
Abstract
Background
The patients with dual oesophageal squamous cell carcinoma (ESCC) and hypopharyngeal cancer (HPC) have poor prognosis; their underlying genetic pathogenesis is unclear. We hypothesise that development of synchronous ESCC/HPC depends on multicentricity or independent origin, rather than multifocality due to local or lateral spreading.
Method
Multiple region whole-exome sequencing (M-WES) and clonality analysis were used to assess clonal relationship and spatial inter- or intra-tumour heterogeneity (ITH) in 62 tumour regions from eight dual ESCC/HPC and ten ESCC patients.
Results
All synchronous ESCC/HPC patients had COSMIC 16 mutation signatures, compared to only 40% ESCC in the current study (p = 0.013) and public data set (n = 165, p = 0.003). This alcohol consumption-related mutation signature 16, commonly involved in multiple alcohol-related cancers, was significantly associated with drinking and alcohol metabolism-related ADH1B rs1229984. The mutational landscape and copy number profiles were completely distinct between the two primary tumours; clonality analysis further suggested the two primary tumours shared no or only one clone accompanying independent subclone evolution. M-WES strategy demonstrated higher sensitivity and accuracy for detection of mutational prevalence and the late branch mutations among different regions in the ESCC tumours, compared to traditional sequencing analysis based on single biopsy strategy. Patients with high ITH assessed by cancer cell fraction analysis after M-WES were significantly associated with both relapse and survival.
Conclusions
Our hypothesis-generating M-WES ITH assessment data have implications for prognostication. Collectively, our findings support multicentric independent clonal evolution, the field cancerisation theory, and suggest novel insights implicating an aetiologic role of alcohol metabolism in dual ESCC/HPC carcinogenesis.
Similar content being viewed by others


Introduction
Oesophageal cancer (EC) is a deadly cancer worldwide [1]. Oesophageal squamous cell carcinoma (ESCC) is the prevalent histological subtype endemic in Asia, Africa, and Europe [1]. The occurrence of multiple primary cancers occurs in 8.3–12.6% of ESCC patients [2,3,4,5,6,7]. The treatment and prognosis of patients with dual ESCC/head and neck cancer (HNC) are primarily based on ESCC stage, which is usually advanced at diagnosis, contributing to its dismal five-year survival and management challenges [3]. Current treatment strategies, including chemoradiation therapy (CRT) and surgery, are often ineffective due to intra-tumoural heterogeneity (ITH) derived from the complex subclonal tumour evolution. The multiple primary HNCs and aerodigestive tract cancers may be driven by “field cancerization” [8]. This concept proposes an aetiologic role of epithelium exposure to common carcinogens associated with alcohol and tobacco consumption, leading to multifocal lesions. To date no comprehensive genomic studies have examined the genetic pathogenesis of multifocal tumours with regards to the ITH and mutational signatures in patients with dual synchronous ESCC/hypopharyngeal cancer (ESCC/HPC) [9, 10]. It is unclear whether synchronous tumours share similar genomic features arising from clonal expansion or if they independently evolve. To understand its underlying aetiology, we applied multiple region-sampling whole-exome sequencing (M-WES) strategy and compared the mutation signatures associated with primary ESCC and synchronous ESCC/HPC. WES analysis demonstrated the quantification of ITH and existence of more than two subclones could be a universal potentially useful survival biomarker across twelve cancer types, but this is unknown for ESCC [11]. We aimed to utilise M-WES analysis to evaluate if degree of ITH carries prognostic information.
Methods
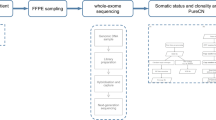
During 2018–2019, ten ESCC and five synchronous ESCC/HPC patients were recruited from Queen Mary Hospital (QMH), University of Hong Kong. Another three synchronous ESCC/HPC patients were recruited from 2020 at Shanghai Chest Hospital (SCH). Diagnosis of synchronous ESCC/HPC was based on the criteria of clear separation of tumours by histological examination and normal mucosa and the exclusion of the second tumour due to metastasis of the primary tumour [12]. A total of 60 tumour tissues, 6 matched normal tissues from oesophagus and hypopharynx and 15 matched fresh bloods from 10 ESCC and 8 ESCC/HPC patients with patient’s informed consent were obtained for M-WES analysis. The study was approved by the Ethics Committee of Shanghai Jiaotong University and Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (HKU/HA HKW IRB number UW 17–187) and was performed according to the principles of the Declaration of Helsinki. All patients were treatment-naive, directly undergoing surgery. M-WES was utilised to understand spatial ITH by obtaining 4–6 ESCC regions from distant regions of each tumour specimen after upfront surgery of 10 Hong Kong ESCC patients. For dual cancer patients, at least one specimen from each primary site was collected, except there was no available HP tumour for HK5 (Table S1). Figure S1 shows good-quality sequencing data with mean target coverage for 81 samples from 18 patients.
Whole-exome sequencing and bioinformatics analysis
WES libraries were prepared with KAPA HTP Library Preparation and SeqCap EZ Exome+UTR Kits (Roche). WES and bioinformatics analysis were performed, as previously described, and detailed in the supplementary methods [13, 14]. The multi-regional sample identities were confirmed using Identity-by-descent (IBD) analysis by PLINK v1.9. A mutational heatmap with the ComplexHeatmap R package showing the protein-altering mutations or mutated genes across regions was drawn for each ESCC patient [15]. Subclonal compositions of multi-regional samples were analysed using the SuperFreq R package considering somatic single nucleotide variants (SNVs) and CNVs [16]. Bamfiles containing the pre-filtered variants including both germline variants and somatic mutations were input into SuperFreq to generate patient river plots. Copy number variation (CNV) gains and losses were above 1 or below −1 after log 2 transformation. Cancer cell fractions (CCFs) were computed for each somatic mutation based on sample purity (predicted by ABSOLUTE v1.2) and local copy-number [17]. Sanger sequencing demonstrated an accuracy of 98.2% (110/112) for selected mutations (Table S2).
Mutation signature analysis
Lists of exonic mutations were uploaded to the web-based tool, Mutational Signatures V3.2 (http://cancer.sanger.ac.uk/signatures/) for mutation signature calling. Reports of somatic mutation prevalence, mutational profiles, COSMIC signature contributions, comparison with cancer signatures, reconstructed mutational profiles, and principal component analysis were downloaded. Mutational signature calling was performed for 165 ESCC patients from previous publications with two data sets from Sequence Read Archive (accession number SRP033394) and European Genome–phenome Archive (accession number EGAS00001000932). Another in-house WES data set included our previous study [13].
Statistical analysis
The differences between mutation signature occurrence and association of survival and relapse with ITH were examined with Fisher’s exact test. Pearson correlation coefficient was calculated for the correlation between mutation signature 16 and drinking, males, smoking, and dual primary cases. A p < 0.05 (2-sided) was considered statistically significant. Analyses were performed with SPSS v26 (SPSS Inc., IBM Corporation, Armonk, NY, USA).
Results and discussion
Synchronous ESCC/HPC and ESCC patients’ clinical parameters
Detailed clinical information of 18 synchronous ESCC/HPC and ESCC patients for unbiased M-WES analysis is summarised in Table 1. All ESCC/HPC and 70% ESCC patients are males; age ranges of ESCC/HPC (49–84, average = 62.9) and ESCC (48–85, average = 62.6) patients are similar. Five (62.5%) ESCC/HPC and one (10%) ESCC patient died, after a median 23.5 months follow-up.
Multi-region sampling captures intra-tumour heterogeneity of SNVs and CNVs of synchronous ESCC/HPC patients
The mean coverage of aligned reads was 52.6×, 72.4× and 88.8× for 15 bloods or 6 matched normal and 60 tumour samples from 18 patients, respectively (Fig. S1). The total numbers of exonic non-silent mutations (including frameshift and non-frameshift indels, stop gain, stop loss, and nonsynonymous) of 18 patients are summarised in Table S3. Similar somatic mutation prevalence was observed among patients with only primary ESCC and with dual cancers, with an average of 4.35 somatic exonic mutations/Mb (range 0.52–11.58) (Fig. S2). TP53 remained the most frequently mutated gene in dual cancer (71.4%, 5/7) and ESCC (70%, 7/10) patients. Surprisingly, both the mutational landscape (Figs. 1c and S3A) and CNV patterns across chromosomes (Fig. 2) of oesophageal and hypopharyngeal tumours of synchronous ESCC/HPC patients are distinct. Two primary tumours from four dual cancer patients (4/7, 57.1%) shared no common exonic non-silent mutations, while a few mutations were shared in three patients (Table S4). The ESCC/HPC tumours showed high inter-tumour heterogeneity in terms of CNVs at ten genes frequently occurring in ESCC (Fig. S4). For the common CCND1 amplification at 11q13, high intra-tumour heterogeneity existed in three dual primary patients, HK3, HK4 and SH2, which was only amplified in either the ESCC or HP tumours, but not present in both. CCND1 amplification was present in both the ESCC and HP tumours in HK1, HK2, SH1 and SH3 although the three ESCC (EA, EB, and EC) and two HP tumours (HA and HC) of HK1, as well as the ESCC and HP tumours of SH1 were amplified to different levels. Further analysis of CNVs between the ESCC and HP tumours of the same patients identified 73 regions differentially amplified or deleted CNVs between the ESCC and HP tumours of the same patient (p < 0.01, fdr <0.15) (Table S5). These included cancer relevant genes involved in actin cytoskeleton remodelling, integrin activation, EMT-Wnt signalling pathway, and metastasis or with potential prognostic role such as SPEN on chromosome 1, PLEKHG4B, and CCT5 on chromosome 5, LRP5 on chromosome 11, RFPL1, SLC5A1, and SHANK3 on chromosome 22 [18,19,20,21,22]. Our WES findings substantiate earlier reports of discordant patterns of inter-tumour allelic losses inferred by microsatellite typing or SNP profiling in multiple ESCC and HNCs patients [9, 10]. The clonality analysis tracking multiple clones from different tumour regions in the same individual further confirmed that no (HK1, HK4, SH1, SH3) or only one clone (HK2, HK3, SH2) was shared between the primary sites, consistent with the GATK-called mutations (Figs. 1d and S3B). Clonality analysis easily differentiates the ESCC/HPC dual cancers from ESCC patients with clonal relationships (Fig. 1). More detailed analyses with respect to the change of dominant subclones (defined as clonality>0.5) and somatic mutations revealed that, in general, the dominant subclones were mutually exclusive between ESCC and HP tumours from dual-cancer patients (Table S6). Among the multiple regions of ESCC, one dominant subclone was shared in at least two ESCC regions in HK5, two dominant subclones were shared in at least two regions in HK8. At least three or four dominant subclones were shared in at least two regions in the remaining 9 ESCC patients without dual cancer. Three (3/7, 42.9%) dual cancer patients had single shared clones containing both CNVs and mutations mostly occurring at intronic, promoter and untranslated regions (Table S7), suggesting the possibility of a minority of tumour cells migrating along the aerodigestive tract. To the best of our knowledge, this is the first WES study reporting the completely distinct genetic landscape, CNV profile and subclones between two patient’s tumours of synchronous dual cancers. The mechanism underlying unique origins of dual cancers is supported by WES mutation landscape and CNV profiling. Their entirely different mutational landscapes and CNV patterns suggest the multicentricity of HPC and ESCC tumours was not clonally related and arose independently. Future studies with larger sample cohorts are warranted, as we cannot rule out the possibility that the differences of copy number variation pattern and genetic mutation profile may result from the small size of the cohort in the study.

Heatmap showing the exonic non-silent mutations and clonality analysis by combining SNVs and CNVs in two primary ESCC (HK8 and HK13) (a, b) and four ESCC-related dual cancer patients (HK1-4) (c, d). HK8 and HK13 showed low and high degree of ITH for comparison of spatial and temporal ITH within single ESCC tumour and the ESCC/HP tumours from dual cancer patients. River plots show several subclones in different colours contributing to one regional sample.

CNV plots showing distinctive inter-tumour heterogeneity pattern between hypopharygngeal and oesophageal regions of synchronous tumour cases (HK1-4) and ITH between multiple regions of ESCC (HK5-HK15).
Alcohol consumption-related mutational signature 16 is dominant in synchronous cancer patients
The mutagenic processes from defective DNA repair, replication and genotoxins exposure of various carcinogens in cancer genomes during tumourigenesis are reflected by the mutational signatures [23, 24]. The M-WES studies in 36 regions from ten primary ESCC identified seven frequently occurring signatures (1, 2, 6, 7, 10, 13, 16) from 30 COSMIC mutational signatures, similar to those observed from 165 single-region ESCC WES public data (Fig. 3) and are concordant to earlier studies [25,26,27]. All patients showed ITH by M-WES in the mutational signatures (1, 2, 6, 7, 13, and 16) detected in at least one-third of all regions (12/36). Significantly, mutation signature 16, which is prevalent in ESCC synchronous cases (100%, 8/8), has a significantly lower frequency in ESCC patients in the current study (40%, 4/10, p = 0.013) and in the publicly available data set (40%, 66/165, p = 0.003) (Fig. 3). Our current study is the first to report such a high prevalence of COSMIC mutation signature 16 in dual ESCC/HPC patients compared to the current study and literature reported frequency ranging from 16–40% in ESCC [26]. Mutation signature 16 correlates with drinking, males, smoking, dual primary cases (Pearson correlation 0.723, p = 0.001, 0.721, p = 0.001, 0.523, p = 0.026, and 0.555, p = 0.017, respectively). By Fisher’s exact test, mutation signature 16 associated significantly with drinking (p = 0.008), smoking (p = 0.047), male gender (p = 0.012) and a trend with relapse (p = 0.101) and overall survival (p = 0.114) (Table 2). The distinct occurrence of drinking-associated mutation signature 16 between ESCC-related dual cancers bridges the gap between field cancerization and alcohol exposure. Past epidemiological studies reported that alcohol consumption is a risk factor of multiple dysplasia lesions in the oesophagus, head and neck, and development of multiple primaries in the upper-GI tract [28]. The alcohol consumption-associated mutational signature 16 was previously identified as one of the common mutational signatures in several alcohol-related cancers including liver cancer, head and neck cancer, and ESCC, highlighting the aetiologic risk factor role of alcohol consumption in tumourigenesis [26, 29, 30]. In the current study, the presence of mutation signature 16 occurs in all dual ESCC/HPC patients and correlates to drinking history. We further verified the relationship between COSMIC signature 16 and alcohol-related SNP ADH1B rs1229984 (T>C) in the public ESCC cohort and further confirmed that this signature was associated with the patients carrying the C allele of rs1229984, especially those patients with homozygous CC (CC vs. TT, 44.4% vs. 26.5%, p = 0.047, Chi-Square test). Consistently, dual synchronous ESCC/HPC patients were more likely to carry genotype CC compared to other ESCC patients with marginal significance (dual cancer vs. ESCC, 50.0% vs. 23.0%, p = 0.0998, Fisher’s exact test). East Asian individuals carrying the TT/TC genotypes of the alcohol metabolism-related functional SNP ADH1B rs1229984 were more likely to experience drinking discomfort, as the T allele causes rapid accumulation of acetaldehyde after alcohol intake. Our association findings of rs1229984 with COSMIC signature 16 were in line with earlier observation of individual carriers with the CC genotype of ADH1B rs1229984 having a significantly higher risk of multiple alcohol-related cancers of the larynx, pharynx, oesophagus, and pancreas [31]. Thus, our study provides new evidence implicating the aetiologic role of alcohol consumption in dual ESCC/HPC tumourigenesis. The molecular pathogenesis of HNC is classified into nonkeratinized HPV-related and keratinised HPV-unrelated forms associated with elderly males, smoking and drinking [32]. HPV-16/18 DNAs were undetectable by PCR-based consensus primer analysis (Fig. S6) [33], suggesting dual primary cancer patients in our cohort belong to the HPV-unrelated subgroup, which was further supported by their high frequency p53 mutations (Table S4) and p16 deletions (Fig. S4). Given the limited clinical sample capacity due to the rarity of dual cancers in ESCC patients, findings in the current study are considered hypothesis-generating and the novel insights between alcohol consumption in dual ESCC/HPC tumourigenesis merit further validation with larger sample cohorts. Despite the limited number of cases analysed, our findings are clinically relevant. The power of our study was 97.52% with the detection of COSMIC 16 mutational signature in 100% of the 8 HP/ESCC cases and 40% of the 165 ESCC from public data set and the type I error rate of 0.05. Given that mutational signature 16 is significantly more prevalent in ESCC-related dual cancers than ESCC patients, the likelihood of developing multiple primaries may be assessed in part with this potential molecular parameter.

The 165 WES samples were analysed by Musica to call COSMIC mutation signature contributions. The most common mutation signatures were signatures 1 (165/165), 13 (144/165), 2 (116/165), 7 (116/165), 10 (95/165), and 6 (91/165). The underlying aetiology of ESCC in both our cohorts (n = 10) and the public data set (n = 165) suggested by the seven dominating mutational signatures: ageing (signature 1), APOPEC enzyme family activity (signatures 2 and 13), mismatch repair (signature 6), POLE (ultra-hypermutation) (signature 10), UV exposure in squamous cancers (signature 7), and signature 16. Mutation signature 16 occurs predominately in all 8 dual ESCC/HPC, but only in 40% of 10 ESCC patients treated by upfront surgery in our cohorts and the WES public data set with 165 ESCC patients.
M-WES sensitively tracks ITH by multiple sampling
M-WES analysis of 12 patients in multiple ESCC regions identified 1981 non-silent mutations in 1749 genes in 44 regions (Table S3). The median number of exonic mutations detected was 117 (non-silent = 88), similar to previous reports [24, 27]. Researchers attempted to investigate inter- and intra-tumour heterogeneity by multi-regional sampling underlying the phylogenetic branched evolution model, providing evidence that such spatial and temporal ITH occurs in various cancers including ESCC [27, 34,35,36,37,38,39]. Our findings of ESCC ITH recapitulate similar observations of high ITH in two earlier studies [25, 27]. The presence of mutations in all regions from an ESCC patient is defined as early “trunk”; otherwise, it is defined as late branch mutations during clonal evolution. The phylogenetic trees constructed based on the somatic mutations in all ten primary ESCC patients to track evolutionary patterns, indicated extensive spatial ITH variation of the somatic non-silent mutations (Fig. S5). The generalisation that driver tumour suppressors occurred early as trunk mutations did not apply to two patients with poor prognosis; two branch missense mutations at TP53 in HK5 and KMT2D in HK13 were detected (Fig. 4a). The within-patient mutation rates of ESCC putative driver genes are higher than within-regions (Table S8). The strategy of M-WES analysis with multiple region sampling resulted in more sensitive detection of late branch mutations such as KMT2A, EP300, and EP400. ITH may impede precision medicine by affecting strategies on biopsy sampling and treatment decision through more in-depth characterisation of actionable drug targets [40, 41]. this study also identified an additional 28 frequently mutated genes, including 5 trunk and 23 branch mutations in 16.7–25% of 12 patients, suggesting the current single biopsy sequencing analysis will underestimate mutational prevalence and miss late branch mutations occurring in tumour evolution. Two of the five newly identified trunk early mutations previously not reported in ESCC, RBMXL2 and TRIP12, occur in 25% (3/12) patients. RBMXL encodes an RNA-binding protein complex with heterogeneous nuclear RNA (hnRNA) involved in pre-mRNA processing and regulating splicing with unknown functions [42]. TRIP12 (thyroid hormone receptor interactor 12) encodes an E3 ubiquitin-protein ligase regulating degradation of the critical ESCC tumour suppressors including FBXW7 and the p19ARF/ARF/TP53 axis [43, 44]. Future M-WES studies are needed to evaluate whether any of these are potential ESCC driver mutations.

Clonal status of putative driver mutations defined by previous large-scale ESCC genomic studies [26, 48,49,50,51] and frequently mutated genes in different regions across patients by a heatmap. Tumour suppressor mutations predominately occurred early in the evolutionary process as trunk mutations (TP53, KMT2D, NOTCH1, FBXW7, CDKN2A, etc.), while most other driver gene mutations occurred as both trunk and branch mutations, including oncogenes (PIK3CA, NFE2L2) and other genes involved in WNT signalling (FAT1, FAT2, FAT3), DNA repair (BRCA2, BAP1), histone modification and chromatin remodelling (EP300, EP400, KMT2C, KMT2A, KDM6A). b The distribution of trunk, private or shared branch mutations in 12 patients. In a total of 1982 somatic non-silent mutations, 709 are clonal trunk and 1273 (64.2%) subclonal branch mutations. c Clonality analysis and the physical distance of multiple ESCC regions of HK5 (dual primary cancers) and HK14 (primary ESCC). Grey and red indicate trunk and branch mutations, respectively. ∗denotes the highest heterogeneity rate of 100 and 90% in two patients, HK5 and HK13.
High ITH associated with poor survival
Various degrees of heterogeneity observed in 12 patients are summarised by numbers and distribution frequencies of non-silent trunk and branch mutations (Fig. 4b and Table S9). The heterogeneity frequency was calculated by dividing total non-silent branch mutations by patient-specific mutations. The median ITH of 12 patients with multiple ESCC regions was 37%, ranging from 15.9–100%, concordant with an earlier Japanese study with 13 patients with higher mean coverage depth of 150X [27]. The highest heterogeneity rate of 100 and 90% were observed in two patients, HK5 and HK13, followed by two other patients (HK6 and HK10) with 71 and 78%, while the majority of the remaining patients had <40% heterogeneity. Patients HK5 and HK13 had the highest number of branch (>300) and private (~250) mutations, compared to <100 branch and 70 private mutations observed in other patients (Fig. 4a, b). The clonal status of putative ESCC driver genes within each region is displayed as CCFs (Fig. 5), calculated after integration of CNVs, VAF and tumour cell purity [45, 46]. HK5, HK6, and HK13 showed the highest proportion of subclonal status among putative ESCC driver genes of 85% (11/13), 57% (4/7), 67% (10/15), respectively, concordant with results when only considering somatic non-silent mutations (Fig. 4a, b). For the eight dual primary cancer patients, both the recurrence and survival rates were 62.5% (5/8). For the ten ESCC patients, the recurrence and survival rates were 20% (2/10) and 10% (1/10), respectively. After a median follow-up of 25.5 months for twelve patients with multiple ESCC regions receiving surgery, two patients with the highest degree of ITH died. In addition to overall survival, the dual primary cancer patient, HK5, and two primary ESCC patients, HK6 and HK13, experienced the highest degree of heterogeneity based on the evidence from clonal status of putative driver mutations and frequently mutated genes in non-silent mutation heatmaps and CCF (Figs. 4a, b and 5) in this ESCC cohort of patients with relapse. HK5, HK6 and HK13 had the highest proportion of subclonal driver mutations associated with poorer overall survival (p = 0.045) and disease relapse (p = 0.005) (Table 2). To the best of our knowledge, earlier M-WES ESCC studies did not address the prognostic role of high tumoural ITH. The evidence presented here supports the hypothesis for the potential clinical usefulness of utilisation of high ITH as a prognostic indicator, but further larger cohort studies are needed to validate our findings.

Clonal and subclonal status of putative ESCC driver and frequently mutated genes across patients. ∗denotes patients HK5, HK6, and HK13 showed the highest proportion of subclonal status among putative ESCC driver genes.
Conclusions
ESCC patients are at high risk to develop multiple primary cancers in the upper aerodigestive tract, especially at the hypopharynx and larynx [3, 47]. Our key findings of the alcohol exposure-related mutational signature present in all eight dual ESCC/HPC patients provide aetiologic insight implicating alcohol consumption being involved in dual ESCC/HPC carcinogenesis, as previously postulated by the field cancerization theory. Future verification studies are needed to substantiate this novel observation about alcohol-related mutational signature COSMIC 16 and analysis of dual synchronous ESCC/HPC cancers related to alcohol exposure. Current data from both clonality analysis and distinctive genomic profiles suggest multicentricity independent origin of clonal evolution rather than the clonal expansion of common neoplastic clones giving rise to dual primary ESCC/HPC. Our data indicate that M-WES assessment of ITH is needed to improve prognostication and lays the groundwork required to unravel the underlying genetic pathogenesis for the identification of novel therapeutic options and biomarkers for early screening, prediction and prognostication of treatment outcomes to improve survival for this deadly cancer.
Data availability
All WES data are deposited at the European Genome-phenome Archive (EGA) with accession number (EGAS00001005966). All other data supporting the findings of this study are available from the corresponding author upon request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Strong MS, Incze J, Vaughan CW. Field cancerization in the aerodigestive tract-its etiology, manifestation, and significance. J Otolaryngol. 1984;13:1–6.
Poon RT, Law SY, Chu KM, Branicki FJ, Wong J. Multiple primary cancers in esophageal squamous cell carcinoma: incidence and implications. Ann Thorac Surg. 1998;65:1529–34.
Shibuya H, Wakita T, Nakagawa T, Fukuda H, Yasumoto M. The relation between an esophageal cancer and associated cancers in adjacent organs. Cancer. 1995;76:101–5.
Shibuya H, Takagi M, Horiuchi J, Suzuki S, Kamiyama R. Carcinomas of the esophagus with synchronous or metachronous primary carcinoma in other organs. Acta Radio Oncol. 1982;21:39–43.
Fogel TD, Harrison LB, Son YH. Subsequent upper aerodigestive malignancies following treatment of esophageal cancer. Cancer. 1985;55:1882–5.
Erkal HS, Mendenhall WM, Amdur RJ, Villaret DB, Stringer SP. Synchronous and metachronous squamous cell carcinomas of the head and neck mucosal sites. J Clin Oncol. 2001;19:1358–62.
Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8.
Califano J, Leong PL, Koch WM, Eisenberger CF, Sidransky D, Westra WH. Second esophageal tumors in patients with head and neck squamous cell carcinoma: an assessment of clonal relationships. Clin Cancer Res. 1999;5:1862–7.
Sunpaweravong S, Bunbanjerdsuk S, Pongrujikorn T, Naktang C, Sunpaweravong P, Nitiruangjaras A, et al. Clonal relationship of synchronous head and neck cancer and esophageal cancer assessed by single nucleotide polymorphism-based loss of heterozygosity analysis. BMC Cancer. 2019;19:1174.
Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22:105–13.
Lo OS, Law S, Wei WI, Ng WM, Wong KH, Tong KH, et al. Esophageal cancers with synchronous or antecedent head and neck cancers: a more formidable challenge? Ann Surg Oncol. 2008;15:1750–6.
Dai W, Ko JMY, Choi SSA, Yu Z, Ning L, Zheng H, et al. Whole-exome sequencing reveals critical genes underlying metastasis in oesophageal squamous cell carcinoma. J Pathol. 2017;242:500–10.
Zheng H, Dai W, Cheung AK, Ko JM, Kan R, Wong BW, et al. Whole-exome sequencing identifies multiple loss-of-function mutations of NF-kappaB pathway regulators in nasopharyngeal carcinoma. Proc Natl Acad Sci USA. 2016;113:11283–8.
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–9.
Flensburg C, Sargeant T, Oshlack A, Majewski IJ. SuperFreq: Integrated mutation detection and clonal tracking in cancer. PLoS Comput Biol. 2020;16:e1007603.
Carter SL, Cibulskis K, Helman E, Mckenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21.
Zhang N, Shi J, Shi X, Chen W, Liu J. Mutational characterization and potential prognostic biomarkers of chinese patients with esophageal squamous cell carcinoma. Onco Targets Ther. 2020;13:12797–809.
Ninomiya K, Ohta K, Yamashita K, Mizuno K, Ohashi K. PLEKHG4B enables actin cytoskeletal remodeling during epithelial cell-cell junction formation. J Cell Sci. 2021;134:jcs249078.
Li Y, Liu C, Zhang X, Huang X, Liang S, Xing F, et al. CCT5 induces epithelial-mesenchymal transition to promote gastric cancer lymph node metastasis by activating the Wnt/beta-catenin signalling pathway. Br J Cancer. 2022;126:1684–94.
Lilja J, Zacharchenko T, Georgiadou M, Jacquemet G, De Franceschi N, Peuhu E, et al. SHANK proteins limit integrin activation by directly interacting with Rap1 and R-Ras. Nat Cell Biol. 2017;19:292–305.
Tian S, Meng G, Zhang W. A six-mRNA prognostic model to predict survival in head and neck squamous cell carcinoma. Cancer Manag Res. 2019;11:131–42.
Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Yan T, Cui H, Zhou Y, Yang B, Kong P, Zhang Y, et al. Multi-region sequencing unveils novel actionable targets and spatial heterogeneity in esophageal squamous cell carcinoma. Nat Commun. 2019;10:1670.
Li XC, Wang MY, Yang M, Dai HJ, Zhang BF, Wang W, et al. A mutational signature associated with alcohol consumption and prognostically significantly mutated driver genes in esophageal squamous cell carcinoma. Ann Oncol. 2018;29:938–44.
Hao JJ, Lin DC, Dinh HQ, Mayakonda A, Jiang YY, Chang C, et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat Genet. 2016;48:1500–7.
Katada C, Yokoyama T, Yano T, Kaneko K, Oda I, Shimizu Y, et al. Alcohol consumption and multiple dysplastic lesions increase risk of squamous cell carcinoma in the esophagus, head, and neck. Gastroenterology. 2016;151:860–9.e867.
Letouze E, Shinde J, Renault V, Couchy G, Blanc JF, Tubacher E, et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat Commun. 2017;8:1315.
Plath M, Gass J, Hlevnjak M, Li Q, Feng B, Hostench XP, et al. Unraveling most abundant mutational signatures in head and neck cancer. Int J Cancer. 2021;148:115–27.
Chang TG, Yen TT, Wei CY, Hsiao TH, Chen IC. Impacts of ADH1B rs1229984 and ALDH2 rs671 polymorphisms on risks of alcohol-related disorder and cancer. Cancer Med. 2022. https://doi.org/10.1002/cam4.4920.
Kobayashi K, Hisamatsu K, Suzui N, Hara A, Tomita H, Miyazaki T. A review of HPV-related head and neck cancer. J Clin Med. 2018;7:241.
Si HX, Tsao SW, Poon CS, Wang LD, Wong YC, Cheung AL. Viral load of HPV in esophageal squamous cell carcinoma. Int J Cancer. 2003;103:496–500.
De Bruin EC, Mcgranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–6.
Cao W, Wu W, Yan M, Tian F, Ma C, Zhang Q, et al. Multiple region whole-exome sequencing reveals dramatically evolving intratumor genomic heterogeneity in esophageal squamous cell carcinoma. Oncogenesis. 2015;4:e175.
Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256–9.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl J Med. 2012;366:883–92.
Grzywa TM, Paskal W, Wlodarski PK. Intratumor and intertumor heterogeneity in melanoma. Transl Oncol. 2017;10:956–75.
Anderson K, Lutz C, Van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–61.
Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–82.
Saunders NA, Simpson F, Thompson EW, Hill MM, Endo-Munoz L, Leggatt G, et al. Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives. EMBO Mol Med. 2012;4:675–84.
Ehrmann I, Crichton JH, Gazzara MR, James K, Liu Y, Grellscheid SN, et al. An ancient germ cell-specific RNA-binding protein protects the germline from cryptic splice site poisoning. Elife. 2019;8:e39304.
Gudjonsson T, Altmeyer M, Savic V, Toledo L, Dinant C, Grofte M, et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 2012;150:697–709.
Khan OM, Almagro J, Nelson JK, Horswell S, Encheva V, Keyan KS, et al. Proteasomal degradation of the tumour suppressor FBW7 requires branched ubiquitylation by TRIP12. Nat Commun. 2021;12:2043.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101.
Landau DA, Carter SL, Stojanov P, Mckenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–26.
Yoshida N, Eto K, Kurashige J, Izumi D, Sawayama H, Horinouchi T, et al. Comprehensive analysis of multiple primary cancers in patients with esophageal squamous cell carcinoma undergoing esophagectomy. Ann Surg. 2020. https://doi.org/10.1097/SLA.0000000000004490.
Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–102.
Song Y, Li L, Ou Y, Gao Z, Li E, Li X, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91–5.
Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46:467–73.
Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2:899–905.
Acknowledgements
All Hong Kong ESCC and dual primary patient specimens and demographics were collected by the Departments of Surgery and Clinical Oncology of Queen Mary Hospital. The Shanghai dual primary specimens and demographics were collected by the Department of Thoracic Surgery, Section of Oesophageal Surgery, Shanghai Chest Hospital, Shanghai Jiao Tong University. We gratefully thank all study participants.
Funding
This work was supported by the Hong Kong Research Grants Council for the financial support of Theme-based Research Scheme (TBRS) funding (T12-701/17R) grant to MLL.
Author information
Authors and Affiliations
Contributions
Conceptualisation, SL and MLL; methodology, CG, WD, and JM-YK; software, WD, CG and LN; validation, CG, CL, and LN; formal analysis, JM-YK, CG, and CL; investigation, JM-YK, CG, LT, AW-IL, CWYW; resources, IY-HW, FS-YC, CL-YW, KKC, TTL, NP-YL ZL, HJ, ZL, and SL; data curation, ZL and SL; writing—original draft preparation, JM-YK and CG; writing—review and editing, WD, JM-YK, and MLL; Figures, CG, CL, JM-YK, and CW-YW; supervision, SL and MLL; project administration, MLL; funding acquisition, MLL. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Informed consent for sample collection from the dual primary ESCC/HPC and ESCC patients was obtained according to protocols approved by the Ethics Committee of Shanghai Jiaotong University and Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (HKU/HA HKW IRB number UW 17–187) and was performed according to the principles of the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ko, J.MY., Guo, C., Liu, C. et al. Clonal relationship and alcohol consumption-associated mutational signature in synchronous hypopharyngeal tumours and oesophageal squamous cell carcinoma. Br J Cancer 127, 2166–2174 (2022). https://doi.org/10.1038/s41416-022-01995-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-022-01995-0
This article is cited by
-
Elucidating the clonal relationship of esophageal second primary tumors in patients with laryngeal squamous cell carcinoma
Infectious Agents and Cancer (2023)
