
Abstract
Small-cell carcinoma of the esophagus (SCCE) is a rare and aggressive cancer. Although several consistent genomic changes were observed previously between SCCE and small-cell lung cancer (SCLC), detailed mutational landscapes revealing discrepancies in genetic underpinnings of tumorigenesis between these two cancers are scarce, and little attention has been paid to answer whether these genetic alterations were related to the prognosis. Herein by performing whole-exome sequencing of 48 SCCE and 64 SCLC tumor samples, respectively we have shown that the number of driver mutations in SCCE was significantly lower than in SCLC (p = 0.0042). In SCCE, 46% of recurrent driver mutations were clonal, which occurred at an early stage during tumorigenesis, while 16 driver mutations were found clonal in SCLC. NOTCH1/3, PIK3CA, and ATM were specifically clonal in SCCE, while TP53 was clonal in SCLC. The total number of clonal mutations differed between two cancers and presented lower in SCCE compared to SCLC (p = 0.0036). Moreover, overall survival (OS) was shorter in patients with higher numbers of clonal mutations for both cancers. In summary, SCCE showed distinct mutational background and clonal architecture compared with SCLC. Organ-specific clonal events revealed different molecular mechanisms underlying tumorigenesis, tumor development, patients’ prognosis, and possible variations in therapeutic outcomes to candidate treatments.
Similar content being viewed by others


Introduction
Small-cell carcinoma of the esophagus (SCCE) is a rare and aggressive carcinoma with a poor prognosis, accounting for only 0.6–2.8% of all esophageal malignant tumors1,2. Without being covered by National Comprehensive Cancer Network Clinical Practice Guidelines (NCCN Guidelines) in Oncology or Chinese Society of Clinical Oncology diagnosis and treatment guidelines, SCCE is often treated by referring to the guidelines and clinical experience of small-cell lung cancer (SCLC). Nevertheless, inaccurate classification and the substantial genetic heterogeneity across cancer types always lead to decreased therapeutic benefits in SCCE compared with SCLC, resulting in dismal prognosis in patients3. Previous study provided a comprehensive genomic profile of SCCE and showed consistent somatic genomic alterations between SCCE and SCLC4; however, little attention was paid to the difference in genomic characteristics, and how these disparities might influence therapeutic outcomes.
Occurrence and relative timings of driver mutations lead to the tumor molecular and phenotypic heterogeneities, which lead to variable clinicopathological characteristics and treatment outcomes5,6. Small-cell carcinoma originated from different histological lesions, had both convergent and organ-specific mutational patterns, and was demonstrated to undergo highly heterogenous evolutionary processes7. Whether the clonal architectures, consisting of clonal driver events (occurring at an early stage during tumorigenesis and found in the majority of tumor cells) and subclonal driver events (occurring at a relatively late stage and found in the small proportion of tumor cells) were consistent between SCCE and SCLC, and whether they are associated with patients’ prognosis remain elusive.
To characterize the clonal architectures and compare the genetic underpinnings of SCCE and SCLC, we performed whole-exome sequencing (WES) of 48 SCCE and 64 SCLC tumor samples, respectively, and analyzed how these genetic features and molecular heterogeneities might affect the clinical outcome in patients.
Results
Patient enrollment characteristics
The details of the clinicopathological characteristics of patients enrolled in this study are listed in Table 1. The SCCE cohort was comprised of 48 patients, with a mean age of 61.9 years (range, 46–79 yrs). The median follow-up of the SCCE cohort was 11.1 months (range, 4.0–58.2 months), with 15 (31.3%) patients lost in the follow-up, and the median overall survival was 11.0 months (range, 4.0–21.5 months). The SCLC cohort consisted of 60 males and 4 females, with a mean age of 62 years (range, 43–84 yrs). Most of SCLC patients (93.75%, 60/64) had a history of smoking. The median follow-up of the SCLC cohort was 12.4 months (range, 3.3–113.2 months), with 2 (3.1%) patients lost to follow-up, and the median overall survival was 11.7 months (range, 3.3–49.6 months). No significant association was observed between disease stage or smoking history with patients’ overall survival in SCLC.
WES sequencing analysis
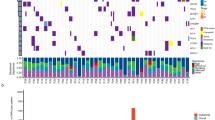
We performed WES sequencing of FFPE samples obtained from 48 SCCE and 64 SCLC patients, with an average sequencing depth of 133× and 157×, respectively (Fig. 1). As shown in Fig. 2, the median number of altered driver oncogenes in the SCCE cohort was five (range, 1–27), while it was seven in the SCLC cohort (range, 0–22). TP53 and RB1 were the most frequently mutated genes in both SCCE and SCLC patients, mutated in 60% and 54% patients, respectively, in SCCEs, and 64% and 77% in SCLCs. Genes in NOTCH family have been validated as important tumor suppressors and master regulators of neuroendocrine tissue differentiation in SCLC and SCCE8,9. Mutations in NOTCH family occurred in 25% SCCEs (12/48), higher than in SCLCs (15.62%, 10/64). Comparison of the prevalence of mutations in SCCE and SCLC showed significantly enriched NOTCH1 mutations in SCCE (two-tailed Fisher’s exact test, p = 0.0339), and LRP1B mutations in SCLC (two-tailed Fisher’s exact test, p = 0.0125). Besides, the number of identified driver mutations was significantly lower in SCCE than that in SCLC (Mann−Whitney test, p = 0.0042).

FFPE of patients with SCCE and SCLC was sequenced using WES. Somatic mutations were evaluated with Pure CN.

A Driver mutation profiling of SCCE (Top20 was shown); B Driver mutation profiling of SCLC (Top20 was shown); C Comparison of mutation frequency between SCCE and SCLC; D Driver gene NO. was significantly lower in SCCE than that in SCLC.
No significant difference in driver mutation number was found between upper and mid-lower SCCE (Mann−Whitney test, p = 0.1451, Fig. S1), due to the patient bias (upper, 6%, 3/48; mid-lower, 94%, 45/48).
Different clonal and subclonal events between SCLC and SCCE
To compare the clonal architectures in SCLCs and SCCEs, the cancer cell fraction (CCF) of each driver mutation was calculated and compared between the two cohorts. Clonality analysis of 29 recurrent driver mutations was performed in SCCE and SCLC subsequently. In total,13 of 29 (46%) recurrent driver mutations were evaluated to be clonal events in SCCE, which occurred at an early stage during tumor development, while 16 clonal driver mutations were found in SCLC (Fig. 3A). Among these recurrent mutations, mutations in RB1, PAX5, FANCD2, MED12, and KDM5C were clonal in both tumors. Notably, mutations in NOTCH1/3, PIK3CA, and ATM were specifically clonal in SCCE, while TP53 mutations were specifically clonal in SCLC. Mutations in POLE, which could lead to the hypermutated and ultramutator phenotypes and possibly improved response to immune therapies in patients, were observed subclonal in both cancers.

A Clonal and subclonal driver gene in SCCE (x-axis) and SCLC (y-axis), clonality of genes was identified using PureCN, and presented with different colors; B the total number of clonal mutations differed between two cancers, and presented lower in SCCE, compared with SCLC (median, 7 v.s. 10 mutations, Mann−Whitney test, p = 0.0036).
Despite the distinct clonalities observed in driver mutations, the total number of clonal mutations also differed between two cancers and presented lower in SCCE compared with SCLC (median, 7 v.s. 10 mutations, Mann−Whitney test, p = 0.0036, Fig. 3B). However, the total number of subclonal mutations showed no difference.
Association between genetic features with prognosis
We next explored whether these characteristics of clonal events were related to the patient outcome. Median values of clonal mutation number were used as the criteria to define high or low clonal mutation number. As a result, overall survival (OS) was shorter in SCCE patients with higher burdens of clonal mutations (median OS, 10.1 months v.s. median OS, 10.7 months; HR, 0.36; 95% CI, 0.11–1.15; p = 0.08; Fig. 4A). This difference in prognosis was also significant in SCLC (HR, 0.39; 95% CI, 0.21–0.72; p = 0.003).

A Kaplan−Meier analysis of overall survival (OS) in SCCE patients with more and less clonal mutation NO. (HR, 0.36; 95% CI, 0.11–1.15; P = 0.08). B Kaplan−Meier analysis of overall survival (OS) in SCLC patients with more and less clonal mutation NO. (HR, 0.39; 95% CI, 0.21–0.72; P = 0.003).
In SCLC, a higher risk of mortality was observed in patients with more driver mutations (1-year mortality rate: 68% v.s. 38%, Fisher’s exact test, p = 0.0473; Fig. 5). In conclusion, a higher mutational burden was associated with the poor prognosis in patients, possibly due to increased tumor malignancy.

SCLC patients with more driver mutation number showed a higher 1-year mortality rate (68%) than those with less driver mutation number (38%, Fisher’s exact test, P = 0.0473).
Discussion
This study indicated that SCCE and SCLC exhibited both convergent and distinct mutational and evolutionary characteristics. Previous study profiled the genomic landscape of SCCE and provided evidence to elucidate the origination of SCCE. To our knowledge, for the first time, we compared the driver events between SCCE and SCLC and found that SCCE showed a relatively simple composition of driver mutations. This finding revealed that less driver events were involved in SCCE tumorigenesis and tumor development, and they might play vital roles in tumor cell differentiation and cell migration10,11,12,13,14,15. The low-density lipoprotein receptor-related protein 1B (LRP1B), which encoding endocytic LDL-family receptor, among the top 10 significantly mutated genes in human cancer, has been demonstrated associated with high tumor mutation burden and reported to be a biomarker indicating prolonged survival in melanoma patients and non-small-cell lung cancer patients receiving immunotherapies16,17, was found frequently mutated in SCLC, and might also serve as a biomarker to predict response to immune therapies in SCLC. In this regard, SCCE harbored recurrent NOTCH1 mutations, contributing to the formation of an immune-suppressive tumor microenvironment (TME) and exhibiting great potential in the development of antitumor immunotherapies18,19. In the immune system, CD8+ T cells can kill tumor cells with cytotoxic molecules, such as granzymes and perforin. An increasing number of reports have demonstrated that the NOTCH pathway was required for CD8+ T-cell activation and homeostasis; it is therefore probable that the alteration of this pathway could be useful in guiding the treatment decisions of ESCC when the immune system is involved.
More obvious differences were presented in the clonalities of these driver mutations. Although TP53 was recurrently mutated in both cancers; mutations in this gene were identified subclonal in SCCE but clonal in SCLC. Loss of TP53 function is well-known to influence cell cycle checkpoint controls and apoptosis, and regulate other key stages of metastatic progression, such as cell migration and invasion. Earlier aberrance in TP53 gene may lead to more drastic migration and invasion of SCLC20. In ESCC and EACC, TP53 mutations were reported to occur before genome-wide copy number alterations and play an early and crucial role in the tumorigenesis and progression. Even normal esophageal mucosa and esophagitis were reported to harbor TP53 mutations21. However, our study showed subclonal TP53 mutations of SCCE, which suggest TP53 mutated relatively later than other clonal genes. This result may distinguish somatic genetic abnormalities in TP53 that were not necessary for tumorigenesis, but capable of clonal expansion of SCCE.
We also identified SCCE-specific clonal mutations in PIK3CA, NOTCH family, and ATM. PIK3CA mutations have been reported to play various roles in tumorigenesis and drug resistance. Especially in breast cancer, activating PI3K mutations may have some general effects upon both proliferation and chemotherapy responses22. ATM gene and its protein product are critically involved with the DNA damage response (DDR) pathway, which orchestrates the detection and repair of DNA damage with transient cell cycle arrest to ensure maintenance of genomic stability and cell viability. Loss function of ATM is associated with the hypersensitivity to ionizing radiation, cancer susceptibility, immunodeficiency, and genomic instability. Carcinoma deficient of ATM frequently displays chemotherapy resistance and poor survival23. These clonal events suggested diverse phenotypic characteristics and heterogeneous response to systemic therapy24,25. On the side, drug development and precision medicine strategies will likely require not only an understanding of cancer genes and mutational processes but also an appreciation of the clonal status of driver events and the timing of mutational processes. Targeted drugs of PI3K pathway and effective immune checkpoint inhibitors that have been approved in other solid tumors may provide clinical benefit for fractional SCCE, especially patients with this clonal gene aberrance26,27,28.
Moreover, in SCCE, patients with less clonal mutations showed improved survival, and this prognostic value of clonal architecture was also verified in the SCLC cohort. In conclusion, tumor heterogeneity results from the occurrence of various driver mutations, and subclonal events that emerged during the process of tumor evolution would affect patients’ prognosis29. As previously reported in NSCLC, heterogeneous driver alterations that occurred later in evolution were found in more than 75% of the tumors and were common in PIK3CA and NF1 and in genes that are involved in chromatin modification and DNA damage response and repair. High proportion of subclonal copy-number alterations was associated with an increased risk of recurrence or death23. A broad understanding of the heterogeneity of driver genes, deciphering the clonal and subclonal frequencies, and the timing of mutational processes involved in SCCE evolution, is lacking.
Despite the long follow-up time in our study, our conclusions may be affected by the small number of patients and retrospective analysis. Second, whether some clonal events would contribute to the chromosomal instability due to aberrant driver activation, needed to be further explored. Third, re-biopsy samples could provide horizontal temporal information of tumor evolution. Therefore, large-scale prospective studies, multi-omics analysis together with functional exploration and serial biopsy samples, will be required to further elucidate the evolutionary mechanisms and validate prognostic factors of SCCE.
In summary, SCCE showed distinct genetic underpinnings and clonal architectures compared with SCLC. Organ-specific clonal events revealed differences in tumorigenesis and predicted patients’ prognosis, showing great potential to translate into clinical practice to guide disease management and inform treatment decisions. Clonal mutations could serve as a potential biomarker related to the prognosis of both SCCE and SCLC patients.
Materials and methods
Patients and samples
Patients with SCCE and SCLC were eligible for this study. The inclusion criteria were as follows: (1) patients with a pathologically confirmed diagnosis of SCCE or SCLC, (2) age ≥ 18 years, (3) Eastern Cooperative Oncology Group performance status of 0−1, (4) without any previous treatment, and (5) provided written informed consent. Tumor samples of 48 SCCE and 64 SCLC patients who were treated at Zhejiang Cancer Hospital, Fujiang Cancer Hospital, and Hebei Cancer Hospital were enrolled in this study. Tumor samples were obtained from resection surgeries or tissue biopsies from August 2009 to August 2017. DNA was isolated from formalin-fixed paraffin-embedded (FFPE) tissue. All patients provided with informed consents and all tumors were reviewed and histopathologically confirmed to be SCCEs or SCLCs. The American Joint Committee on Cancer (AJCC) 7th edition TNM classification of esophageal and lung carcinoma was used for the tumor staging. This study was approved by the institutional review board (IRB) of all hospitals and was conducted in accordance with the Helsinki’s Declaration.
WES sequencing
In all, 10−15 10 μm-thick FFPE sections were subjected to DNA extraction using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Tumor samples were carefully dissected to avoid contamination with normal parenchymal and stromal tissues. The concentrations of DNA were measured by Qubit fluorometer (Invitrogen, Carlsbad, CA, USA) and the Qubit dsDNA BR (Broad-Range) Assay Kit (Invitrogen, Carlsbad, CA, USA). A total of 1 µg of DNA was fragmented into 200–250bp segments using a Covaris S2 instrument (Woburn, MA, USA). The KAPA DNA Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA) was used to construct sequencing libraries according to the manufacturer’s protocol and the libraries were hybridized to SeqCap EZ Exome 64 M (Roche NimbleGen, Madison, WI, USA). In brief, the fragments were end-repaired, A-tailed and adapter-ligated, amplified, hybridized to the SeqCap EZ library for 72 h, and then washed. The captured DNA was recovered using Streptavidin Dynabeads (Life Technologies). The captured DNA was then amplified by PCR. The purified captured DNA was then clustered using the cBot (Illumina, San Diego, CA, USA), and sequenced using the HiSeq 3000 Sequencing System (Illumina, San Diego, CA, USA) with 2×101-bp paired-end reads.
Data processing
Exome sequence data were analyzed employed Sentieon Genomics software (version 201711.05)30. Adaptor sequences and low-quality reads that included high proportions of Ns (>10%) and low-quality bases (>50% bases with quality <5) were first filtered out. Eligible sequencing reads were aligned to the human reference genome (build GRCh37) with BWA (version 0.7.12, http://bio-bwa.sourceforge.net/). Sentieon’s quality control algorithms based on Picard’s alignment metrics were performed to calculate the aligned sequencing data. Subsequently, Picard’s CollectHsMetrics was used to collect key metrics of the aligned reads, the average coverage, as well as other metrics. BAM-matcher was utilized to ensure that paired samples came from the same patient by comparing the tumor and adjacent normal tissue BAMs31.
Identification of somatic mutations
Somatic single-nucleotide variations (SNVs) and small insertions/deletions (Indels) were confirmed by Sentieon’s TNsnv. Mutations were filtered as follows to exclude common single-nucleotide polymorphisms (SNPs). Variants with frequencies ≤0.0002 in ExAC (The Exome Aggregation Consortium), ≤0.001 in ESP6500 (The Exome Sequencing Project), ≤0.01 in dbSNP (The Single-Nucleotide Polymorphism database), and 1000 G (The 1000 Genomes Project Consortium) databases were retained. Meanwhile, variants (except hot spots TP53 and RB1) with supported reads ≥5 (3), variant allele fraction ≥5% (3%), and coverage ≥30 in tumors were kept as well. To filter out germline polymorphisms detected in tumor samples without paired control, mutations were furthermore filtered by highly (5% or above) mutated genes in previous research8,32 in SCLCs, while a gene list containing highly altered genes in a former study31 and genes mutated over two patients in the paired SCCE patients in this cohort was used for mutation filtration in SCCEs with a single-tumor sample. In addition, mutations in genes reported to possibly affect the benefits of immunotherapies were also kept. Driver mutations were ascertained based on a comprehensive analysis of oncogenic driver genes33. The candidate variants were all manually verified in the Integrative Genomics Viewer (IGV, http://www.broadinstitute.org/igv/). Nonsynonymous mutations were annotated by ANNOVAR (http://www.openbioinformatics.org/annovar/).
CCF and clonality analysis
CCF and clonality of detected mutations were analyzed with PureCN software (https://github.com/lima1/PureCN) using default settings34.
Statistical analysis
Mann−Whitney test was employed to compare the median of different groups. The Fisher’s exact test was performed to test the mutational frequencies between groups. The Wilcoxon test and Fisher’s exact test were employed to compare the significant differences between two groups. The cutoff value of clonal mutation number equaled to median of each cohort. Kaplan−Meier survival plots were generated for studying the associations between genomic features with patients’ prognosis using log-rank tests. All statistical analyses were performed with SPSS (v.21.0; STATA, College Station, TX, USA) or GraphPad Prism (v. 6.0; GraphPad Software, La Jolla, CA, USA) software. Statistical significance was defined as a two-sided p-value of <0.05.
References
Hudson, E. et al. Small cell oesophageal carcinoma: an institutional experience and review of the literature. Br. J. Cancer 96, 708–711 (2007).
Ku, G. Y. et al. Small-cell carcinoma of the esophagus and gastroesophageal junction: review of the Memorial Sloan-Kettering experience. Ann. Oncol. 19, 533–537 (2008).
Brenner, B., Tang, L. H., Klimstra, D. S. & Kelsen, D. P. Small-cell carcinomas of the gastrointestinal tract: a review. J. Clin. Oncol. 22, 2730–2739 (2004).
Wang, F. et al. The genomic landscape of small cell carcinoma of the esophagus. Cell Res. 28, 771–774 (2018).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
McGranahan, N. et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 7, 283ra254 (2015).
Chang, M. T. et al. Small-cell carcinomas of the bladder and lung are characterized by a convergent but distinct pathogenesis. Clin. Cancer Res. 24, 1965–1973 (2018).
George, J. et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015).
Wang, F. et al. The genomic landscape of small cell carcinoma of the esophagus. Cell Res. 28, 771–774 (2018).
Lobry, C., Oh, P., Mansour, M. R., Look, A. T. & Aifantis, I. Notch signaling: switching an oncogene to a tumor suppressor. Blood 123, 2451–2459 (2014).
Agrawal, N. et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333, 1154–1157 (2011).
Yang, J. et al. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol. Cancer 18, 26 (2019).
Samuels, Y. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004).
Cremona, C. A. & Behrens, A. ATM signalling and cancer. Oncogene 33, 3351–3360 (2014).
Choi, M., Kipps, T. & Kurzrock, R. ATM mutations in cancer: therapeutic implications. Mol. Cancer Ther. 15, 1781–1791 (2016).
Lan, S. et al. Somatic mutation of LRP1B is associated with tumor mutational burden in patients with lung cancer. Lung Cancer (Amst., Neth.) 132, 154–156 (2019).
Chen, H. et al. Association of LRP1B mutation with tumor mutation burden and outcomes in melanoma and non-small cell lung cancer patients treated with immune check-point blockades. Front. Immunol. 10, 1113 (2019).
Qiu, H., Zmina, P. M., Huang, A. Y., Askew, D. & Bedogni, B. Inhibiting Notch1 enhances immunotherapy efficacy in melanoma by preventing Notch1 dependent immune suppressive properties. Cancer Lett. 434, 144–151 (2018).
Tsukumo, S. I. & Yasutomo, K. Regulation of CD8(+) T cells and anti-tumor immunity by Notch signaling. Front. Immunol. 9, 101 (2018).
Muller, P. A., Vousden, K. H. & Norman, J. C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 192, 209–218 (2011).
Yuan, W. et al. Clonal evolution of esophageal squamous cell carcinoma from normal mucosa to primary tumor and metastases. Carcinogenesis 40, 1445–1451 (2019).
Chen, L. et al. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat. Commun. 9, 1357 (2018).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. New England J. Med. 376, 2109–2121 (2017).
Choi, M., Kipps, T. & Kurzrock, R. ATM mutations in cancer: therapeutic implications. Mol. Cancer Ther. 15, 1781–1791 (2016).
Wang, Q. et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 9, 739 (2018).
Zammataro L. et al. Whole-exome sequencing of cervical carcinomas identifies activating ERBB2 and PIK3CA mutations as targets for combination therapy. 116, 22730−22736 (2019).
Xing Y. et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. 21, 78 (2019).
Chen, Y. et al. Association of tumor protein p53 and Ataxia-Telangiectasia mutated comutation with response to immune checkpoint inhibitors and mortality in patients with non-small cell lung cancer. JAMA Netw. Open 2, e1911895 (2019).
Andor N., Graham T. A. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. 22, 105−113 (2016).
Freed D., Aldana R., Weber J. A., Edwards J. S. The Sentieon Genomics Tools——a fast and accurate solution to variant calling from next-generation sequence data. bioRxiv https://doi.org/10.1101/115717 (2017).
Wang, P. P., Parker, W. T., Branford, S. & Schreiber, A. W. BAM-matcher: a tool for rapid NGS sample matching. Bioinformatics 32, 2699–2701 (2016).
Jiang, L. et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS Genet. 12, e1005895 (2016).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 174, 1034–1035 (2018).
Riester, M. et al. PureCN: copy number calling and SNV classification using targeted short read sequencing. Source Code Biol. Med. 11, 13 (2016).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (grant number 81802276) and Science and Technology Program of Fujian Province, China, (ganrt number 2018Y2003, 2019L3018 and 2019YZ016006).
Author information
Authors and Affiliations
Contributions
M.C., Z.S., and G.C. conceived the study and designed the experiments. G.C. and Z.S. prepared the research materials. L.C. and Z.Y. did the bioinformatic analysis and performed initial exploratory analysis. Y.L. and G.C. provided insight into methodological approaches and analysis. M.C., Z.S., and G.C. supervised the study. Z.S. and L.C. drafted the paper. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Ethics approval
The study was approved by the Ethics Committees of Zhejiang Cancer Hospital. All patients had signed the informed consent form.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by S. Tait
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, Z., Liu, Y., Cheng, G. et al. Distinct mutational backgrounds and clonal architectures implicated prognostic discrepancies in small-cell carcinomas of the esophagus and lung. Cell Death Dis 12, 472 (2021). https://doi.org/10.1038/s41419-021-03754-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03754-0
