
Abstract
In nature, ion channels play key roles in controlling ion transport between cells and their surroundings. Calcium ion (Ca2+)-induced Ca2+ release (CICR), a critical control mechanism for Ca2+ channels, occurs due to a Ca2+ concentration gradient working in synergy with ryanodine receptors, which are famously known as “calcium sparks”. Inspired by this self-regulated biological process, a smart Ca2+ concentration-modulated nanochannel system was developed by integrating a poly{N-isopropylacrylamide-co-acrylamide-[4-(trifluoromethyl) phenyl]-2-thiourea0.2-co-acrylamide-DDDEEKC0.2} (denoted as PNI-co-CF3-PT0.2-co-DDDEEKC0.2) three-component copolymer onto the nanochannels of a porous anodic alumina (PAA) membrane. In this smart polymer design, the DDDEEKC hepta-peptide unit has an extraordinary binding affinity with Ca2+ through coordination bonds, while CF3-PT functions as a hydrogen bond mediation unit, facilitating the remarkable conformational transition of the PNI main chain in response to Ca2+-specific adsorption. Due to these futures, the dynamic gating behaviors of the modified nanochannels could be precisely manipulated by the Ca2+ concentration. In addition, the sensitive Ca2+ response, as low as 10 pM with a high specificity toward Ca2+ capable of discriminating Ca2+ from other potential interference metal ions (e.g., K+, Cu2+, Mg2+, Zn2+, Fe3+, and Al3+), remarkable morphological change in the nanochannel and satisfactory reversibility indicate the great potential of Ca2+-responsive polymers for the fabrication of biodevices and artificial nanochannels.
Similar content being viewed by others

Introduction
Natural ion channels, with high fidelity to specific ions, act as a pivotal part in supporting the metabolism of living cells (Scheme 1), and their dysfunction can lead to various diseases1,2; 40% of clinical drugs are designed to target ion channel proteins. Among ion channels, Ca2+ channels are prerequisites for an extensive range of life processes3,4, such as functions of nervous systems, cell proliferation, genetic transcription, and muscular contraction5,6. In particular, ryanodine receptors (RyRs) on sarcoplasmic reticulum (SR) membranes, which are in charge of the release of intracellular Ca2+ stores, are sensitive to the concentration of Ca2+ (Scheme 1, the lower part). Once the concentration reaches a threshold (10−6–10−5 mol L−1 (M)), conformational switching of RyRs will occur, which increases the helicity of the protein and shortens the distance and dihedral angle between two S6-peptide chains (Scheme 2a)7,8,9, resulting in the opening of nanopores of RyRs and the release of a large number of Ca2+ ions from the SR. These released Ca2+ will bind with calmodulin protein, subsequently leading to the closure of the Ca2+ channel. Thus, a Ca2+-induced Ca2+ release (CICR) process proceeds, which is crucial to the maintenance of the intracellular Ca2+ concentration and controls muscle contraction, particularly the cardiac muscle. The malfunction of the CICR process can lead to a great number of serious diseases10,11. This unique biological event has inspired chemists to develop various Ca2+-sensitive ion channels and nanodevices 12,13,14.

Illustration of the types of calcium-ion (Ca2+) channels in cells, including voltage-gated calcium channels, ligand-gated calcium channels (e.g., cAMP-modulated Ca2+ channels), inositol-1,4,5-triphosphate receptor (IP3R)-modulated Ca2+ channels, and RyR-modulated Ca2+ channels (CICR process). CICR is typically a positive-feedback system. First, voltage-gated Ca2+ channels are activated to allow a small amount of Ca2+ to pass through the cell membrane. Ryanodine receptors (RyRs) on the sarcoplasmic reticulum (SR) are sensitive to the above Ca2+ influx and open the nanochannels, which cause a large amount of Ca2+ stored in the SR to be released into the cytosol. A remarkable increase in cytosolic Ca2+ activates muscle contraction. Adapted with permission2

Design idea of biomimetic calcium-actuated nanochannel. a Structural transition of the S6 linker of the RyR1 protein activated by the binding of Ca2+. The outward movements of the U-motif and C-terminal domains (CTDs) trigger an outward movement of the inner S6 helices, presenting as an increase in the dihedral angle (θ). Thus, the hydrogen bond (E4948-Q4949), which helps to keep the closed state of the ionic channel, will be broken, and the nanochannel is opened. b, c Schematic illustration of Ca2+-adsorption-induced globule-to-coil transition of the copolymer chains (b) determines the open and closed state of the ionic channel (c). Strong coordination binding between hepta-peptide DDDEEKC and Ca2+ destroys the initial hydrogen bond network that is constructed by the hepta-peptide and the neighboring [4-(trifluoromethyl) phenyl]-2-thiourea (CF3-PT), which triggers the conformational transition of the copolymer chains. The expansion of the copolymer chains will block the nanochannel and substantially decrease the ionic flux
In the past decade, several elegant approaches have been applied to construct Ca2+-modulated nanochannels15,16,17. As typical examples, Siwy et al.18 reported a Ca2+-induced voltage gating, ionic current oscillations19, and charge inversion20, on the basis of track-etched asymmetric nanopores on polyethylene terephthalate (PET) films. Meng et al.21 utilized calcein-modified PET nanochannels to achieve a Ca2+-responsive nanogating. These modulation strategies mainly rely on surface charge changes of the nanochannels to achieve “on–off” switching, which are different from the ion channels in vivo whose gating behaviors are mainly achieved by the conformational transition of channel proteins, as illustrated in Scheme 2a. Within this context, much attention has been devoted to smart polymer-based22 nanochannels, which bear the obvious advantages of high controllability, satisfactory reversibility, and remarkable conformational transition in response to external stimuli23,24. By producing diverse external stimuli, such as temperature, pH, light, or redox potential25,26,27, reversible tuning of ion transport across smart polymer-modified nanochannels can be achieved, which has resulted in wide applications for both material sciences and life sciences28,29,30,31,32.
Nevertheless, to the best of our knowledge, smart polymer-based nanochannels that can be manipulated by Ca2+ concentration have rarely been reported. Building biomimetic Ca2+-sensitive ion channels is important for simulating the self-regulating and gating behaviors of RyR channels, the largest known calcium ion channel in humans. These channels will contribute to a more comprehensive understanding of Ca2+ signal pathways and promote numerous interesting applications in tissue engineering, controllable drug release, bioseparation, biosensors, microfluidics, and microreactors33.
Herein, inspired by the CICR process, a biomimetic Ca2+ self-regulated nanochannel system is constructed based on a Ca2+-responsive polymer design. A three-component copolymer PNI-co-CF3-PT0.2-co-DDDEEKC0.2 was designed according to a “recognition–mediation–main chain” concept34,35, in which the hepta-peptide DDDEEKC works as a Ca2+-recognition unit, CF3-PT functions as a hydrogen-bond mediation unit and PNI provides a flexible polymer main chain. As the core recognition unit for Ca2+, DDDEEKC with an α-helix conformation was first found as an N-terminal region of statherin36 with a strong adsorption capacity toward hydroxyapatite, which inspired us to utilize this natural peptide to design the Ca2+-binding material37,38. Subsequently, CF3-PT was introduced as a mediation unit owing to its strong hydrogen bond donating capacity. The thiourea group in CF3-PT can combine with the carboxyl groups in the above hepta-peptide utilizing multiple hydrogen bonding interactions to build a compact hydrogen bond network within the copolymer, as shown in Scheme 2b in the left panel. When the copolymer film is exposed to Ca2+, multiple and strong chelation bindings between Ca2+ and the carboxyl groups in the hepta-peptide gradually destroy the initial hydrogen bond network. When the Ca2+ concentration reaches a threshold, the copolymer chains undergo a dramatic globule-to-coil transition, leading to an obvious expansion of the copolymer film (Scheme 2b, right panel). This smart polymer design not only improves the grafting density of the core hepta-peptide to provide more binding sites for Ca2+ but also supplies an ideal platform to amplify the Ca2+ recognition signal by taking advantage of the conformational transition of the copolymer chain. The combination of these two merits can substantially improve the sensitivity of the material.
Then, porous anodic alumina (PAA) was chosen as a substrate to build the nanochannel system. Compared with other porous materials, PAA exhibits remarkable advantages, such as adjustable film thickness, high porosity, and tunable parameters, and has been widely used in controlling ion transportation, as well as constructing various functional devices39,40,41. By using surface-initiated atom transfer radical polymerization (SI-ATRP)42,43, the designed copolymer was then immobilized onto the straight-through nanochannel of a PAA membrane with an average diameter ranging from 70 to 100 nm. The affinity test, quartz crystal microbalance with dissipation (QCM-D) binding analysis, electrochemical impedance spectroscopy (EIS) analysis, morphological observations, and transmembrane ionic current measurements clearly demonstrated that the well-developed nanochannel displayed controllable and reversible gating abilities toward Ca2+ adsorption or desorption, as illustrated in Scheme 2c. It is worth noting that such a gating effect was highly specific to Ca2+, and other metal ions, such as K+, Mg2+, Al3+, Zn2+, Fe3+, and Cu2+, did not induce any evidential changes. This work provides a smart polymer-based design concept to mimic the crucial CICR process; precise Ca2+-modulated gating performance might facilitate the development of various bioseparation membranes, nanochannels, and microfluidic devices44.
Materials and methods
Materials
N-Isopropylacrylamide (NIPAAm, 98%) was purchased from Sigma-Aldrich (China) and was recrystallized in n-hexane three times before being used in the copolymerization. The peptide sequence DDDEEK(Dde)C was purchased from ChinaPeptides Co., LTD with high purity (>99%). 2-Bromoisobutyryl bromide, 2-mercaptoethylamine, and N,N,N′,N″,N″- pentamethyldiethylenetriamine (PMDETA) (Sigma-Aldrich) were used as received.
Synthesis of acrylamide hepta-peptide
The synthesis of acrylamide hepta-peptide was illustrated in Scheme 3a. DDDEEK(Dde)C (0.204 g, 0.2 mmol) and 0.2 mL triethylamine were dissolved in 15 mL anhydrous N,N′-dimethylformamide (DMF), and the mixture was stirred for 15 min in an ice bath. Then, 5 mL of DMF containing acryloyl chloride (0.035 mL, 0.4 mmol) was added to the mixture dropwise and stirred for 0.5 h in an ice bath. The reaction mixture was then stirred at room temperature for 24 h. Most DMF was evaporated under reduced pressure. The crude product was purified on a Shimadzu UFLC 20A purity system with a C18 reverse-phase semipreparative chromatographic column. The pure product was obtained as a white powder (0.142 g, yield: 66%). 1H NMR (d6-DMSO) δ (ppm): 13.23 (s, 2H, COOH), 12.00–12.50 (br, 4H, COOH), 9.35–9.53 (m, 6H, CONH), 6.23 (d, J = 17.2 Hz, 1H, CH = C), 6.05, 6.09 (dd, J1 = J2 = 10 Hz, 1H, CH = C), 5.86 (d, J = 10.4 Hz, 1H, CH = C), 4.46–4.58 (m, 2H, *CH), 4.30–4.39 (m, 2H, *CH), 4.19–4.28 (m, 3H, *CH), 3.06–3.10 (m, 16H, CH2C, 3H, CCH3), 2.47 (s, 2H, Dde-COCH2), 2.27 (s, 2H, Dde-COCH2), 1.85–1.93 (m, 2H, CH2C), 1.67–1.77 (m, 2H, CH2C), 1.53–1.60 (m, 2H, CH2C), 1.32–1.39 (m, 2H, CH2C), 0.94 (s, 6H, Dde-CCH3). Elemental analysis, calcd. (%) for C44H62N8O21S: C, 49.34; H, 5.83; N, 10.46; found C, 49.21; H, 5.75; N, 10.63; MALDI-MS: m/z calcd. for C44H62N8O21S: 1070.38; found: 1071.37 [M + H]+.

a Preparation of acrylamide DDDEEK(Dde)C. b Modification of porous anodic alumina (PAA) membrane, gold surface of QCM-D resonator, or electrode with poly{N-isopropylacrylamide-co-acrylamide 1-[4-(trifluoromethyl)phenyl]-2-thiourea0.2-co-acrylamide-DDDEEKC0.2} (denoted as PNI-co-CF3-PT0.2-co-DDDEEKC0.2) via surface-initiated atom transfer radical polymerization
Synthesis of PNI-co-CF3-PT0.2-co-DDDEEKC0.2-modified PAA membrane
For the modification of the PAA membrane (Scheme 3b), the PAA membrane was first immersed in distilled water and ethanol for 10 min, followed by a quick dip in a hydrochloric acid aqueous solution (5%, v/v) for 35 s and subsequently in hydrogen peroxide heated at 100 °C for 1 h to generate surface hydroxyl groups. After that, the membrane was washed with an excess of distilled water and ethanol and dried under a nitrogen flow. Then, it was heated at 65 °C in 40 mL toluene containing 1.0 mL APTES for 3 h to obtain chemically bonded-NH2 groups on the membranes. The reaction was performed in a nitrogen atmosphere. Ethanol was used to wash out the remaining APTES. After drying under a flow of nitrogen gas, the PAA membrane was immersed in 50 mL dry dichloromethane containing 0.4 mL pyridine. Then, 2-bromoisobutyryl bromide (0.4 mL) was added dropwise into the solvent at 0 °C for 1 h, and then at 25 °C for 12 h. After rinsing with CH2Cl2, a bromine-modified PAA membrane was received.
The modifications of PNI-co-CF3-PT0.2-co-DDDEEKC0.2 were synthesized by the method of ATRP according to the literature43. The bromine-modified PAA membrane was immersed in a degassed solution of NIPAM (0.1371 g, 1.2 mmol), acrylamide CF3-PT (0.1096 g, 0.4 mmol), acrylamide-DDDEEKC (0.4284 g, 0.4 mmol) in 10 mL DMF containing CuBr (0.0143 g, 0.1 mmol), and N,N,N’′,N″,N′-PMDETA (0.1 mL, 0.47 mmol). The reaction was carried out at 60 °C for 15 h with nitrogen protection. Subsequently, the copolymer-modified PAA membrane was sequentially cleaned with DMF, water, and ethanol and subsequently dried under a nitrogen flow. The PNIPAAm-modified and CF3-PT-co-DDDEEKC-modified PAA membranes were prepared through a method similar to that described above.
Electrical measurements
Electrochemical impedance spectroscopy measurement
EIS experiments were performed in 0.1 mmol L−1 KCl solution containing [Fe(CN)6]3−/4− (5 mmol L−1), and the experimental conditions were as follows: open circuit potential, 0.3 V; alternative voltage, 5 mV; frequency range, 0.1−105 Hz. The PNI-co-CF3-PT0.2-co-DDDEEKC0.2-modified Au electrode was prepared using the same method described above. The working electrode, which was a Au electrode modified with PNI-co-CF3-PT0.2-co-DDDEEKC0.2, a Ag/AgCl reference electrode and a graphite auxiliary electrode made up the three-electrode system. Temperature: 20 °C.
Ionic current measurement
A piece of PAA membrane (bare or modified) was mounted between a two-compartment electrochemical cell according to the literature45. Ag/AgCl electrodes were used to apply a transmembrane potential across the membrane. The transmembrane ionic current was measured with a Keithley 6487 picoammeter/voltage source (Keithley Instruments) through Ag/AgCl electrodes. The effective area for the ionic conduction measurements was ~20 mm2. The electrolyte was 0.01 M sodium chloride (NaCl) solution.
Results
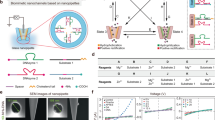
First, the binding affinity of the hepta-peptide with CaCl2 was evaluated by isothermal titration calorimetry (ITC), which has been widely used as a label-free and quantitative technique to detect the thermodynamic parameters of interactions between a small molecule and a biomacromolecule in solution46. A classical isothermal calorimetric titration profile of 20 mM Ca2+ with 1 mM hepta-peptide in pure water at 25 °C is shown in Fig. 1a. Strong heat release was observed, corresponding to an ~5:1 binding stoichiometry between Ca2+ and hepta-peptide with stepwise binding constants of Ka1, 773; Ka2, 1030; Ka3, 763; Ka4, 1050; and Ka5, 700 L mol−1 (Fig. 1b), as well as a accumulative Ka (Ka1 × Ka2 × Ka3 × Ka4 × Ka5) of 4.46 × 1014 L mol−1, which suggested strong and specific complexation. Carbon nuclear magnetic resonance (13C NMR) titration experiments validated this complexation. As shown in Fig. 1c–f, upon the addition of different amounts of CaCl2 to the hepta-peptide in deuterated water, six sets of carbon atom signals for carboxyl groups in the hepta-peptide display clear downfield shifts. By comparison, no obvious chemical shift change was observed for the carbon atom signals of the amide groups. This difference indicated that multiple coordination bonds between Ca2+ and the carboxyl groups in the hepta-peptide were the main driving forces for the strong complexation.

a, b Isothermal titration calorimetric data when various equivalents of CaCl2 (20 mM in H2O) were added to a DDDEEKC aqueous solution (1.0 mM) at 20 °C. The red line in b represents a nonlinear fitting curve based on a sequential binding site model (N = 5). Partial carbon nuclear magnetic resonance (13C NMR) spectra of DDDEEKC (c) and the mixture of DDDEEKC and CaCl2 at molar ratios of 1:1 (d), 1:3 (e), and 1:5 (f) in d6-DMSO at 20 °C; chemical shift changes of carboxylic acids and amides of the hepta-peptide are indicated by black dashed arrows. The NMR attribution of each group is determined by quantum chemistry calculation [Gassiness, density functional theory (DFT), at 6–31 g level of theory]
A possible binding model was provided by molecular dynamic calculations by using the density function theory. Figure 2a, b shows three-dimensional structures of the hepta-peptide before and after interacting with five Ca2+, and some remarkable changes in peptide conformation and dihedral angle (θ) of the carboxyl group could be observed. Specifically, the distance between the amine in the side chain of the sixth Lys and the thiol in the seven Cys decreases from 9.98 to 9.59 Å, while the distance between the carboxyl groups in the fourth Glu and the fifth Glu decreases from 10.97 to 10.61 Å. The dihedral angle of the carboxyl group in the fifth Glu displays a noteworthy increase from −31° to −39° after binding with Ca2+. Circular dichroism (CD) was applied to further discuss the conformation change of the hepta-peptide. The CD spectrum of the hepta-peptide was similar to that observed for α-helical polypeptides according to the literature47,48. Although the banding intensity may vary with solvents due to the different polarity and hydrogen bonding strength, a strong negative adsorption peak centered at 200 nm and a weak shoulder peak at ~225 nm could be observed, which represent the exciton split of the π–π* and n–π at the long wavelength component, respectively (Fig. 2c). Upon the addition of CaCl2 to the hepta-peptide solution, the CD intensity of the peak at 200 nm decreases slightly, while the signal at 188 nm increases, which indicated a decrease in the proportion of the α-helix structure and the appearance of a random coil structure due to competitive binding interactions between hepta-peptide and the CaCl2 solute. The remarkable conformational change (Fig. 2d) and high affinity measured by the ITC and 13C NMR titration experiments revealed that this hepta-peptide can serve as an ideal binding unit for Ca2+.

Three-dimensional structures of DDDEEKC before (a) and after binding with five Ca2+ ions (b), obtained by molecular dynamic calculation. c Circular dichroism (CD) spectra of the hepta-peptide (0.5 mM) upon additions of a gradient concentration of CaCl2 from 0.5 to 2.5 mM in H2O. d Graphic illustration of the secondary conformation changes of the hepta peptide interacting with Ca2+. e Fluorescence spectra of fluorescein-labeled CF3-PT (5 × 10−6 mol L−1) upon the addition of hepta-peptide in a Tris–HCl buffer solution (pH = 7.4, 10 mM) at 20 °C. The inset shows the fluorescence intensity change (at 517 nm) upon the addition of the hepta-peptide. [G]/[H] is an abbreviation of the molar ratio of guest to host, and the red line in the inset is a nonlinear-fitted curve for calculating the association constant (Ka). f Fluorescence spectra of a mixture of fluorescein-labeled CF3-PT (5 × 10−6 mol L−1) and equimolar amount of hepta-peptide upon the addition of CaCl2 in a Tris–HCl buffer solution at 20 °C. Recovery of fluorescence intensity indicated that the addition of Ca2+ ions destroyed the complexation between CF3-PT and the hepta-peptide copolymer
For the purpose of improving the binding affinity with Ca2+ and amplifying the recognition signal, the acrylamide hepta-peptide was prepared to copolymerize with acrylamide CF3-PT and NIPAAm through SI-ATRP, generating a smart PNI-co-CF3-PT0.2-co-DDDEEKC0.2 copolymer film (average thickness of 20 nm) on a QCM resonator gold electrode surface. The grafting ratios of 0.2 for hepta-peptide and CF3-PT units were determined according to the elemental analysis and integration ratios of the characteristic peaks in the 1H NMR spectrum of the copolymer (Supplementary Fig. 2a in SI). To test the three-component copolymer design (Scheme 2b), a fluorescence titration experiment was performed to evaluate the binding affinity (Ka)49 between a fluorescein-labeled CF3-PT (Supplementary Fig. 1 in SI) and the hepta-peptide. As shown in Fig. 2e, when 35 equivalents of hepta-peptide were added into the fluorescein-labeled CF3-PT (5 × 10−6 mol L−1) in Tris–HCl buffer solution (pH 7.4, 10 mM), and the fluorescence intensity of the solution decreased by 14%, corresponding to a Ka of 7969 ± 480 L mol−1 calculated from a nonlinear fitting. It is worth noting that such a fluorescence intensity decrease was not caused by the acidity of the carboxyl groups in the hepta-peptide because the solution pH was maintained at a constant value by a buffer solution. Then, an equimolar mixture of the hepta-peptide and the fluorescein-labeled CF3-PT in Tris–HCl buffer solution was prepared, and the fluorescence spectra were recorded (Fig. 2f). Interestingly, when different amounts of CaCl2 capable of binding with the hepta-peptide were added to the mixture, the fluorescence intensity of CF3-PT increased gradually and returned to its initial value prior to the hepta-peptide addition. The recovery of fluorescence intensity could be reasonably attributed to the competitive binding of the hepta-peptide with Ca2+, which was substantially stronger than that of the hepta-peptide with CF3-PT50,51. Attenuated total reflection flourier transform infrared spectroscopy (ATR-IR) further validated the interaction between the hepta-peptide and CF3-PT (Supplementary Fig. 3 in SI).
The above control experiment demonstrated the feasibility of our smart polymer design. Then, the adsorption dynamics of various metal ions on the PNI-co-CF3-PT0.2-co-DDDEEKC0.2 copolymer surface were monitored using a QCM-D to measure the frequency (Δƒ) and energy dissipation (ΔD)52. As shown in Fig. 3a by the red line, CaCl2 (10 μM in pure water) displays slow and strong adsorption on the copolymer surface and reaches a maximum after 45 min (Δƒ = 32.5 Hz); according to the Sauerbrey equation53, the adsorption quantity is 191.5 ng cm−2. Under the same conditions, the FeCl3-induced frequency change was only −3.8 Hz, and NaCl, KCl, or AlCl3-induced frequency changes were negligible. Interestingly, the adsorption of MgCl2 and ZnCl2 was quite similar to that of CaCl2 in electric charge, leading to abnormal weight loss of the copolymer film, and the QCM frequency slightly increased to 3 Hz. This high Ca2+-ion selectivity was further validated by a time-dependent dissipation curve, and the real-time information of the changes in viscoelasticity and thickness of the copolymer layer was recorded. As shown in Fig. 3b (red line), a prominent dissipation increase (ΔD: 5.6 × 10−6) was only observed for CaCl2 adsorption on the copolymer film. Based on the QCM adsorption theory54, a softer and swollen copolymer film after interacting with CaCl2 was demonstrated by these data, reflecting a relaxed state of the copolymer chains. By comparison, the NaCl, KCl, ZnCl2, or AlCl3 adsorption-induced dissipation changes were negligible. MgCl2 or FeCl3 adsorption led to a continuous decrease in the dissipation value (ΔD: −3.6 × 10−6 or −6.1 × 10−6), revealing remarkable shrinkage of the copolymer film. Distinct adsorption dynamics of various metal ions on the copolymer surfaces and the corresponding dissipation curve variations demonstrated the excellent Ca2+ selectivity of the copolymer film. The 1H NMR titration experiment provided auxiliary evidence for complexation between the copolymer and Ca2+, in which clear chemical shift changes of the carboxylic acids and amides of the copolymer were observed after the addition of CaCl2 (Supplementary Fig. 2b in SI).

Time dependency of frequency a dissipation and b variations during MgCl2, CaCl2, FeCl3, ZnCl2, AlCl3, or KCl (10 μM in pure water) adsorbed on the copolymer-modified quartz crystal resonator surface at 20 °C. c Nyquist plots of EIS obtained at the copolymer-modified electrode surface in 0.1 M KCl solution containing 5 mM Fe (CN)63−/4− upon additions of CaCl2 from 10−10 to 10−4 M. Inset, equivalent circuit of the electrochemical sensor to study the impedance spectra. d CaCl2 concentration dependence of the impedance change ratio (Rct) of the copolymer-modified gold electrodes in 5 mM [Fe(CN)6]3−/4−. Inset: the Ca2+-triggered transition of the copolymer chains promoted the mass transport of [Fe(CN)6]3−/4− through the copolymer brush. e Nyquist plots of EIS obtained at the copolymer-modified electrode surface upon addition of different metal ion solutions (CaCl2, ZnCl2, MgCl2, CuCl2, or AlCl3; 0.1 mM). f Comparison of the Rct decrease caused by the adsorption of different metal ions (0.1 mM). Error bars represent standard error measurements
The shrinking-to-swelling transition of the copolymer brushes might strongly influence the electrochemical process on the surface of a copolymer-modified gold electrode, resulting in remarkable alteration of the mass transport of [Fe(CN)6]3−/4− through the copolymer brush, from bulk solution to the electrode surface (Inset of Fig. 3d)35. Based on this presumption, the Ca2+-adsorption-induced conformational transition could be monitored by EIS. Figure 3c displays the Nyquist plots obtained for the copolymer-modified electrode before and after being treated with different amounts of CaCl2 in 5 mM K3Fe(CN)6/K4Fe(CN)6 solution for 5 min. Initially, the electrochemical activity was blocked by the copolymer on the electrode with a charge transfer resistance (Rct) of 160 Ω, and this value gradually dropped down to 75 Ω when 100 μM of CaCl2 was added. This Rct decrease easily avoided the potential interference of nonspecific adsorption, which often results in the increase of Rct. As shown in Fig. 3d, the Rct value decreased gradually with the increasing CaCl2 concentration, and a 53% Rct decrease was measured upon the addition of 100 μM of CaCl2. Notably, even when the concentration of CaCl2 was only 100 pM, the decrease in Rct (5%) was still obvious and readily detected. In addition, the ion selectivity of the copolymer film was evaluated by EIS measurements, as shown in Fig. 3e, f. The most obvious changes in the Nyquist plot and corresponding Rct were observed when CaCl2 (100 μM) was tested, while the NaCl, ZnCl2, MgCl2, CuCl2, or AlCl3-induced Rct changes were substantially weaker under the same conditions. These data further illustrated the highly selective response of the copolymer toward CaCl2.
To construct biomimetic ionic nanochannels, PNI-co-CF3-PT0.2-co-DDDEEKC0.2 was grafted onto the porous channel of PAA membranes with uniform straight nanochannels (average pore size: 90–110 nm). The ATR-IR and thermal gravimetric analysis tests validated the modification of the copolymer on the PAA membrane (Supplementary Figs. 4 and 5 in SI). Then, atomic force microscopy (AFM) and scanning electron microscopy (SEM) were adopted to observe the morphological changes of the copolymer-modified PAA membranes after being immersed in a CaCl2 aqueous solution (20 μM) for 10 min. From the top view of the membrane, as observed by AFM (Fig. 4a, b), the initially well-defined nanopores became indistinct, and a few nanopores were blocked by the expanded copolymers according to the section profiles of the AFM images. According to the statistical analysis of the AFM images, the average pore size decreased from 82.5 to 52 nm, while the root mean square roughness (Rq) of the PAA membrane increased from 7.23 to 10.85 nm, which indicated conspicuous macroscopic surface changes in the copolymer-modified PAA caused by Ca2+ adsorption. In addition, based on the SEM cross section images (Fig. 4c, d), the average wall thickness of the nanochannels increased from 55 to 81 nm, resulting in a remarkable decrease in the average size of the nanochannels from 67 to 40 nm. These obvious morphological changes provided direct evidence for the expansion of the copolymer chains.

a, b and scanning electron microscopy (SEM) cross-sectional images (c, d) of the copolymer-modified porous anodic alumina (PAA) membrane (a, c) and after treatment with CaCl2 solutions (20 μM) for 10 min at 20 °C (b, d); green lines in the corresponding AFM section profiles. Narrow scan X-ray photoelectron spectra of the copolymer-modified PAA membrane before (f) and after (e, g) treatment with CaCl2 solution (20 μM) for 10 min at 20 °C. e Ca 2p; f, g C 1s. These surfaces were rinsed with water before measurements to eliminate the physical adsorption of Ca2+
To analyze the elementary composition of the copolymer film, X-ray photoelectron spectroscopy (XPS) was performed. Compared with the XPS spectrum of the bare PAA membrane, signals of C 1s, N 1s, S 2p, and F 1s with binding energies of 285.0, 399.9, 168.4, and 689.0 eV, respectively, could be clearly observed for the copolymer-modified PAA membrane, which validated the successful immobilization of the copolymer on the PAA membrane. When the copolymer-modified membrane was treated with CaCl2 solution (20 μM) for 10 min and then rinsed with water, Ca 2p signals with binding energies of 347.3 and 350.8 eV appeared (Fig. 4e), which indicated that Ca2+ had been chemically adsorbed onto the copolymer film. From the perspective of C 1s signals, Ca2+-adsorption remarkably strengthened the carbonyl signal of C 1s, as shown by the blue dashed lines in Fig. 4f, g, which is a contribution from chelation binding. In addition, the C–H, C–C and C–N signals of C 1s increased substantially, indicating that these functional groups were fully exposed due to the expansion of the copolymer chains55.
With the expansion of the copolymer chains, the size of the nanochannels decreased sharply, which further led to a decrease in the ionic flux of the nanochannels. This ionic transport property of the nanochannel was examined by current–voltage measurements using a Keithley 6487 picoammeter (Fig. 5a). All PAA membranes were separately mounted in an electrochemical cell (inset of Fig. 5a). A NaCl solution served as the electrolyte to measure the ionic current across the nanochannels at a constant volume of 1 mL56. The initial transmembrane ionic current was 100 μA (at +0.2 V), indicating the good permeability of the copolymer-modified PAA membrane for ion transport. Then, electrolytes with different concentrations of CaCl2 were separately added to the cell exposed to the PAA membrane. Figure 5b displays the CaCl2 concentration dependence of the ionic current (at +0.2 V) change ratio [defined as (I − I0)/I0, where I0 is the initial current] for the bare, PNIPAAm and PNI-co-CF3-PT0.2-co-DDDEEKC0.2-modified PAA membranes45. The detection range for Ca2+ extended from 1 × 10−4 mol L−1 to an ultratrace level of 1 × 10−11 mol L−1, which fully covered the intracellular and extracellular Ca2+ / 1 × 10−8 to 1 × 10−3 mol L−157,58 and makes it possible to further utilize this artificial nanochannel for medical treatment. For the copolymer-modified PAA membrane, an elegant negative correlation curve between (I − I0)/I0 and the Ca2+ concentration was observed, suggesting the powerful and flexible regulatory capacity of Ca2+ for this nanochannel. The ionic current change ratio gradually increased from 12.3% at 10−11 mol L−1 to 47.5% at 10−4 mol L−1, reflecting a satisfactory gating performance. In contrast, a negligible change in the ionic current was observed for the bare or PNIPAAm-modified PAA, which indicated the strong Ca2+ binding capacity and high grafting density of the hepta-peptide in the three-component copolymer contributed to the remarkable sensibility and excellent gating efficiency.

a Transmembrane ionic current curves of DDDEEKC0.2-co-CF3-PT0.2–modified PAA membranes in NaCl aqueous solution (0.01 mol L−1) with (red) or without (black) 10 μM CaCl2 at 20 °C. Inset, electrical measurement apparatus. Current change ratios of PNI-co-DDDEEKC0.2-co-CF3-PT0.2 (black), bare (red)- or PNIPAAm (blue)-modified PAA (b) or PNI-co-DDDEEKC0.2-co-CF3-PT0.2 (black), DDDEEKC monolayer (red)- or poly(CF3-PT-co-DDDEEKC0.5) (blue)-modified PAA (c) in 0.01 mol L−1 NaCl solutions with different amounts of CaCl2. d Schematic illustration of different Ca2+ response modes corresponding to three kinds of PAA membranes shown in c. e Comparison of ionic current change ratios of PNI-co-DDDEEKC0.2-co-CF3-PT0.2-modified PAA membrane upon the addition of different metal ions (CaCl2, KCl, ZnCl2, MgCl2, FeCl3, AlCl3, and CuCl2; 0.01 mM). f Ionic current cycling measurement of the copolymer-modified membrane through alternate treatments with 0.01 mol L−1 NaCl with CaCl2 (10 μM) or individual NaCl solution for 10 min at 20 °C, separately. All data are shown as the mean ± standard error
A control experiment was performed to determine the rationality of the three-component copolymer design. Using hepta-peptide single monolayer and poly(CF3-PT-co-DDDEEKC0.5)-modified PAA membranes, the maximal ionic current change ratios caused by Ca2+ were only 13.4% and 20%, respectively (Fig. 5c), both of which are substantially lower than that of PNI-co-CF3-PT0.2-co-DDDEEKC0.2-modified PAA. We presumed that Ca2+ adsorption on the hepta-peptide monolayer surface only changed the surface charge of the nanochannels owing to the chelation binding between Ca2+ and the carboxylic acid groups in the hepta-peptide (Fig. 5d, left panel). For the poly(CF3-PT-co-DDDEEKC0.5) two-component copolymer with the absence of PNIPAAm, the flexibility of the copolymer chains was substantially reduced, resulting in a more contracted state owing to strong hydrogen bonding interactions between CF3-PT and the hepta-peptide units. Under this condition, the introduction of Ca2+ could not destroy such a compact hydrogen bond network, and the conformational change was limited (Fig. 5d, middle panel). Only the integration of hepta-peptide, CF3-PT, and flexible PNIPAAm into one system allowed the Ca2+-triggered globule-to-coil transition of the copolymer to occur and contributed to the remarkable ionic current change (Fig. 5d, right panel).
Satisfactory reversibility of the ionic gating behaviors was also displayed. As shown in Fig. 5e, the ionic current switches between 100 and 50 μA through alternate treatment by the electrolyte with or without CaCl2 (10 μM), and the reversibility was well maintained after seven cycles.
Due to the complexity of the cellular environment, a high demand is set not only for sensitivity and reversibility but also for selectivity of natural ion channels. Thus, the selectivity of an artificial nanochannel system is a vital evaluation criterion. A series of selectivity tests was performed using a control variable method. As expected, not only a sensitive and reversible response to the target Ca2+ was achieved; this nanochannel system also displayed accurate discrimination capacity among Ca2+, K+, Zn2+, Mg2+, Fe3+, Al3+, and Cu2+ (Fig. 5f). The Ca2+ adsorption-induced ionic current change ratio (47.5%) was significantly larger than that of K+ (12.4%) or Zn2+ (12%) and was distinct from that of Mg2+, Fe3+, Al3+, and Cu2+, the adsorption of which led to an increase in the ionic current. This indicates the remarkable advantage of our material with a high specificity toward Ca2+ that conventional artificial nanochannels have difficulty achieving.
Discussion
In conclusion, inspired by the CICR process, we constructed a smart (PNI-co-CF3-PT0.2-co-DDDEEKC0.2)-based calcium-actuated nanochannel. With the intense conformational transitions of the copolymer chains from globule to coil, the gating behavior of the smart nanochannel is closer to that of an ion channel protein, facilitating sensitive monitoring of Ca2+ concentrations as low as 10 pM and achieving a wide stimulus response range to Ca2+ that fully covers Ca2+ levels in human cells. Excellent selectivity among various multivalent metal ions and satisfactory reversibility are attractive features, making it possible to recognize Ca2+ and modulate the gating of nanochannels in a complex environment. Moreover, this work demonstrates the feasibility of utilizing a biomimetic strategy for building artificial nanochannels with the help of biomolecule-responsive polymer design, which will give rise to more attractive work on biomimetic nanochannels.
In addition to Ca2+ channels, as one of the most widespread secondary messengers used in signal transduction, Ca2+ ions play key roles in the physiology and biochemistry of organisms and cells2. Particularly, the calcium levels in mammals are tightly regulated, and channels determine the release of Ca2+ from bone into bloodstream, reabsorption of Ca2+ in the kidney back into circulation, activation of vitamin D3 to calcitriol capable of promoting calcium absorption, and participate in blood-clotting cascade and muscular contraction34. Therefore, the concentration of Ca2+ in the human body has become a vital index in research on tissue engineering and biomedicine59, increasing demand for Ca2+ detection and Ca2+ controllable release in interdisciplinary fields. In this context, our smart material may have potential applications in high-sensitivity Ca2+ detection and portable devices in therapy, as well as promising Ca2+-actuated bioseparation membranes and microfluidic devices60.
References
Noskov, S. Y., Bernèche, S. & Roux, B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature 431, 830 (2004).
Dong, Z., Saikumar, P., Weinberg, J. M. & Venkatachalam, M. A. Calcium in cell injury and death. Annu. Rev. Pathol. Mech. Dis. 1, 405–434 (2006).
Nonner, W., Catacuzzeno, L. & Eisenberg, B. Binding and selectivity in L-type calcium channels:a mean spherical approximation. Biophys. J. 79, 1976–1992 (2000).
Matulef, K. & Zagotta, W. N. Cyclic nucleotide-gated ion channels. Annu. Rev. Cell Dev. Biol. 19, 23–44 (2003).
Sun, Z. H., Barboiu, M., Legrand, Y. M., Petit, E. & Rotaru, A. Highly selective artificial cholesteryl crown ether K+- channels. Angew. Chem. Int. Ed. 54, 14473–14477 (2015).
Gao, Y., Szymanowski, J. E., Sun, X., Burns, P. C. & Liu, T. Thermal responsive ion selectivity of uranyl peroxide nanocages: an inorganic mimic of K+ ion channels. Angew. Chem. Int. Ed. 55, 6887–6891 (2016).
Guo, T., Gillespie, D. & Fill, M. Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ. Res. 111, 28–36 (2012).
Zalk, R. et al. Structure of a mammalian ryanodine receptor. Nature 517, 44 (2014).
Wei, R. et al. Structural insights into Ca2+-activated long-range allosteric channel gating of RyR1. Cell Res. 26, 977 (2016).
Zhou, Q. et al. Impairment of PARK14-dependent Ca2+ signalling is a novel determinant of Parkinson’s disease. Nat. Commun. 7, 10332 (2016).
Immler, R., Simon, S. I. & Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Invest. 48, e12964 (2018).
Hoffman, A. S. Stimuli-responsive polymers: biomedical applications and challenges for clinical translation. Adv. Drug Deliv. Rev. 65, 10–16 (2013).
Pérez-Mitta, G., Albesa, A. G., Trautmann, C., Toimil-Molares, M. E. & Azzaroni, O. Bioinspired integrated nanosystems based on solid-state nanopores: “iontronic” transduction of biological, chemical and physical stimuli. Chem. Sci. 8, 890–913 (2017).
Zhang, X. et al. Visual and highly sensitive detection of cancer cells by a colorimetric aptasensor based on cell-triggered cyclic enzymatic signal amplification. Anal. Chem. 86, 5567–5572 (2014).
Siwy, Z. S., Powell, M. R., Kalman, E., Astumian, R. D. & Eisenberg, R. S. Negative incremental resistance induced by calcium in asymmetric nanopores. Nano Lett. 6, 473–477 (2006).
Vilozny, B., Actis, P., Seger, R. A., Vallmajo-Martin, Q. & Pourmand, N. Reversible cation response with a protein-modified nanopipette. Anal. Chem. 83, 6121–6126 (2011).
Ali, M. et al. Calcium binding and ionic conduction in single conical nanopores with polyacid chains: model and experiments. ACS Nano. 6, 9247–9257 (2012).
Siwy, Z. S. et al. Calcium-induced voltage gating in single conical nanopores. Nano. Lett. 6, 1729–1734 (2006).
Powell, M. R. et al. Nanoprecipitation-assisted ion current oscillations. Nat. Nanotechnol. 3, 51 (2007).
He, Y. et al. Tuning transport properties of nanofluidic devices with local charge inversion. J. Am. Chem. Soc. 131, 5194–5202 (2009).
Meng, Z., Jiang, C., Li, X. & Zhai, J. Calcein-modified multinanochannels on PET films for calcium-responsive nanogating. ACS Appl. Mater. Interfaces 6, 3794–3798 (2014).
Secker, C., Brosnan, S. M., Luxenhofer, R. & Schlaad, H. Poly(α-Peptoid) s revisited: synthesis, properties, and use as biomaterial. Macromol. Biosci. 15, 881–891 (2015).
Wen, L. & Jiang, L. Bio-inspired smart gating nanochannels based on polymer films. Sci. China Chem. 54, 1537 (2011).
Hou, X., Zhang, H. & Jiang, L. Building bio-inspired artificial functional nanochannels: from symmetric to asymmetric modification. Angew. Chem. Int. Ed. 51, 5296–5307 (2012).
Guo, W., Tian, Y. & Jiang, L. Asymmetric ion transport through ion-channel-mimetic solid-state nanopores. Acc. Chem. Res. 46, 2834–2846 (2013).
Cobo, I., Li, M., Sumerlin, B. S. & Perrier, S. Smart hybrid materials by conjugation of responsive polymers to biomacromolecules. Nat. Mater. 14, 143 (2014).
Sun, Y. et al. A light-regulated host–guest-based nanochannel system inspired by channelrhodopsins protein. Nat. Commun. 8, 260 (2017).
Sun, Z. et al. Fabrication of cysteine-responsive biomimetic single nanochannels by a thiol-yne reaction strategy and their application for sensing in urine samples. Adv. Mater. 26, 455–460 (2014).
Mamad-Hemouch, H. et al. Biomimetic nanotubes based on cyclodextrins for ion-channel applications. Nano. Lett. 15, 7748–7754 (2015).
Huang, C., Yang, G., Ha, Q., Meng, J. & Wang, S. Multifunctional “smart” particles engineered from live immunocytes: toward capture and release of cancer cells. Adv. Mater. 27, 310–313 (2015).
Li, X. et al. Integrated solid-state nanopore electrochemistry array for sensitive, specific, and label-free biodetection. Langmuir 34, 14787–14795 (2018).
Li, L. et al. High Performance field-effect ammonia sensors based on a structured ultrathin organic semiconductor film. Adv. Mater. 25, 3419–3425 (2013).
Li, W., Yang, C.-X. & Yan, X.-P. A versatile covalent organic framework-based platform for sensing biomolecules. Chem. Commun. 53, 11469–11471 (2017).
Qing, G. & Sun, T. Chirality-triggered wettability switching on a smart polymer surface. Adv. Mater. 23, 1615–1620 (2011).
Ding, S., Cao, S., Zhu, A. & Shi, G. Wettability switching of electrode for signal amplification: conversion of conformational change of stimuli-responsive polymer into enhanced electrochemical chiral analysis. Anal. Chem. 88, 12219–12226 (2016).
Long, J. R., Shaw, W. J., Stayton, P. S. & Drobny, G. P. Structure and dynamics of hydrated statherin on hydroxyapatite as determined by solid-state NMR. Biochemistry 40, 15451–15455 (2001).
Roy, D., Cambre, J. N. & Sumerlin, B. S. Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci. 35, 278–301 (2010).
Stuart, M. A. C. et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 9, 101 (2010).
Masuda, H. & Fukuda, K. Ordered metal nanohole arrays made by a two-step replication of honeycomb structures of anodic alumina. Science 268, 1466 (1995).
Yuan, J. H., He, F. Y., Sun, D. C. & Xia, X. H. A simple method for preparation of through-hole porous anodic alumina membrane. Chem. Mater. 16, 1841–1844 (2004).
Wu, S. et al. Facile fabrication of nanofluidic diode membranes using anodic aluminium oxide. Nanoscale 4, 5718–5723 (2012).
Zhang, Z. et al. A Bioinspired multifunctional heterogeneous membrane with ultrahigh ionic rectification and highly efficient selective ionic gating. Adv. Mater. 28, 144–150 (2016).
Hui, C. M. et al. Surface-initiated polymerization as an enabling tool for multifunctional (nano-)engineered hybrid materials. Chem. Mater. 26, 745–762 (2014).
Okuyama, H., Oshiba, Y., Ohashi, H. & Yamaguchi, T. Control of target molecular recognition in a small pore space with biomolecule-recognition gating membrane. Small 14, 1702267 (2018).
Lu., Q. et al. Developing an inositol-phosphate-actuated nanochannel system by mimicking biological calcium ion channels. ACS Appl. Mater. Interfaces 9, 32554–32564 (2017).
Roselin, L. S., Lin, M.-S., Lin, P.-H., Chang, Y. & Chen, W.-Y. Recent trends and some applications of isothermal titration calorimetry in biotechnology. Biotech. J. 5, 85–98 (2010).
Raj, P. A., Johnsson, M., Levine, M. J., Nancollas, G. H. & Salivary, statherin Dependence on sequence, charge, hydrogen bonding potency, and helical conformation for adsorption to hydroxyapatite and inhibition of mineralization. J. Biol. Chem. 267, 5968–5976 (1992).
Vacogne, C. D., Wei, C., Tauer, K. & Schlaad, H. Self-assembly of α-helical polypeptides into microscopic and enantiomorphic spirals. J. Am. Chem. Soc. 140, 11387–11394 (2018).
Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 40, 1305–1323 (2011).
Caltagirone, C. & Gale, P. A. Anion receptor chemistry: highlights from 2007. Chem. Soc. Rev. 38, 520–563 (2009).
Ragusa, A., Hayes, J. M., Light, M. E. & Kilburn, J. D. A combined computational and experimental approach for the analysis of the enantioselective potential of a new macrocyclic receptor for N-protected α-amino acids. Chemistry 13, 2717–2728 (2007).
Zhang, G. & Wu, C. Quartz crystal microbalance studies on conformational change of polymer chains at interface. Macromol. Rapid Commun. 30, 328–335 (2009).
Sauerbrey, G. Verwendung von schwingquarzen zur wägung dünner schichten und zur mikrowägung. Z. Phys. 155, 206–222 (1959).
Satoh, M., Kawashima, T. & Komiyama, J. Competitive counterion binding and dehydration of polyelectrolytes in aqueous solutions. Polymer 32, 892–896 (1991).
Weng, Y. et al. Polyethyleneimine-modified graphene oxide nanocomposites for effective protein functionalization. Nanoscale 34, 7 (2015).
Tian, Y. et al. A biomimetic zinc activated ion channel. Chem. Commun. 46, 1682–1684 (2010).
Boittin, F.-X., Gribi, F., Serir, K. & Bény, J.-L. Ca2+-independent PLA2 controls endothelial store-operated Ca2+ entry and vascular tone in intact aorta. Am. J. Physiol. Heart Circ Physiol. 295, H2466–H2474 (2008).
Vig, M. et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220 (2006).
Brini, M., Ottolini, D., Calì, T. & Carafoli, E. Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 15, 2787–2814 (2014).
Zhang, Q. et al. Redox switch of ionic transport in conductive polypyrrole-engineered unipolar nanofluidic diodes. Nano. Res. 11, 3715–3725 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51473131, 51533007, and 21775116), DICP Innovation Funding (DICP-RC201801), and LiaoNing Revitalization Talents Program (XLYC1802109). G.Q. acknowledges Wuhan Morning Light Plan of Youth Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Xiong, Y., Wang, D. et al. Smart polymer-based calcium-ion self-regulated nanochannels by mimicking the biological Ca2+-induced Ca2+ release process. NPG Asia Mater 11, 46 (2019). https://doi.org/10.1038/s41427-019-0148-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-019-0148-4
