
Abstract
Variable responsiveness to zileuton, a leukotriene antagonist used to treat asthma, may be due in part to genetic variation. While individual SNPs were previously associated with zileuton-related lung function changes, specific quantitative trait loci (QTLs) and biological pathways that may contribute have not been identified. In this study, we investigated the hypothesis that genetic variation within biological pathways is associated with zileuton response. We performed an integrative QTL mapping and pathway enrichment study to investigate data from a GWAS of zileuton response, in addition to mRNA expression profiles and leukotriene production data from lymphoblastoid cell lines (LCLs) (derived from asthmatics) that were treated with zileuton or ethanol (control). We identified 1060 QTLs jointly associated with zileuton-related differential LTB4 production in LCLs and lung function change in patients taking zileuton, of which eight QTLs were also significantly associated with persistent LTB4 production in LCLs following zileuton treatment (i.e., ‘poor’ responders). Four nominally significant trans-eQTLs were predicted to regulate three candidate genes (SELL, MTF2, and GAL), the expression of which was significantly reduced in LCLs following zileuton treatment. Gene and pathway enrichment analyses of QTL associations identified multiple genes and pathways, predominantly related to phosphatidyl inositol signaling via PI3K. We validated the PI3K pathway activation status in a subset of LCLs demonstrating variable zileuton-related LTB4 production, and show that in contrast to LCLs that responded to zileuton, the PI3K pathway was activated in poor responder LCLs. Collectively, these findings demonstrate a role for the PIK3 pathway and its targets as important determinants of differential responsiveness to zileuton.
Similar content being viewed by others

Introduction
The development of innovative approaches that integrate genomic, proteomic, and metabolomic data has expanded the number of high-priority candidate genes and biomarkers identified for complex diseases, including asthma [1,2,3,4,5]. These strategies, which combine RNA and/or protein and metabolite expression profiling with genetic association studies, are a breakthrough in the search for the biological mechanisms that underlie complex diseases. SNPs that are tightly linked to or capable of directly influencing gene expression represent QTLs that regulate expression of mRNAs (i.e., eQTLs), or production of metabolites and small molecules (i.e., pQTLs) [6,7,8], respectively. Through QTL mapping, variation in molecular, transcriptomic, and phenotypic traits can be correlated with variation in DNA sequences [7, 9,10,11]. Correlation of molecular and phenotypic traits can also be informative for inferring molecular networks that contribute to asthma pathogenesis or drug treatment responses.
In this study, we applied integrative methods to investigate the relationship between patient genotype and treatment response to zileuton, a leukotriene modifying agent [12,13,14]. Leukotrienes are synthesized from arachidonic acid (AA) by 5-lipoxygenase (5-LO) [15, 16]. After export from the cell, leukotrienes modulate important immunological activities including neutrophil activation, chemotaxis, eosinophil migration and smooth muscle contraction [16, 17]. Zileuton exerts its effects through inhibition of 5-LO-mediated production of leukotrienes, in particular leukotriene B4 (LTB4) [18]. Clinical response to leukotriene modifiers, including zileuton, is highly variable and repeatable within individuals, suggesting that polymorphic variation in genes in the leukotriene pathway may play a role in zileuton response [19,20,21]. In particular, SNPs that are significantly associated with both treatment-related clinical outcomes, as well as changes in leukotriene levels may represent important genetic biomarkers.
We hypothesized that genetic variation at the level of unique biological pathways is associated with variability in lung function in response to zileuton. In order to identify sets of genes and pathways that were associated with zileuton response, we first performed a pathway-based analysis using GWAS data obtained from a recent genome-wide investigation of zileuton response [22]. The advantage of using data identified through GWAS to interrogate biological pathways is that this method enriches for potentially functionally important candidate genes and SNPs that may not have been individually strongly associated with clinical response phenotypes. Having identified a set of candidate genes and pathways related to clinical response to zileuton, we then sought to identify specific QTLs associated with changes in LTB4 concentrations post-treatment, as well as changes in global mRNA expression. We next integrated pathway enrichment and QTL information to identify potentially causal candidate genes. Finally, we experimentally validated the expression of selected candidate pathway genes, and determined the activation status of a key pathway of interest.
Materials and methods
Genomic datasets
Clinical cohort
Summary statistics, genome-wide genotype and phenotypic information from a recent GWAS [22] investigating two placebo-controlled clinical trials of zileuton response (309 subjects) were available for interrogation. For this study, 226,890 SNPs with concordant effect size estimates (β coefficient sign) in both the discovery and replication GWAS [22] were pursued for pathway analysis.
Cell lines
Lymphoblastoid cell lines (LCLs) were available from 800 probands from the Childhood Asthma Management Program (CAMP), a 4 year National Heart Lung and Blood Institute-sponsored clinical trial comparing the efficacy of budesonide vs. nedocromil vs. placebo in 1041 patients [23, 24]. Genotypes, microarray expression data, and LTB4 concentrations from 345 LCLs derived from these patients were used to generate the QTL datasets.
Approach to hypothesis testing
Statistical approaches for hypothesis testing for each of the core analyses performed in this study are described in Supplemental Table 1.
Pathway analysis methods
Gene-based pathway analysis
For gene-based pathway analysis, genes were analyzed for their enrichment in pathways from KEGG, Pathway Commons and WikiPathways. Query genes were analyzed relative to all genes annotated in the human genome using the hypergeometric test for evaluation of enrichment, and correcting the resulting P-values for multiple testing by the Benjamini and Hochberg method [25]. The resulting pathways were then ranked according to the lowest P-value (or FDR q-value).
SNP-based pathway analysis
For SNP-based pathway analysis, “Improved Gene Set Enrichment Analysis for Genome-Wide Association Studies” (iGSEA-4-GWAS) [26] (http://gsea4gwas.psych.ac.cn/), was used to generate pathways from SNP and P-value data. The iGSEA-4-GWAS approach applies an improved gene set enrichment analysis (i-GSEA) to GWAS data (SNP P-value, odds ratio or other statistics) using SNP label permutation instead of phenotype to enrich for gene sets with high proportion of significant genes [26]. SNP rsIDs and corresponding –log10 P-values were mapped to +/− 500 KB from genes annotated to canonical pathways in the Molecular Signatures Database (MSigDB). A nominal iGSEA P value cutoff of 0.01 was specified, and FDR was used to correct for multiple testing [26]. Pathways meeting a significance threshold of FDR < 0.25 (to denote the confidence of ‘possible’ or ‘hypothesis’) were prioritized.
Cell studies and functional validation methods
Cell culture and incubation experiments
Cellular analyses were performed using lymphoblastoid cell lines (LCLs) that were derived from CAMP. For this study, 100 paired LCLs were cultured in growth medium (RPMI 1640, 5% FBS, 1% L-Glutamine, and 1% antibiotic–antimycotic) in a T25 flask in a 37 °C, 5% CO2 incubator. The cells were grown to a density of ~200,000 viable cells/mL. For stimulation experiments, 10–13 million cells were resuspended in 32 ml (~350 K/ml) regular growth medium and 2.5 ml were dispensed into each well of two 6-well culture plates (~850 K/well). Three wells of each 6- well plate were treated with 10 mM arachidonic acid or 2.5 uL of 100% ethanol. After incubating for 12 hours, the supernatant was removed and cells were treated with 1 µM zileuton (ZIL) or 2.5 uL of 100% ethanol for 6 hours. After treatment, the cells were harvested and spun down for stimulation. Cell stimulations were performed by resuspending cell pellets in phosphate buffered saline (PBS ( + Ca++, + Mg++)) containing 0.1 mM calcium ionophore and 4 mM arachidonic acid and incubated at 37̊ °C for 5 minutes. Cells were placed on ice and supernatants were harvested for quantification of LTB4 concentration using the LTB4 Express EIA kit (Caymen Chemical), while the cell pellets were lysed for RNA extraction using the RNeasy Mini Kit (Qiagen).
LTB4 enzyme-linked immunosorbent assay (ELISA)
An LTB4-specific ELISA was performed using a commercial Leukotriene B4 Enzyme Immunoassay Kit, according to the manufacturer’s instructions. LTB4 concentrations in each sample were determined using fluorimetry at 405 nm wavelength, and normalized to a standard curve.
Microarray analysis
For microarray, 200 ng RNA specimens were amplified using Illumina’s TotalPrep RNA Amplification kit (Ambion) according to the manufacturer’s protocol, and 750 ng of each amplified specimen were hybridized onto a Sentrix BeadChip Array for Gene Expression HumanRef12_V4, according to the manufacturer’s protocol. The BeadChip Array was scanned using Illumina’s BeadArray Reader. Quality control analysis of array data was performed using Illumina’s GenomeStudio v2009.1 Software, Gene Expression Module version 1.1.1 and iCheck_0.1.4.
Western analysis
For Western blot experiments, cell cultures were harvested after 24 hours of treatment (as described above) and lysed using RIPA buffer with protease and phosphatase inhibitors added. Cell lysates were prepped under reducing conditions per the Life Technologie’s iBind™ Western System protocol. Western blots were either probed with a mouse anti-PI3K primary antibody (Abcam), a rabbit anti-AKT primary antibody (Cell Signaling Technology) or a rabbit anti-phospho-AKT primary antibody (Cell Signalling Technology). AKT and phospho-AKT blots were probed with an HRP conjugated goat anti-rabbit IgG secondary antibody (Abcam), while PI3K blots were probed with an HRP conjugated goat anti-mouse IgG secondary antibody (Life Technologies). β-actin was used as a protein loading control and was probed using a mouse anti- β-actin primary antibody (Life Technologies) and an HRP conjugated goat anti-mouse IgG secondary antibody (Life Technologies). Results were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and captured using a G:Box (Syngene). Quantitative analysis was performed using GeneTools image analysis software (Syngene).
Quantitative trait loci (QTL) analyses
LTB4 QTL (pQTL) Analysis
LTB4 concentrations from 100 LCLs were obtained as described above. A linear regression model was used to associate individual SNP genotypes (>500,000 SNPs) from each sample with LTB4 concentration values, in corresponding zileuton-treated and ethanol-treated samples. PLINK v.1.0.7 was used to exclude SNPs with >10% missing data, a minor allele frequency <5%, and a paired r 2 > 0.9, resulting in 463,791 SNPs remaining for analysis. The effect of each locus on LTB4 levels was analyzed using PLINK v.1.0.7, assuming an additive genetic model, and adjusting for sex and race. Significance of association with LTB4 concentration values was assessed for each SNP using the Chi-square test, and a Bonferroni corrected significance threshold was set at 1.1 × 10−07 for 463,791 tests. SNPs with nominally significant associations (P < 0.05) in the ethanol-treated group were excluded from further analysis. For comparative analysis in samples from a relevant clinical population taking zileuton, a subset of 22,505 pQTLs that were unique to the zileuton treatment group, and that were at least nominally significant based on association P-values < 0.05, were also investigated for overlap using data from a recent GWAS of zileuton response [22]. SNPs that were at least nominally significant for both pQTL and GWAS analyses (pQTL and GWAS P values < 0.05) were selected for pathway analyses.
Expression QTL (eQTL) analysis
To evaluate the effect of zileuton treatment on gene expression, we evaluated 345 paired LCLs treated with zileuton or ethanol. After stimulation with arachidonic acid, each pair was treated with zileuton or ethanol (sham), as described above. The expression levels of 47,131 gene probes were measured using Illumina’s HumanHT-12_V4 chip, and 36,993 gene probes were retained after quality control procedures. The probe expression levels were log2-transformed and quantile normalized, and the log2 difference in expression level between zileuton treated and sham treated cell lines was obtained for each sample. Paired t-test was used to evaluate differential gene expression in samples treated with zileuton vs. sham, and the false discovery rate (FDR) was applied to correct for multiple testing. Of 36,993 probes, 5123 probe sets achieved nominal significance (P < 0.05) based on paired t-test, of which 918 probes surpassed the FDR threshold of 0.05. We next extracted genotypes (505,066 SNPs) from 345 samples, using the genome-wide genotype data from the subjects from whom the individual LCLs were derived (CAMP dataset). SNPs that failed tests for Hardy-Weinberg, missingness, or with minor allele frequency < 0.05 were excluded from the final data set, resulting in 459,261 remaining SNPs. No population substructure was detected by EIGENSTRAT. To identify eQTLs, we used the MatrixEQTL R package to test the association of SNP genotype with the log2 difference in expression for a gene probe, adjusting for age, gender, and the top four principal components of the expression data. The human genome version 19 annotation for both genes and SNPs was used. For each gene probe, all SNPS within a 50 KB range from flanking genic regions wherein a given probe resided were used to test for cis-effects. All SNPs outside this range, and on other chromosomes, were used to test for trans-effects. To increase the scope of potential inputs that could better inform pathway analyses, with a priori knowledge that we could generate functional models to validate the subsequent results, we chose a raw P value cut-off of 1 × 10−08, and applied a FDR < 0.05 as the multiple test correction threshold.
Results
Pathway analysis of zileuton response GWAS
An overview of the study design is shown in Fig. 1. To identify molecular pathways related to zileuton clinical response, we first evaluated existing data from a recent GWAS of zileuton response in 309 asthmatics (Abbott-Zileuton GWAS) [22]. Zileuton response was defined as a change in lung function, as measured by forced expiratory volume in one second from baseline to 8 weeks of zileuton therapy (∆FEV1) [22]. Data from 226,890 SNPs having the same direction of effect (β) in the discovery and replication GWAS were evaluated using a SNP-based pathway analysis and gene set enrichment analysis. A total of 212,364 SNPs were annotated to 15,117 genes and mapped to 523 gene sets, resulting in four enriched pathways (FDR < 0.25): “PLC PATHWAY”, “CASPASE PATHWAY” and “SIG IL4 RECEPTOR IN B LYMPHOCYTES” and “INNATE IMMUNE RESPONSE” (Table 1)).

Overview of study design
QTL mapping
QTL identification and annotation
In an effort to characterize variability in the leukotriene production pathways that could potentially underlie differential responsiveness to zileuton, we evaluated global gene expression and/or LTB4 production in LCLs treated with zileuton or ethanol (control) in paired LCLs derived from asthmatic patients, and mapped QTLs for both treatment groups. LCLs were first stimulated with arachidonic acid (AA) to stimulate LTB4 production, and were then treated with either zileuton or ethanol. For pQTL analyses, LTB4 concentrations in 100 paired LCLs were determined by ELISA. As expected, following activation with AA, LCLs treated with zileuton showed a significant but variable decrease in LTB4 production ([LTB4]mean_etoh = 270 ± 271 ng/mL vs. [LTB4]mean_zileuton = 74.5 ± 66.9 ng/mL; P < 0.0001) (Supplemental Fig. 1A). We next integrated genome-wide genotypes (463,791 QC-validated SNPs) from each of the LCL samples with LTB4 concentrations from both treatment groups to identify zileuton-related pQTLs. A set of 22,505 SNPs specific to the zileuton-treated group were nominally associated (unadjusted P < 0.05) with differential LTB4 production, of which eight SNPs remained significant following correction for multiple testing; the top 100 pQTLs, ranked by their P-values, are listed in Supplemental Table 2. Of the significant pQTLs, all were associated with persistent LTB4 production despite treatment with zileuton (i.e., poorer responsiveness) (Supplemental Table 2). Two of the top pQTLs (rs10743992 and rs6485597) were annotated to introns within cAMP responsive element binding protein-like 2 (CREBL2) and PR domain-containing protein 11 (PRDM11) genes, while a third SNP (rs2823730) was found within a long intergenic non-protein coding RNA, LINC00478 (Supplemental Table 2).
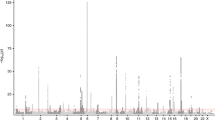
For eQTL mapping, 345 paired LCLs were treated with AA, followed by treatment with zileuton or ethanol as described above and in Methods, and gene expression levels for both treatment groups were measured using microarray. We identified 262 trans-eQTLs that met an unadjusted significance threshold of P < 1 × 10−08. The top four eQTLs also met FDR < 0.05. The 262 eQTLs regulated the expression of 161 unique genes, of which 132/262 eQTLs (50.3%) were annotated to 81 genes. The top 50 eQTLs are listed in Supplemental Table 4, and the four eQTLs meeting FDR < 0.05 are shown in Figs. 2b–e. Four of the genes in which the 262 eQTLs resided were also significantly differentially expressed in LCLs following zileuton treatment (FDR < 0.05). A quantile-quantile (QQ) plot (Fig. 2a) of the log-difference in gene expression related to zileuton vs. ethanol treatment revealed a significant deviation from the expected for the trans-eQTL analysis (Figs. 2b–e, boxplots). Both rs12762462 and rs12251868 were annotated to G protein-coupled receptor 158 (GPR158), and are predicted to regulate the expression of selectin L (SELL) (Supplemental Table 4). rs10495426 is an intergenic SNP that is predicted to regulate metal response element binding factor 2 (MTF2) (Supplemental Table 4). rs12414020 was annotated to neuregulin 3 (NRG3) and is predicted to regulate the expression of galanin and galanin message-associated peptide (GMAP) prepropeptide (GAL). In LCLs, all of these eQTLs significantly reduced the expression of the genes they are predicted to regulate following treatment with zileuton (Figs. 2b–e). However, these signals appeared to be driven by a small number of subjects who carried homozygous variant genotypes. We identified 10 individuals who carried the homozygous variant genotypes for the four eQTLs (three for rs10495426, three for rs12762462 and rs12251868 (the same individuals for both), and four for rs12414020) whose removal resulted in P-values that remained nominally significant, but were no longer significant after controlling for multiple testing (FDR < 0.05) (data not shown).

. a The Q–Q plot shows the relationship between observed vs. expected (diagonal; gray line) P-values for 735,488 local (red line/dots) and 13,009,811,306 distant (blue line/dots) gene-SNP P values. Jittered boxplots b-e show individual data points (red dots) and medians (±IQR) of log2 mRNA expression values (zileuton:ethanol) (y-axis), stratified by genotype category (“0” = reference allele; “1” = heterozygous variant; “2” = homozygous variant) (x-axis): b Expression of probe 6940358 (gene symbol = SELL) by rs12762462 genotype; c Expression of probe 2760255 (gene symbol = MTF2) by rs10495426 genotype; d Expression of probe 6940358 (gene symbol = SELL) by rs12251868 genotype; e Expression of probe 60452 (gene symbol = GAL) by rs12414020 genotype
QTL integration and pathway analysis
There were 22,505 nominally significant pQTLs that were unique to the zileuton treatment group; these were evaluated to determine whether any were also associated with zileuton-related changes in lung function, using data from a GWAS of zileuton response in 309 asthmatics [22]. A total of 1060 SNPs were nominally associated with differential LTB4 production, as well as zileuton-related 8-week ∆FEV1 based on pQTL and GWAS P values < 0.05. The top 50 pQTLs are shown in Supplemental Table 3. Pathway analysis of the 1060 pQTLs identified two major ‘hubs’ inclusive of the top 10 significantly enriched pathways: phosphatidyl inositol signaling (via phosphatidylinositide 3-kinase (PI3K)) and catenin signaling. The phosphatidyl inositol signaling (via PI3K) hub included pathways for “FCγR-mediated phagocytosis”, “FcεR1 signaling pathway”, “Focal adhesion”, “Neurotrophin signaling pathway”, “Regulation of actin cytoskeleton”, “Pathways in cancer” and “Type II diabetes mellitus”, while the catenin signaling hub included pathways for “Adherens junction”, “Endometrial cancer”, “Pathways in cancer”, “Arrhythmogenic right ventricular cardiomyopathy (ARVC)” (shown in Table 2). These pathways comprise 31 unique genes, of which PI3K isoforms (PIK3R1 and PIK3R5) were most highly enriched relative to all other genes (Supplemental Fig. 2).
We next evaluated whether any of the 262 eQTLs were also present (overlapping) within the set of 22,505 pQTLs related to LTB4 production. We identified 22 overlapping eQTL/pQTL relationships, including three of the top-ranked eQTLs (rs12762462, rs10495426 and rs12251868 (Supplemental Table 4)); 18/22 eQTLs/pQTLs were annotated to 7 genes (Supplemental Table 5). However, none of the 262 eQTLs were also included among the set of 1060 pQTLs that were jointly associated with LTB4 production and zileuton clinical response; as such, they could represent unique determinants of zileuton pharmacological response vs. clinical response.
Pathway analysis of the 262 eQTLs identified seven significantly enriched pathways (Table 3). Notably, these pathways also coincided with those for the pQTLs (Table 3, Supplemental Table 4). Among the pathways, several genes that directly or indirectly interacted with the PIK3 signaling pathway hub were also present, e.g., calcium voltage-gated channel auxiliary subunit beta 4 (CACNB4) and fibroblast growth factor (FGF) isoforms within the “MAPK Signaling Pathway”, and F GF isoforms and integrin subunit beta 6 (ITGB6) within the “Regulation of the actin cytoskeleton” pathway, which act upstream of the phosphatidylinositol signaling pathway (Table 3).
To determine the functional importance of these pathways in LCLs, the expression of 68 genes that were collectively identified through pathway analyses (listed in Tables 1–3 ) was validated by microarray analysis of zileuton treated vs. ethanol treated LCLs. Of these, 8 genes were differentially expressed (zileuton vs. ethanol) at a nominal significance threshold, with one gene, suppressor of cytokine signaling 1 (SOCS1), remaining significant following multiple test correction (Supplemental Table 7).
Experimental validation of candidate pathway and genes
Pathway prioritization
A pathway analysis of 1060 joint pQTLs had identified phosphatidyl inositol signaling via PI3K as a major hub, where PI3K isoforms were more frequently enriched among pathway genes (Table 2; Supplemental Fig. 2). In addition, by integrating pQTL and eQTL data, we also identified several genes that interact with PIK3 (Table 3 and Supplemental Table 4), namely phosphodiesterase 4B (PDE4B), fragile histidine triad (FHIT), myosin heavy chain 9 (MYH9), cadherin 2 (CDH2), and others. Furthermore, we show that among the 8 differentially expressed pathway genes, PIK3CA expression was reduced, albeit at a nominal significance level (Supplemental Table 7). As the collective analyses pointed toward a rationale for prioritizing the PI3K signaling pathway (which was further emphasized due to its relevance as a potential therapeutic target for asthma [27, 28]), we hypothesized that differential zileuton responsiveness could be related to changes in the expression and activation of genes within this pathway. We therefore next validated the PI3K pathway using a cellular model of zileuton response.
Pathway validation
To validate the PI3K pathway in a clinically relevant cellular model, we first selected LCLs that had demonstrated different magnitudes of treatment responses to zileuton, based on LTB4 production. Four LCLs that had demonstrated significant down-regulation of LTB4 production following zileuton treatment (mean fold change = 6.8 (termed “good responders”)) and four LCLs that demonstrated less than 2-fold change in LTB4 concentrations following zileuton treatment (mean fold change = 1.2 (termed “poor responders”)) were selected for profiling (Supplemental Fig. 1B).
A consequence of PI3K pathway activation is an increase in expression of phosphorylated AKT (phospho-AKT), relative to the un-phosphorylated AKT protein [29, 30]. To evaluate the activation status of this pathway, we compared the expression of AKT, phospho-AKT and PI3K proteins following treatment with fetal bovine serum (FBS), zileuton, or sham (ethanol), in the two groups of LCLs possessing differential LTB4 production following zileuton treatment (i.e., ‘good’ and ‘poor’ responders). For both LCL groups, treatment with fetal bovine serum (FBS), a pathway activator (Fig. 3, red bars), increased expression of all three proteins relative to sham (Fig. 3, green bars), with the greatest increase in AKT and phospho-AKT expression values in the ‘poor’ responders (Fig. 3). Compared with sham treatment, zileuton treatment did not significantly increase expression of phospho-AKT in the ‘good’ responder LCLs (Fig. 3, blue bars). In contrast, relative to ‘good’ responders, expression of phospho-AKT was selectively increased by 64% in the ‘poor’ responder group following zileuton treatment (P = 0.037) (Fig. 3). Otherwise, the expression levels of AKT, phospho-AKT and PI3K did not significantly differ across treatment groups (Fig. 3). These results suggest that, compared with LCLs that responded well to zileuton inhibition, the PI3K pathway is activated in ‘poor’ responder LCLs, at basal levels, as well as in response to zileuton treatment.

. The barplots show normalized mean expression levels ± SEM for AKT (left plot), phospho-AKT (center plot) and PIK3CA (right plot) in ‘good’ and ‘poor’ responders (labels, x-axis), by treatment category (FBS = red; Sham (ethanol) = green; Zileuton = blue)
Differential gene expression related to zileuton treatment
The top 50 differentially expressed probe sets (genes) are listed in Supplemental Table 6. Among the 918 probe sets, PIK3CA (T-statistic = −2.0; P = 0.044) and AKT1 (T-statistic = −3.2; P = 0.0015) demonstrated an overall reduction of expression in the zileuton treated LCLs relative to sham, across 345 LCLs. However, expression of these genes from the subset of ‘good’ and ‘poor’ responder LCLs was congruent with protein expression values for zileuton vs. ethanol treated LCLS (Supplemental Fig. 3). Notably, the mRNA and protein expression of AKT (zileuton:ethanol) was reduced in both ‘good’ and ‘poor’ responder groups, while the expression of PIK3CA (zileuton:ethanol) was reduced only in the ‘good’ responders (Supplemental Fig. 3).
Discussion
We applied pathway analysis in an integrative approach, combined with experimental validation in a relevant cellular model, to identify candidate genes and pathways underlying clinical and pharmacological (cellular) response to zileuton, a leukotriene inhibitor prescribed for asthma treatment. Using data from a recently published GWAS of zileuton response in asthmatics [22], we first utilized an improved gene set enrichment analysis (i-GSEA) algorithm, which accounts for the combined effects of SNPs/genes with modest effect sizes, and identified four significantly-enriched pathways containing 23 unique genes that collectively represent coordinated pathways related to lipid mediator signaling, leading to cellular growth and survival. Due to the high level of cross talk among these pathways, multiple genes with important regulatory roles in lipid mediator signaling appeared in more than one pathway (e.g., PIKC3A), suggesting that they may be biologically relevant for zileuton treatment responses. The most highly ranked pathway (FDR = 0.022, Table I) comprised phosphoinositide 3-kinases (PI3K) lipid signaling molecules. The PI3K family members phosphorylate the inositol rings of phosphoinositide lipids, influencing vesicle trafficking, cell proliferation, and migration [31]. Akt is also an important co-regulator of PI3K signaling leading to leukotriene production, and the activity of PLA2 is necessary for conversion of AA to 5-LO substrates. Together, these results suggest that key regulatory genes within molecular signaling cascades that regulate the production and signaling of lipid mediators are related to zileuton response in patients.
We subsequently replicated these pathways and identified a set of SNPs that are strongly associated with zileuton-related LTB4 production in LCLs derived from asthmatic patients (pQTLs). Among the top SNP associations that surpassed FDR, we identified rs10743992 and rs6485597 in cAMP responsive element binding protein-like 2 ( CREBL2) and PR/SET domain 11 (PRDM11) that were associated with a dramatic increase in LTB4 production following zileuton treatment (Table 2). Both rs6485597 and rs10743992 are in complete LD with regulatory and non-synonymous coding SNPs that are also present within these genes, suggesting a potential functional role for CREBL2 and PRDM11 (data not shown). Through pathway analysis of pQTL data, we identified numerous genes within the CREB signaling pathway, including CREB1 (Table 2). These data suggest that CREB-mediated signaling events are involved in zileuton pharmacological response.
Pathway analysis revealed an enrichment of pathways for lipid signaling and cellular migration in response to immune activation. From the 68 genes identified by pathway analysis (shown in Tables 1–3) eight were significantly differentially expressed in zileuton vs. ethanol treated LCLs (Supplemental Table 7). Included with these, as well as a majority of the pathways in Tables 1–3, is PIK3CA, a key mediator of phospholipid signaling that is crucial to the production of leukotrienes. PIK3CA encodes the 110 kDa catalytic subunit of PI3K and relies on ATP to phosphorylate PtdIns, PtdIns4P and PtdIns [4, 5]P2. These phosphorylated lipids activate downstream targets that in turn activate transcriptional regulators such as CREB, leading to the transcription of genes involved in cell proliferation and survival. We also identified PLC activity as a shared pathway in both pQTL analyses. PLC directly affects LTB4 production through conversion of PI to AA, the substrate of 5-LO and precursor of LTB4. Together, these data suggest that PIK3CA pathway activity may represent a functional network that determines the pharmacological and clinical response to leukotriene modifiers. Furthermore, the production of LTB4 in zileuton-treated cells demonstrated high variability (Supplemental Fig. 1). We subsequently validated the PIK3CA pathway in asthmatic LCLs with differential production of LTB4 in response to zileuton. We found that there was significant expression of the activated target of PI3K, phospho-AKT, which was selectively upregulated only in the ‘poor’ responder group (Fig. 3). Relative to ethanol, zileuton-treated LCLs also showed increased mRNA and protein expression of PIK3CA, in ‘poor’ responders only (Supplemental Fig. 3). Collectively, these data suggest that zileuton responses can be regulated by PI3K pathway activity, and that the pathway is differentially activated in individuals with poorer zileuton responses. From a mechanistic perspective, the ‘poor’ responders would be expected to have increased activation of PIK3CA and its targets, in parallel with increased LTB4 production that is resistant to zileuton inhibition. Higher concentrations of LTB4 would be available to bind to cell surface, G-protein coupled receptors, leading to increased PIK3CA signaling and resulting in increased proliferation of pro-inflammatory responses mediated by leukotriene activity. In contrast, our data indicate that ‘good’ responders would appropriately limit LTB4 production following zileuton inhibition, showing correspondingly diminished activity of this pathway due to reduced LTB4 concentrations.
This study is the first to report a role for zileuton in modulating the PI3K pathway in adults with asthma, for which zileuton is clinically used. However, a recent study showed that zileuton could selectively up-regulate phosphorylated Akt expression and down-regulate the over-expression of NF-KB p65 and COX-2, in a rat model of ischemic brain damage [32]. This finding is consistent with our observation that zileuton boosted AKT and phospho-AKT expression in samples from ‘poor’ responders (Fig. 3) and provides additional evidence for interaction of zileuton and the PI3K pathway.
While our results pointed toward a role for the PI3K pathway, we also identified four eQTLs that significantly reduced the expression of SELL, MTF2, and GAL, none of which have been annotated to the canonical PI3K pathway. While these signals were driven by a small number of subjects with homozygous variant genotypes, removal of these subjects did not alter the nominal significance of the four eQTLs. There is evidence that these genes may interact with the PI3K pathway, as well as with the CREB signaling pathway. For example, SELL is part of a co-expression network that includes PI3KCA, PIK3CG and PIK3R1 [33], and its protein product physically interacts with PRKCQ, which in turn interacts with PIK3CA through targeting CD28 and RASGRP3 [34]. MTF2 and CREB1 are also predicted to physically interact [35]; in addition, MTF2 interacts with PIK3R3 [36] which directly targets PIK3CA [36, 37]. Finally, as both GAL and PIK3CA are both activated by binding guanosine triphosphate (GTP), they are co-regulated through the G protein coupled receptor signaling cascade and may also have synergistic molecular roles. For example, GAL receptors can activate the phosphatidylinositol (PtdIns) signaling pathway, and there is evidence that PC12 apoptotic cell death mediated through Gal2R may be dependent on PI3K [38]. Further work is needed to ascertain the roles of these genes in P13K signaling in the context of leukotriene modifier response in asthma.
In summary, we used a multi-tiered, integrative pathway analysis of GWAS, eQTL, pQTL, and clinical data to prioritize pathways of interest, and functionally validated a candidate pathway with implications in asthma biology. This approach provides a means to identify novel biology for complex diseases and drug response phenotypes.
References
Deng FY, Lei SF, Zhu H, Zhang YH, Zhang ZL. Integrative analyses for functional mechanisms underlying associations for rheumatoid arthritis. J Rheumatol. 2013;40:1063–8.
Hicks C, Koganti T, Brown AS, Monico J, Backus K, Miele L. Novel integrative genomics approach for associating GWAS information with intrinsic subtypes of breast cancer. Cancer Inform. 2013;12:125–42.
Jia P, Wang L, Fanous AH, Pato CN, Edwards TL, Zhao Z, et al. Network-assisted investigation of combined causal signals from genome-wide association studies in schizophrenia. PLoS Comput Biol. 2012;8:e1002587
Hu VW. From genes to environment: using integrative genomics to build a “systems-level” understanding of autism spectrum disorders. Child Dev. 2013;84:89–103.
Dahlin A, Tantisira KG. Integrative systems biology approaches in asthma pharmacogenomics. Pharmacogenomics. 2012;13:1387–404.
Damerval C, Maurice A, Josse JM, de Vienne D. Quantitative trait loci underlying gene product variation: a novel perspective for analyzing regulation of genome expression. Genetics. 1994;137:289–301.
Jansen RC, Tesson BM, Fu J, Yang Y, McIntyre LM. Defining gene and QTL networks. Curr Opin Plant Biol. 2009;12:241–6.
Doerge RW. Mapping and analysis of quantitative trait loci in experimental populations. Nat Rev Genet. 2002;3:43–52.
Jansen RC, Nap JP. Genetical genomics: the added value from segregation. Trends Genet. 2001;17:388–91.
Jansen RC. Studying complex biological systems using multifactorial perturbation. Nat Rev Genet. 2003;4:145–51.
Gilad Y, Rifkin SA, Pritchard JK. Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 2008;24:408–15.
Zileuton for asthma. Med Lett Drugs Ther. 1997;39:18–9.
DuBuske LM, Grossman J, Dubé LM, Swanson LJ, Lancaster JF. Randomized trial of zileuton in patients with moderate asthma: effect of reduced dosing frequency and amounts on pulmonary function and asthma symptoms. Zileuton Study Group. Am J Manag Care. 1997;3:633–40.
Lazarus SC, Lee T, Kemp JP, Wenzel S, Dubé LM, Ochs RF, et al. Safety and clinical efficacy of zileuton in patients with chronic asthma. Am J Manag Care. 1998;4:841–8.
Salmon JA, Higgs GA. Prostaglandins and leukotrienes as inflammatory mediators. Br Med Bull. 1987;43:285–96.
Hammarström S. Biosynthesis and metabolism of leukotrienes. Monogr Allergy. 1983;18:265–71.
Hammarström S. Leukotrienes. Annu Rev Biochem. 1983;52:355–77.
Bell RL, Young PR, Albert D, Lanni C, Summers JB, Brooks DW, et al. The discovery and development of zileuton: an orally active 5-lipoxygenase inhibitor. Int J Immunopharmacol. 1992;14:505–10.
Drazen JM, Yandava CN, Dubé L, Szczerback N, Hippensteel R, Pillari A, et al. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat Genet. 1999;22:168–70.
Drazen JM, Silverman EK, Lee TH. Heterogeneity of therapeutic responses in asthma. Br Med Bull. 2000;56:1054–70.
Lima JJ. Treatment heterogeneity in asthma: genetics of response to leukotriene modifiers. Mol Diagn Ther. 2007;11:97–104.
Dahlin A, Litonjua A, Irvin CG, Peters SP, Lima JJ, Kubo M, et al. Genome-wide association study of leukotriene modifier response in asthma. Pharmacogenomics J. 2015.
Childhood Asthma Management Program Research Group (CAMP): design, rationale, andmethods. Childhood Asthma Management Program Research Group Control Clin Trials. 1999;20:91–120.
Raby BA, Lazarus R, Silverman EK, Lake S, Lange C, Wjst M, et al. Association of vitamin D receptor gene polymorphisms with childhood and adult asthma. Am J Respir Crit Care Med. 2004;170:1057–65.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B (Methodological) 1995;57:289–300.
Zhang K, Cui S, Chang S, Zhang L,Wang J, i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res. 2010;38:W90–5.
Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. 2009;228:253–72.
Yoo EJ, Ojiaku CA, Sunder K, Panettieri RA. Phosphoinositide 3-Kinase in asthma: novel roles and therapeutic approaches. Am J Respir Cell Mol Biol. 2016. https://doi.org/10.1165/rcmb.2016-0308TR.
Mendez R, Kollmorgen G, White MF, Rhoads RE. Requirement of protein kinase C zeta for stimulation of protein synthesis by insulin. Mol Cell Biol. 1997;17:5184–92.
Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–76.
Rameh LE, Cantley LC. The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem. 1999;274:8347–50.
Tu XK, Zhang HB, Shi SS, Liang RS, Wang CH, Chen CM, et al. 5-LOX inhibitor zileuton reduces inflammatory reaction and ischemic brain damage through the activation of PI3K/Akt signaling pathway. Neurochem Res. 2016;41:2779–87.
Boldrick JC, Alizadeh AA, Diehn M, Dudoit S, Liu CL, Belcher CE, et al. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc Natl Acad Sci USA. 2002;99:972–7.
Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–9. https://doi.org/10.1093/nar/gkj109.
Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010;140:744–52.
Grossmann A, Benlasfer N, Birth P, Hegele A, Wachsmuth F, Apelt L, et al. Phospho-tyrosine dependent protein-protein interaction network. Mol Syst Biol. 2015;11:794.
Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–23.
Tofighi R, Joseph B, Xia S, Xu ZQ, Hamberger B, Hökfelt T, et al. Galanin decreases proliferation of PC12 cells and induces apoptosis via its subtype 2 receptor (GalR2). Proc Natl Acad Sci USA. 2008;105:2717–22.
Acknowledgements
We thank the study participants and clinical investigators who collected the samples evaluated in this study.
Funding
This work was supported by the National Institutes of Health [R01 HL092197 to KT, U01 HL65899 to KT and STW, and K01 HL130629-01 to AD].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dahlin, A., Qiu, W., Litonjua, A.A. et al. The phosphatidylinositide 3-kinase (PI3K) signaling pathway is a determinant of zileuton response in adults with asthma. Pharmacogenomics J 18, 665–677 (2018). https://doi.org/10.1038/s41397-017-0006-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41397-017-0006-0
