
Zhang, J., Li, D., Chen, R. & Xiong, Q. Laser cooling of a semiconductor by 40 kelvin. Nature 493, 504–508 (2013).
Li, D., Zhang, J. & Xiong, Q. Laser cooling of CdS nanobelts: thickness matters. Opt. Express 21, 19302–19310 (2013).
Li, D., Zhang, J., Wang, X., Huang, B. & Xiong, Q. Solid-state semiconductor optical cryocooler based on CdS nanobelts. Nano Lett. 14, 4724–4728 (2014).
Ha, S.-T., Shen, C., Zhang, J. & Xiong, Q. Laser cooling of organic–inorganic lead halide perovskites. Nat. Photonics 10, 115–121 (2016).
Fontenot, R. S., Mathur, V. K., Barkyoumb, J. H., Mungan, C. E. & Tran, T. N. Measuring the anti-stokes luminescence of CdSe/ZnS quantum dots for laser cooling applications. Proc. SPIE 9821, 982103 (2016).
Fontenot, R. S., Mathur, V. K. & Barkyoumb, J. H. New photothermal deflection technique to discriminate between heating and cooling. J. Quant. Spect. Rad. Trans. 204, 1–6 (2018).
Abazovic, N. D. et al. Photoluminescence of anatase and rutile TiO2 particles. J. Phys. Chem. B 110, 25366–25370 (2006).
Abazovic, N. D. et al. Photon energy up-conversion in colloidal TiO2 nanorods. Opt. Mater. 30, 1139–1144 (2008).
Sercel, P. C., Efros, A. L. & Rosen, M. Intrinsic gap states in semiconductor nanocrystals. Phys. Rev. Lett. 83, 2394 (1999).
Ezhov, A. A. et al. Liquid-crystalline polymer composites with CdS nanorods: structure and optical properties. Langmuir 27, 13353–13360 (2011).
Morozov, Y. V. et al. Defect-mediated CdS nanobelt photoluminescence up-conversion. J. Phys. Chem. C 121, 16607–16616 (2017).
Poles, E., Selmarten, D. C., Micic, O. I. & Nozik, A. J. Anti-Stokes photoluminescence in colloidal semiconductor quantum dots. J. Chem. Phys. 75, 224708 (1999).
Rakovich, Y. P. et al. Anti-stokes photoluminescence in II–VI colloidal nanocrystals. Phys. Stat. Sol. B 229, 449–452 (2002).
Rakovich, Y. P. et al. Up-conversion luminescence via a below-gap state in CdSe/ZnS quantum dots. Phys. E Low. Dimens. Syst. Nanostruct. 17, 99–100 (2003).
Rusakov, K. I. et al. Control of efficiency of photon energy up-conversion in CdSe/ZnS quantum dots. Opt. Spectrosc. 94, 859–863 (2003).
Li, X. et al. Photoluminescence up-conversion of CdSe/ZnS core/shell quantum dots under high pressure. J. Phys. Chem. C 113, 4737–4740 (2009).
Rakovich, Y. P. & Gladyshchuk, A. Anti-stokes luminescence of cadmium telluride nanocrystals. J. Appl. Spectrosc. 69, 383–387 (2002).
Filonovich, S. A. et al. Up-conversion luminescence in colloidal CdTe nanocrystals. MRS Proc. 737, E13.12 (2002).
Wang, X. et al. Photoluminescence upconversion in colloidal CdTe quantum dots. Phys. Rev. B. 68, 125318 (2003).
Ragab, A. E., Gadallah, A. -S., Mohamed, M. B. & Azzouz, I. M. Photoluminescence and upconversion on Ag/CdTe quantum dots. Opt. Laser Technol. 63, 8–12 (2014).
Xiong, Y., Liu, C., Wang, J., Han, J. & Zhao, X. Near-infrared anti-Stokes photoluminescence of PbS QDs embedded in glasses. Opt. Express 25, 6874–6882 (2017).
Zhang, Y. et al. Near-infrared-emitting colloidal Ag2S quantum dots exhibiting upconversion luminescence. Superlattices Microstruct. 102, 512–516 (2017).
Ye, S. et al. Mechanistic investigation of upconversion photoluminescence in all-inorganic perovskite CsPbBrI2 nanocrystals. J. Phys. Chem. C 122, 3152–3156 (2018).
Ouyang, J. et al. Upconversion luminescence of colloidal CdS and ZnCdS semiconductor quantum dots. J. Phys. Chem. C. 111, 16261–16266 (2007).
Wu, W. Z. et al. Upconversion luminescence of CdTe nanocrystals by use of near-infrared femtosecond laser excitation. Opt. Lett. 32, 1174–1176 (2007).
Fernee, M. J., Jensen, P. & Rubinsztein-Dunlop, H. Unconventional photoluminescence upconversion from PbS quantum dots. Appl. Phys. Lett. 91, 043112 (2007).
Roman, B. J. & Sheldon, M. The role of mid-gap states in all-inorganic CsPbBr3 nanoparticle one photon up-conversion. Chem. Commun. 54, 6851–6854 (2018).
Seidel, W., Titkov, A., Andre, J. P., Voisin, P. & Voos, M. High-efficiency energy up-conversion by an “Auger-Fountain” at an InP-AlInAs type-II heterojunction. Phys. Rev. Lett. 73, 2356–2359 (1994).
Diener, J. et al. Strong low-temperature anti-stokes photoluminescence from coupled silicon nanocrystals. Opt. Mater. 17, 135–139 (2001).
Pelant, I. & Valenta, J. Luminescence Spectroscopy of Semiconductors (Oxford Univ. Press, Oxford, 2012).
Vietmeyer, F., Frantsuzov, P. A., Janko, B. & Kuno, M. Carrier recombination dynamics in individual CdSe nanowires. Phys. Rev. B 83, 115319 (2011).
Kuno, M., Lee, J. K., Dabbousi, B. O., Mikulec, F. V. & Bawendi, M. G. The band edge luminescence of surface modified CdSe nanocrystallites: probing the luminescing state. J. Chem. Phys. 106, 9869–9882 (1997).
Brennan, M. C. et al. Origin of the size-dependent stokes shift in CsPbBr3 perovskite nanocrystals. J. Am. Chem. Soc. 139, 12201–12208 (2017).
Würth, C., Grabolle, M., Pauli, J., Spieles, M. & Resch-Genger, U. Comparison of methods and achievable uncertainties for the relative and absolute measurement of photoluminescence quantum yields. Anal. Chem. 83, 3431–3439 (2011).
Resch-Genger, U. & Rurack, K. Determination of the photoluminescence quantum yield of dilute dye solutions (IUPAC Technical Report). Pure Appl. Chem. 85, 2005–2026 (2013).
Würth, C., Grabolle, M., Pauli, J., Spieles, M. & Resch-Genger, U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 8, 1535–1550 (2013).
Brouwer, A. M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 83, 2213–2228 (2011).
Greben, M., Fucikova, A. & Valenta, J. Photoluminescence quantum yield of PbS nanocrystals in colloidal suspensions. J. Appl. Phys. 117, 144306 (2015).
Grabolle, M. et al. Determination of the fluorescence quantum yield of quantum dots: Suitable procedures and achievable uncertainties. Anal. Chem. 81, 6285–6294 (2009).
Laverdant, J. et al. Experimental determination of the fluorescence quantum yield of semiconductor nanocrystals. Materials 4, 1182–1193 (2011).
Würth, C., Geißler, D. & Resch-Genger, U. Quantification of anisotropy-related uncertainties in relative photoluminescence quantum yield measurements of nanomaterials—semiconductor quantum dots and rods. Z. Phys. Chem. 229, 153–165 (2015).
Geißler, D., Würth, C., Wolter, C., Weller, H. & Resch-Genger, U. Excitation wavelength dependence of the photoluminescence quantum yield and decay behavior of CdSe/CdS quantum dot/quantum rods with different aspect ratios. Phys. Chem. Chem. Phys. 19, 12509–12516 (2017).
Resch-Genger, U. et al. Traceability in fluorometry: Part II. Spectral fluorescence standards. J. Fluoresc. 15, 315–336 (2005).
Ahn, T.-S., Al-Kaysi, R. O., Müller, A. M., Wentz, K. M. & Bardeen, C. J. Self-absorption correction for solid-state photoluminescence quantum yields obtained from integrating sphere measurements. Rev. Sci. Instrum. 78, 086105 (2007).
Wang, C. et al. GaAs/GaInP double heterostructure characterization for laser cooling of semiconductors. Proc. SPIE 7951, 79510D (2011).
Wang, C. & Sheik-Bahae, M. Determination of external quantum efficiency in semiconductors using pulsed power-dependent photoluminescence. Opt. Eng. 56, 011106 (2016).
Draguta, S. et al. Spatially non-uniform trap state densities in solution-processed hybrid perovskite thin films. J. Phys. Chem. Lett. 7, 715–721 (2016).
Imangholi, B. Investigation of Laser Cooling in Semiconductors. Ph.D. thesis, The University of New Mexico, Albuquerque (2006).
Robel, I., Gresback, R., Kortshagen, U., Schaller, R. D. & Klimov, V. I. Universal size-dependent trend in auger recombination in direct-gap and indirect-gap semiconductor nanocrystals. Phys. Rev. Lett. 102, 177404 (2009).
Klimov, V. I., Mikhailovsky, A. A., McBranch, D. W., Leatherdale, C. A. & Bawendi, M. G. Quantization of multiparticle auger rates in semiconductor quantum dots. Science 287, 1011–1013 (2000).
Tomasini, E. P., San Román, E. & Braslavsky, S. E. Validation of fluorescence quantum yields for light-scattering powdered samples by laser-induced optoacoustic spectroscopy. Langmuir 25, 5861–5868 (2009).
Olmsted, J. Calorimetric determinations of absolute fluorescence quantum yields. J. Phys. Chem. 83, 2581–2584 (1979).
Santhi, A. et al. Thermal lens technique to evaluate the fluorescence quantum yield of a schiff base. Appl. Phys. B 79, 629–633 (2004).
Wang, C., Li, C.-Y., Hasselbeck, M. P., Imangholi, B. & Sheik-Bahae, M. Precision, all-optical measurement of external quantum efficiency in semiconductors. J. Appl. Phys. 109, 093108 (2011).
Swarnkar, A. et al. Colloidal CsPbBr3 perovskite nanocrystals: luminescence beyond traditional quantum dots. Angew. Chem. Int. Ed. 54, 15424–15428 (2015).
Harbold, J. M. & Wise, F. W. Photoluminescence spectroscopy of PbSe nanocrystals. Phys. Rev. B 76, 125304 (2007).
Morozov, Y. V., Zhang, S., Brennan, M. C., Janko, B. & Kuno, M. Photoluminescence up-conversion in CsPbBr3 nanocrystals. ACS Energy Lett. 2, 2514–2515 (2017).
Jakubek, Z. J., DeVries, J., Lin, S., Ripmeester, J. & Yu, K. Exciton recombination and unconverted photoluminescence in colloidal CdSe quantum dots. J. Phys. Chem. C 112, 8153–8158 (2008).
Rakovich, Y. P., Jäckel, F., Donegan, J. F. & Rogach, A. L. Semiconductor nanowires self-assembled from colloidal CdTe nanocrystal building blocks: optical properties and application perspectives. J. Mater. Chem. 22, 20831–20839 (2012).
Ye, S., Yu, M., Zhao, M., Song, J. & Qu, J. Low temperature synthesis of high-quality all-inorganic cesium lead halide perovskite nanocrystals in open air and their upconversion luminescence. J. Alloy. Compd. 730, 62–70 (2018).
Pant, A. et al. Optomechanical thermometry of nanoribbon cantilevers. J. Phys. Chem. C 112, 7525–7532 (2018).
Zhang, Q. et al. Phonon-assisted anti-stokes lasing in ZnTe nanoribbons. Adv. Mater. 28, 276–283 (2016).
Chen, W., Joly, A. G. & McCready, D. E. Upconversion luminescence from CdSe nanoparticles. J. Chem. Phys. 122, 224708 (2005).
Wu, W. et al. Upconversion luminescent characteristics and peak shift of CdSeS nanocrystals under different wavelength laser excitation. J. Nanopart. Res. 13, 1049–1061 (2011).
Wu, W., Yu, D., Ye, H., Gao, Y. & Chang, Q. Temperature and composition dependent excitonic luminescence and exciton-phonon coupling in CdSeS nanocrystals. Nanoscale Res. Lett. 7, 301 (2012).
Ananthakumar, S. et al. Size-independent peak shift between normal and upconversion photoluminescence in MPA-capped CdTe nanoparticles. Pramana – J. Phys. 82, 353–358 (2014).
Ananthakumar, S. et al. Size dependence of upconversion photoluminescence in MPA capped CdTe quantum dots: existence of upconversion bright point. J. Lumin. 169, 308–312 (2016).
Sun, G., Chen, R., Ding, Y. J. & Khurgin, J. B. Upconversion due to optical-phonon-assisted anti-Stokes photoluminescence in bulk GaN. ACS Photonics 2, 628–632 (2015).
Loranger, S. et al. Spectroscopic and life-time measurements of quantum dot doped glass for optical refrigeration: a feasibility study. Proc. SPIE 8638, 863801 (2013).
Zhu, H. et al. Organic cations might not be essential to the remarkable properties of band edge carriers in lead halide perovskites. Adv. Mater. 29, 1603072 (2017).
Makarov, N. S. et al. Spectral and dynamical properties of single excitons, biexcitons, and trions in cesium–lead-halide perovskite quantum dots. Nano Lett. 16, 2349–2362 (2016).
Scheuerpflug, P., Hauck, M. & Fricke, J. Thermal properties of silica aerogels between 1.4 and 330 K. J. Non-Cryst. Solids 145, 196–201 (1992).
Burden, R. L. & Faires, J. D. Numerical Analysis 9th edn (Cengage Learning, Boston, 2015).
Morozov, Y. V., Zhang, S., Janko, B. & Kuno, M. Charge and thermal modeling of a semiconductor-based optical refrigerator. Appl. Phys. Lett. 113, 181105 (2018).
Morozov, Y. V. et al. Can lasers really refrigerate CdS nanobelts? Nature 570, E60–E61 (2019).
Madelung, O., Rössler, U. & Schulz, M. II–VI and I–VII Compounds; Semimagnetic Compounds (Springer, Berlin, Germany, 1999).
Onoda-Yamamuro, N., Matsuo, T. & Suga, H. Calorimetric and IR spectroscopic studies of phase transitions in methylammonium trihalogeno-plumbates (II). J. Phys. Chem. Solids 51, 1383–1395 (1990).
Baikie, T. et al. Synthesis and crystal chemistry of the hybrid perovskite (CH3NH3)PbI3 for solid-state sensitised solar cell applications. J. Mater. Chem. A 1, 5628–5641 (2013).
Yu, W. W., Qu, L., Guo, W. & Peng, X. Experimental determination of the extinction coefficient of CdTe, CdSe, and CdS nanocrystals. Chem. Mater. 15, 2854–2860 (2003).
Weidman, M. C., Beck, M. E., Hoffman, R. S., Prins, F. & Tisdale, W. A. Monodisperse, air-stable PbS nanocrystals via precursor stoichiometry control. ACS Nano 8, 6363–6371 (2014).
Dai, Q. et al. Size-dependent composition and molar extinction coefficient of PbSe semiconductor nanocrystals. ACS Nano 3, 1518–1524 (2009).
Maes, J. et al. Light absorption coefficient of CsPbBr3 perovskite nanocrystals. J. Phys. Chem. Lett. 9, 3093–3097 (2018).
Iaru, C. M., Geuchies, J. J., Koenraad, P. M., Vanmaekelbergh, D. & Silov, A. Y. Strong carrier–phonon coupling in lead halide perovskite nanocrystals. ACS Nano 11, 11024–11030 (2017).
Swarnkar, A. et al. Quantum dot–induced phase stabilization of α–CsPbI3 perovskite for high-efficiency photovoltaics. Science 354, 92–95 (2016).
Gao, Y. & Peng, X. Photogenerated excitons in plain core CdSe nanocrystals with unity radiative decay in single channel: the effects of surface and ligands. J. Am. Chem. Soc. 137, 4230–4235 (2015).
Greytak, A. B. et al. Alternating layer addition approach to CdSe/CdS core/shell quantum dots with near-unity quantum yield and high on-time fractions. Chem. Sci. 3, 2028–2034 (2012).
Saha, A. et al. Near-unity quantum yield in semiconducting nanostructures: structural understanding leading to energy efficient applications. J. Phys. Chem. Lett. 4, 3544–3549 (2013).
Chen, O. et al. Compact high-quality CdSe–CdS core–shell nanocrystals with narrow emission linewidths and suppressed blinking. Nat. Mater. 12, 445–451 (2013).
Pu, C. & Peng, X. To battle surface traps on CdSe/CdS core/shell nanocrystals: shell isolation versus surface treatment. J. Am. Chem. Soc. 138, 8134–8142 (2016).
Coropceanu, I., Rossinelli, A., Caram, J. R., Freyria, F. S. & Bawendi, M. G. Slow-injection growth of seeded CdSe/CdS nanorods with unity fluorescence quantum yield and complete shell to core energy transfer. ACS Nano 10, 3295–3301 (2016).
Koscher, B. A., Swabeck, J. K., Bronstein, N. D. & Alivisatos, A. P. Essentially trap-free CsPbBr3 colloidal nanocrystals by postsynthetic thiocyanate surface treatment. J. Am. Chem. Soc. 139, 6566–6569 (2017).
Li, G. et al. Surface ligand engineering for near-unity quantum yield inorganic halide perovskite QDs and high performance QLEDs. Chem. Mater. 30, 6099–6107 (2018).
Liu, F. et al. Highly luminescent phase-stable CsPbI3 perovskite quantum dots achieving near 100% absolute photoluminescence quantum yield. ACS Nano 11, 10373–10383 (2017).
Dai, S.-W. et al. Perovskite quantum dots with near unity solution and neat-film photoluminescent quantum yield by novel spray synthesis. Adv. Mater. 30, 1705532 (2018).
Amani, M. et al. Near-unity photoluminescence quantum yield in MoS2. Science 350, 1065–1068 (2015).
Acknowledgements
M.K. thanks the MURI:MARBLe project under the auspices of the Air Force Office of Scientific Research (Award No. FA9550-16-1-0362) for their financial support of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, S., Zhukovskyi, M., Jankó, B. et al. Progress in laser cooling semiconductor nanocrystals and nanostructures. NPG Asia Mater 11, 54 (2019). https://doi.org/10.1038/s41427-019-0156-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-019-0156-4
This article is cited by
-
Electron-phonon interactions in halide perovskites
NPG Asia Materials (2022)
-
Highly-efficient thermoelectric-driven light-emitting diodes based on colloidal quantum dots
Nano Research (2022)
-
Phonon-assisted up-conversion photoluminescence of quantum dots
Nature Communications (2021)
