
Abstract
Variability in response to short-acting β2-agonists (e.g., albuterol) among patients with asthma from diverse racial/ethnic groups may contribute to asthma disparities. We sought to identify genetic variants associated with bronchodilator response (BDR) to identify potential mechanisms of drug response and risk factors for worse asthma outcomes. Genome-wide association studies of bronchodilator response (BDR) were performed using TOPMed Whole Genome Sequencing data of the Asthma Translational Genomic Collaboration (ATGC), which corresponded to 1136 Puerto Rican, 656 Mexican and 4337 African American patients with asthma. With the population-specific GWAS results, a trans-ethnic meta-analysis was performed to identify BDR-associated variants shared across the three populations. Replication analysis was carried out in three pediatric asthma cohorts, including CAMP (Childhood Asthma Management Program; n = 560), GACRS (Genetics of Asthma in Costa Rica Study; n = 967) and HPR (Hartford-Puerto Rico; n = 417). A genome-wide significant locus (rs35661809; P = 3.61 × 10–8) in LINC02220, a non-coding RNA gene, was identified in Puerto Ricans. While this region was devoid of protein-coding genes, capture Hi-C data showed a distal interaction with the promoter of the DNAH5 gene in lung tissue. In replication analysis, the GACRS cohort yielded a nominal association (1-tailed P < 0.05). No genetic variant was associated with BDR at the genome-wide significant threshold in Mexicans and African Americans. Our findings help inform genetic underpinnings of BDR for understudied minority patients with asthma, but the limited availability of genetic data for racial/ethnic minority children with asthma remains a paramount challenge.
Similar content being viewed by others

Introduction
Asthma, a common chronic respiratory disease among children, is a serious global health burden1. Inhaled short-acting β2-agonists (SABAs), such as albuterol, have been the first-line bronchodilator therapy for acute treatment of asthma symptoms. SABAs act on β2-adrenergic receptors to cause a prompt relaxation of bronchial smooth muscle cells (i.e., bronchodilatation) via generation of intercellular cAMP and subsequent activation of cAMP-dependent protein kinase A (PKA)2. Despite their widespread use, however, patients’ responses to SABAs are highly variable3. Identifying the sources of this variability among patients is important to tailor preventive and treatment strategies.
Bronchodilator response (BDR), measured as the percent change in forced expiratory volume in 1 s (FEV1) after inhalation of a SABA, is an important phenotype used for asthma diagnosis, assessing its severity, and determining whether symptoms are under control. There has been a growing recognition that inter-individual variability in BDR is due in part to genetic factors4. Previous studies have shown that BDR in people with asthma substantially varies by race/ethnicity5,6,7. In patients with moderate-to-severe persistent asthma, BDR was lower in African Americans compared to whites5. Among Hispanic groups, Puerto Ricans were less responsive to albuterol than Mexicans, had decreased lung function, greater morbidity, and longer asthma duration6,7. Genetic association studies have reported several BDR-associated genes, including ADRB28,9, ADCY910, ADCYAPR111, ARG112, COL22A113, CRHR214, GLCCI115, GSNOR16, THRB17, and SPATS2L18. However, to date, most of these genetic studies were performed in populations of European ancestry. There are likely other genes that contribute to BDR variability that can be identified in non-European populations of diverse genetic ancestry. Indeed, there is evidence of population-specific variants associated with BDR19,20, underscoring the need for additional research among underrepresented racial/ethnic minority groups. A recent genome-wide association study (GWAS) among minority children with asthma using the tails of high and low BDR as outcome found population-specific and shared risk variants, although replication of primary findings was limited due to lack of additional independent cohorts representing the same minority populations4. In this study, we conducted the largest BDR GWAS to date using as outcome its continuous distribution.
Methods
Study cohorts and sample details
SAGE II (Study of African Americans, Asthma, Genes, and Environments) and GALA II (Genes-environments and Admixture in Latino Americans) are parallel studies that aim to investigate gene-environmental interactions in asthma focusing on African American and Latino children, respectively21. Participants were eligible if they were 8–21 years old at time of recruitment. Parents and grandparents must have self-identified as African American (SAGE II) or Latino (GALA II). Exclusion criteria included the following: (1) 10 or more pack-years of smoking; (2) any smoking within one year of recruitment date; (3) pregnancy in the third trimester; or (4) history of lung diseases other than asthma or chronic illness. Asthma status was confirmed with a history of physician-diagnosed asthma and asthma controller or rescue medication use, and report of symptoms within the last two years preceding enrollment.
Additional African American patients with asthma were drawn from the SAPPHIRE (Study of Asthma Phenotypes and Pharmacogenomic Interactions by Race-Ethnicity)22 and GCPD-A (Genetic Causes of Complex Pediatric Disorders—Asthma)23 studies. SAPPHIRE subjects met the following criteria: (1) self-identified as African American; (2) age 12–56 years; (3) a prior clinical diagnosis of asthma; (4) no record of chronic obstructive pulmonary disease or congestive heart failure. GCPD-A subjects consisted of children and young adults aged 5–21 years seen and followed in the outpatient clinics of the Children’s Hospital of Philadelphia (CHOP). These patients have detailed longitudinal clinical information which includes records of clinical diagnoses. Asthma cases had a documented diagnosis of asthma in the medical record, whereas GCPD-A controls had no documented asthma diagnosis. GCPD-A participants with asthma who completed pulmonary function tests were included in this study.
Replication analyses were conducted in 560 white participants from the CAMP (Childhood Asthma Management Program) clinical trial, in 967 participants from the GACRS (Genetics of Asthma in Costa Rica), and in 417 participants from the HPR (Hartford-Puerto Rico) study. CAMP was a randomized, placebo-controlled trial of budesonide, nedocromil, or placebo for mild-to-moderate childhood asthma followed by three phases of observational follow-up24, where asthma was defined by having 2 or more symptoms per week, using an inhaled bronchodilator at least twice weekly or asthma medication daily, and airway responsiveness to methacholine < 12.5 mg/ml. BDR assessed at randomization in the CAMP clinical trial was used in this study. GACRS was a family-based genetics study of childhood asthma comprised of Costa Rican schoolchildren with asthma ages 6 to 14 years and their parents25,26. Children had a high probability of having at least six great-grandparents born in the Central Valley of Costa Rica and were defined as having asthma if they had a doctor’s diagnosis of asthma and at least two respiratory symptoms or asthma attacks in the year prior to enrollment in the study25,26. HPR was a case–control study of childhood asthma in Puerto Ricans recruited in Hartford, CT and San Juan, PR with asthma defined as physician-diagnosed asthma and at least one episode of wheeze in the year prior to recruitment27. All participants had to have four Puerto Rican grandparents.
Ethics approval and consent to participate
All Human Subjects data utilized was de-identified and previously collected via studies in which participants and/or their parents provided written informed consent. The SAGE II and GALA II studies were approved by the University of California, San Francisco Human Research Protection Program Committee; the SAPPHIRE study was approved by the Institutional Review Board of Henry Ford Health System; the GCPD-A study was approved by the Institutional Review Board of the Children’s Hospital of Philadelphia. The CAMP study was approved by the Institutional Review Boards of Brigham and Women’s Hospital and the other participating centers. The GACRS study was approved by the Institutional Review Boards of the Hospital Nacional de Niños (San José, Costa Rica) and Brigham and Women’s Hospital. The HPR study was approved by the Institutional Review Boards of the University of Puerto Rico (San Juan, PR), Brigham and Women’s Hospital, and the University of Pittsburgh. All research was performed in accordance with relevant guidelines and regulations.
BDR assessments
BDR was evaluated according to American Thoracic Society/European Respiratory Society Guidelines28. Briefly, FEV1 was measured before administration of albuterol (pre-FEV1) and again after two doses of albuterol (post-FEV1), with a 15-min waiting period between two doses. Four puffs of albuterol were administered in the first dose and two (subjects < 16 years old) to four (subjects ≥ 16 years old) puffs were given in the second dose. Spirometry was performed fifteen minutes after each dose of albuterol to obtain post-FEV1 measurements and the higher of those was used to determine BDR. For CAMP, GACRS and HPR, spirometry was conducted following American Thoracic Society recommendations. The best FEV1 and FVC were selected for data analysis. After completing baseline spirometry, subjects were given 200 µg (2 puffs) of an albuterol metered-dose inhaler using a spacer, and spirometry was repeated after 15 min11,18,29. For all subjects, BDR was calculated as the percent change in post-FEV1 compared to pre-FEV1; that is BDR (ΔFEV1) = (post-FEV1 – pre-FEV1) / pre-FEV1.
Trans-omics for precision medicine (TOPMed) whole-genome sequencing (WGS) data
DNA samples for SAGE II, GALA II, SAPPHIRE, and GCPD-A subjects were sequenced as part of the Trans-Omics for Precision Medicine (TOPMed) WGS program30. WGS was conducted at the New York Genome Center and Northwest Genomics Center using a HiSeq X system (Illumina, San Diego, CA) with a mean depth of at least 30X (paired end, 150-bp reads). Full details of DNA sample handling, quality control, library construction, clustering and sequencing, read processing, and sequence data quality control are available in the TOPMed website (https://www.nhlbiwgs.org/). Briefly, demultiplexing of sequencing data was performed with bcl2fastq (Illumina, San Diego, CA, USA), sequencing data was aligned to human reference build 38 (GRCh38 with decoy) using BWA-MEM31, and data were further processed with the GATK best-practices pipeline32. Quality control procedures included marking of duplicate reads using Picard tools33, realignment around indels, and base quality recalibration. Single-sample genotypes were called using the GATK haplotype caller followed by joint genotyping of all subjects. The resulting multi-sample Variant Call Format (VCF) file was used for variant quality control score recalibration (VQSR). A 99.8% truth sensitivity trache level was used for SNPs and 99.0% for indel variants. SNP calls were used to check for sample contamination, and sample identity was confirmed by requiring > 99.5% concordance with SNP array (HumanCoreExome-24 array) genotypes. As part of the TOPMed program, BAM files were submitted to the Informatics Resource Center (IRC) at the University of Michigan and all samples passed TOPMed’s IRC quality control metric (mean genotype coverage > 30X; > 95% of genome covered at > 10X; and < 3% contamination). Genotype consistency between WGS data and previously published Axiom Genome-Wide LAT 1 array (Affymetrix, Santa Clara, CA) genotype data (dbGaP phs000920.v1.p1 and phs000921.v1.p1) was assessed using VCFtools34. Samples with percentage consistency three standard deviations below the mean were excluded. Downstream analyses were performed only on variants that passed quality control filters.
Primary BDR GWAS
Genetic data for each racial/ethnic group was analyzed separately to avoid confounding due to major differences in population stratification. For subjects from the GALA II, SAGE II, and GCPD-A cohorts, association analyses were performed on the ENCORE server (https://encore.sph.umich.edu/) using TOPMed freeze 8 data (DP0 PASS) with SAIGE linear mixed model35 to account for relatedness and fine-scale population structure. The inverse-normalize-response option was enabled to address non-normality of BDR distributions. Age, sex, body mass index (BMI) category, sequencing batch, and the first four genetic principal components were included as covariates. For participants under 20 years old, standardized age- and sex-specific growth charts were used to calculate BMI percentiles and categorize BMI as follows: underweight (BMI percentile < 5th), normal (5th ≤ BMI percentile < 85th), overweight (85th ≤ BMI percentile < 95th) and obese (BMI percentile ≥ 95th). For participants aged 20 years and older, BMI categories were assigned as follows: underweight (BMI < 18), normal (18 ≤ BMI < 25), overweight (25 ≤ BMI < 30) and obese (BMI ≥ 30). Subjects were excluded if they had any missing covariate or BDR was not within 3 standard deviations of the mean. At the genotype level, variants with minor allele frequency (MAF) < 0.01 were excluded. For subjects from the SAPPHIRE cohort, GWAS was performed using the R package GENESIS36 with TOPMed freeze 7 data (DP10 PASS) based on the same model and covariates as used with GALA II, SAGE II, and SAPPHIRE. To evaluate variant-level associations in African Americans, a meta-analysis was performed with the SAGE II, SAPPHIRE, and GCPD-A results using METASOFT37 with the fixed effects model based on inverse-variance-weighted effect sizes. A trans-ethnic meta-analysis was also conducted across the three minority populations using the GWAS results of all cohorts.
Replication analysis of BDR-associated variants
For CAMP, genotyping was performed at the Channing Division of Network Medicine using either the Illumina Quad 610 or Illumina 550 microarray chips (Illumina, Inc., San Diego, CA). For GACRS, genotyping was performed at the Channing Division of Network Medicine using the Illumina BeadStation 500G platform (Illumina Inc., San Diego CA). Genotype imputation was performed with the Michigan Imputation Server38 using the Haplotype Reference Consortium (HRC) r1.1 201639 as the reference panel, and only SNPs with ≥ 0.05 and imputation quality r2 ≥ 0.3 were kept. For HPR, genotyping was conducted using the HumanOmni2.5 BeadChip platform (Illumina Inc., San Diego, CA) and imputation of non-genotyped variants was performed with IMPUTE2 using data from Phase I of the 1000 Genomes Project as the reference panel, as previously described27. Genetic associations were evaluated with linear regression models, adjusting for genetic principal components, age, sex, BMI category, and (if appropriate) study site.
Region-based gene association tests using GWAS summary statistics
We performed region-based gene association tests with the snpsettest package (https://cran.r-project.org/package=snpsettest)40 in R 4.0.4 (R Core Team 2021) using gene annotations from the GENCODE project41. We considered genes in the following biotypes: protein-coding, immunoglobulin variable chain and T-cell receptor genes. For each gene, any variants within 20 kb of the 5’ and 3’ UTRs were selected for association tests, and the 1000 Genomes Project phase 3 African Americans and Admixed American panels42 were used to infer relationships among markers. A significant association was defined as having a Bonferroni-corrected P < 0.05.
Predicted quantitative trait loci (QTL) analysis of GWAS data
We used the S-PrediXcan framework43 to correlate BDR to monocyte gene expression levels as predicted using data from the Multi-Ethnic Study of Atherosclerosis (MESA) cohort, which was optimized to predict gene expression within and across diverse populations44. For testing the association between the predicted gene expression levels of participants in each racial/ethnic group (i.e., African American, Mexican, Puerto Rican) and BDR, we used the MESA ALL model (https://doi.org/10.5281/zenodo.3610513) trained using pooled samples of African American, Hispanic, and European participants in the MESA cohort.
Validation with multiomics data
To investigate functional target genes for association signals that lie distant to protein-coding genes, publicly available chromatin interaction data were explored using the 3D Genome browser45. Specifically, capture Hi-C data were utilized to identify long-range interactions that involved GWAS hits with promoters of distant gene candidates46. Transcriptomic analysis was carried out to investigate whether potential candidate genes identified via GWAS were differentially expressed by asthma-related conditions. Publicly available transcriptomic datasets related to asthma and asthma drug response were retrieved from the Gene Expression Omnibus (GEO)47 and analyzed with the RAVED pipeline48. Differential expression results comparing 1) patients with asthma versus healthy controls, or 2) asthma-related drug exposure versus vehicle control in vitro were obtained for three datasets. The Benjamini–Hochberg approach was used to adjust for multiple comparisons of genes/probes tested within each dataset and a q-value < 0.05 was considered significant. Single-tissue eQTL data from the Genotype-Tissue Expression (GTEx) v8 release49 were explored to identify GWAS hits that were also eQTL in lung, whole blood, and skeletal muscle of candidate genes.
Results
Subject characteristics are provided in Table 1. A total of 1,136 Puerto Ricans, 656 Mexicans and 4,337 African Americans were included in the primary cohorts. The SAPPHIRE subjects were different from the other cohorts in that they were older and had a lower proportion of males. Principal components analysis of genotype data separated the study subjects according to racial/ethnic groups, demonstrating differences in genetic ancestry (Supplementary Fig. S1). The replication cohorts included 560 white, 967 Costa Rican, and 417 Puerto Rican subjects. White and Costa Rican patients had a lower proportion of obese subjects compared to the remainder of the cohorts.
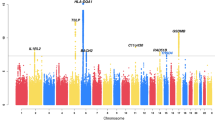
The Manhattan plots for BDR GWAS are shown in Fig. 1. In Puerto Ricans, we found one genome-wide significant locus at 5p15.2 (Table 2). The lead variant, rs35661809 (P = 3.61 × 10–8), was located in the non-coding RNA gene LINC02220 and was > 700 kb away to DNHA5, the nearest coding gene (Fig. 2A). Capture Hi-C data revealed a long-range interaction between this locus and the promoter of DNAH5 in lung tissue, suggesting that DNAH5 may be a functional target gene. The GTEx data demonstrated that several variants around the promoter region of DNAH5 were eQTL in lung. According to transcriptomic data, DNAH5 had decreased expression in bronchial epithelium of asthma patients versus controls and in airway smooth muscle treated with budesonide 100 nM for 24 h (Fig. 2B). The other highly ranked regions included loci at 16q22.1 (rs201865968, P = 1.51 × 10–7), 17q25.3 (rs4890030, P = 5.14 × 10–7), and 4q28.3 (rs2634863, P = 5.27 × 10–7).

Manhattan plots of BDR GWAS results for (a) Puerto Ricans, (b) Mexicans, (c) African Americans, and (d) trans-ethnic meta-analysis. Top 5 loci of each result were annotated with the nearest protein-coding genes. The green horizontal dashed line indicates a genome-wide significance threshold of 5 × 10–8.

Evidence supporting DNAH5 as a BDR-associated gene in Puerto Ricans. (a) eQTL (top) from the GTEx v8 release, regional associations in the GWAS (middle), and a chromatin interaction in lung tissue (bottom) from the Capture Hi-C data. (b) differential expression results for DNAH5 using the GEO data.
While no association achieved genome-wide significance in Mexicans and African Americans, several variants were associated with BDR at P < 1 × 10–6. Among Mexicans, a locus at 6p22.1 (rs138319258, P = 6.08 × 10–8) showed the strongest association with BDR, followed by loci at 2p21 (rs72797438, P = 5.15 × 10–7) and 5p14.3 (rs145081988, P = 6.10 × 10–7). Among African Americans, the top loci were found at 3q25.2 (rs16824202, P = 2.83 × 10–7), 9p21.3 (rs7866009, P = 5.44 × 10–7), and 2p16.3 (rs28657436, P = 5.58 × 10–7).
No loci reached genome-wide significance in the trans-ethnic meta-analysis. The lowest p values were observed in loci at 2p16.3 (rs13077362, P = 1.66 × 10–6), 18q11.2 (rs2239215, P = 1.75 × 10–6), and 5p15.2 (rs1017451, P = 1.93 × 10–6).
A total of 50 variants were selected for replication based on having a genome-wide significant association (i.e., 5p15.2 region found in Puerto Ricans) or P value of < 10–5 in the trans-ethnic meta-analysis (Supplementary Table S1). Several variants at 5p15.2, including the lead variant rs35661809 in Puerto Ricans, were nominally associated (1-tailed P < 0.05) with BDR in the GACRS cohort. None of the other top associations were replicated in the independent cohorts.
The region-based gene association tests showed no significant associations at the Bonferroni threshold (Supplementary Fig. S2). In addition, none of the predicted monocyte expression levels were associated with BDR, although LIN7A (P = 1.27 × 10–5) approached the Bonferroni significance level in Mexican patients with asthma (Supplementary Fig. S3).
Finally, we examined previously reported BDR-associated variants in/near ADRB2, ADCY9, ARG1, COL22A1, CRHR2, GLCCI1, GSNOR, THRB, and SPATS2L, but observed no significant associations in our study (Supplementary Table S2).
Discussion
Our racial/ethnic population-specific GWAS identified one genome-wide significant locus at 5p15.2 in Puerto Ricans, for which the lead variant (rs35661809) was located in the non-coding RNA gene LINC02220. This region was previously identified as a BDR-associated locus in a study of GALA II and SAGE II subjects that used as outcome extreme responders to albuterol sampled from the tails of the BDR distribution, although that study did not provide evidence that linked the associated region to the gene other than physical proximity4. Overall, our results are largely consistent with this previous study, but by using a greater number of subjects and a continuous distribution of BDR to identify this association, we additionally found that 1) capture Hi-C data illustrated a long-range interaction with the promoter of DNAH5 in lung tissue, suggesting its potential role in the transcription of DNAH5; and 2) several variants in the promoter region of DNAH5 were significant eQTL in lung tissue consistent with the Hi-C finding. A recent GWAS based on UK Biobank data identified an association between an intronic DNAH5 variant and the lung function measure FEV1/FVC ratio (FVC: Forced vital capacity) among adults50, and a GWAS from the COPDGene Study reported suggestive associations between DNAH5 variants and post-bronchodilator FEV1/FVC among smokers of European and African ancestry that were more pronounced (P value < 5 × 10–6) when restricting the analysis to those with COPD51.
DNAH5 encodes a dynein protein, which functions as a force-generating protein of respiratory cilia and plays an important role in cilia mobility, especially in respiratory epithelial cells, where ciliary motility is indispensable to mucus transport and airway clearance52,53. Mutations in DNAH5 have been linked to primary ciliary dyskinesia (PCD), which is characterized by marked airway dysfunction54,55. Our transcriptomic results provide evidence of the relevance of DNAH5 to asthma, given that its gene expression levels were decreased in bronchial epithelium of patients with asthma versus controls and in airway smooth muscle cells after budesonide exposure. Although our replication results found that rs35661809 and several LINC02220 intronic variants were nominally associated with BDR with a consistent direction of effect in the GACRS cohort, the replication was not observed in CAMP or HPR. Taken these together, our findings suggest that DNAH5 contributes to variability of BDR, but a more precise mechanistic link between the associated locus and DNAH5 with BDR has yet to be elucidated.
Although we did not observe genome-wide significant associations in Mexicans and African Americans, the top loci may be of interest for future studies. In Mexicans, rs138319258 near TRIM27 at 6p22.1 had the lowest P value with BDR. The tripartite motif containing protein 27 (TRIM27) functions as an E3 ligase to ubiquitinate and inhibit Phosphatidlyinositol-3-kinase C2 beta (PI3KC2β), which plays an important role in the IgE receptor (FcεRI) and mast cell activation56. In allergic asthma, mast cells are activated mainly through IgE-mediated cross-linking of FcεRI with allergens57. The role of TRIM27 in mast cell regulation indicates the potential relationship between genetic variations in TRIM27 and asthma. Along the same lines, previous GWAS have identified an association between a locus near TRIM27 and asthma58,59, and previous studies have shown that IgE levels are associated with BDR3,60. In African Americans, the most strongly associated variant was rs16824202 adjacent to GPR149 at 3q25.2. Variants in the GPR149 gene, which encodes a seven-transmembrane G protein-coupled receptor (GPCR) class A family member, have been associated with coronary artery disease, HDL cholesterol level, and apolipoprotein A1 level61,62.
We did not identify significant associations in the trans-ethnic meta-analysis. The top variant, rs13000632 at 2p16.3, was located near the NRXN1 gene that has been associated with smoking behaviors across diverse populations63,64,65. Given that smokers with asthma have worse outcomes compared to non-smokers with asthma66,67, there may be an interaction between polymorphisms in smoking-related genes and BDR. Another top independent locus was found at at 18q11.2. near AQP4, a gene whose expression was up-regulated in bronchial epithelial cells from patients with asthma68, and whose altered regulation was also observed in allergen and IL-13-induced mouse models of asthma69. Future validation studies are needed to substantiate the functional relationships between NRXN1 and AQP4 with BDR.
Our region-based gene association tests that evaluated the combined effects of variants within prespecified gene boundaries did not find statistically significant associations. To overcome the limitations inherent in assigning a potential functional link to a gene based on proximity of variants alone, we additionally performed predicted QTL analysis to investigate whether a set of variants regulated the expression of a specific gene that was associated with BDR. Due to the paucity of gene expression data for people of non-European ancestry, we were only able to examine associations with monocyte gene expression levels predicted from the MESA study44. Expanding gene expression data to include more diverse people and more asthma-related tissues (e.g., lung, airway smooth muscle, bronchial epithelium) would be of interest to elucidate tissue-specific BDR-related pathways.
Genetic variation of the β2-adrenergic receptor (ADRB2) gene, which encodes the direct target of SABAs, has been of particular interest in BDR studies, but associations between ADRB2 variants and BDR have not been consistently observed70. Our results did not identify BDR-associated variants in ADRB2 or other previously identified candidate genes, which may be in part explained by differences in study design and the fact that results from candidate gene studies have been difficult to replicate71. Several asthma GWAS have been identified strong and consistently replicated genetic associations72, including novel loci in a recent study focused on moderate-to-severe asthma73, but thus far, the asthma susceptibility loci identified have not been linked to BDR pathways. The top BDR-associated loci identified in this study are also not among the previously identified asthma-associated variants.
Our study has several limitations. First, the sample size of the current study was not sufficient to detect modest effect sizes of single-variant associations that might show marginal levels of significance, although it is one of the largest pharmacogenetic studies of minority populations using whole-genome sequencing data to date. Second, there was heterogeneity in subject characteristics across our primary cohorts. Notably, subjects from SAPPHIRE were older than the other cohorts, and therefore, may have different clinical characteristics that influenced BDR. Finally, we were not able to identify additional asthma cohorts of Mexican and African American populations to replicate our primary findings. Future studies that consider population-specific differences in BDR heterogeneity, are better able to account for differences in patterns of linkage disequilibrium among populations of different genetic ancestry, and are of larger sample sizes, may uncover more robust BDR associations and find overlap among genetic risk factors of asthma and BDR.
In summary, by leveraging multiomics data, including capture Hi-C, gene expression, and eQTL, we found that DNAH5 may influence BDR, and its genetic variation may contribute to differences in asthma outcomes among Puerto Ricans. Future mechanistic studies should be pursued to identify how DNAH5 influences BDR.
Data availability
The datasets used and/or analyzed during the current study are available from the PIs of the respective studies upon reasonable request. The TOPMed WGS data analyzed during the current study are available in dbGaP with the following accession numbers: phs000920 (GALA II), phs000921 (SAGE II), phs001467 (SAPPHIRE), and phs001661 (GCPD-A). MESA PrediXcan prediction model: https://doi.org/10.5281/zenodo.3610513. 3D Genome Browser Capture Hi-C: http://3dgenome.fsm.northwestern.edu. Gene Expression Omnibus (GEO) repository: https://www.ncbi.nlm.nih.gov/geo (GED ID: SRP098649; GSE67472; GSE63142) Genotype-Tissue Expression (GTEx) v8 release: https://www.gtexportal.org/home/datasets.
Abbreviations
- BDR:
-
Bronchodilator response
- SABAs:
-
Short-acting β2-agonists
- PKA:
-
CAMP-dependent protein kinase A
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- GWAS:
-
Genome-wide association study
- QTL:
-
Quantitative trait loci
- TOPMed:
-
Trans-Omics for Precision Medicine
- GEO:
-
Gene Expression Omnibus
- GTEx:
-
Genotype-Tissue Expression
- SAGE II:
-
Study of African Americans, Asthma, Genes, and Environments
- GALA II:
-
Genes-environments and Admixture in Latino Americans
- SAPPHIRE:
-
Study of Asthma Phenotypes and Pharmacogenomic Interactions by Race-Ethnicity
- GCPD-A:
-
Genetic Causes of Complex Pediatric Disorders—Asthma
- CAMP:
-
Childhood Asthma Management Program
- GACRS:
-
The Genetics of Asthma in Costa Rica Study
- HPR:
-
Hartford-Puerto Rico
- ATGC:
-
Asthma Translational Genomic Collaboration
- MESA:
-
Multi-Ethnic Study of Atherosclerosis
References
Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention (2020 Update) [Internet] (2020). https://ginasthma.org/wp-content/uploads/2020/04/GINA-2020-full-report_-final-_wms.pdf
Cazzola, M., Page, C. P., Rogliani, P. & Matera, M. G. β2-agonist therapy in lung disease. Am J Respir Crit Care Med 187, 690–696 (2013).
Sharma, S. et al. Clinical predictors and outcomes of consistent bronchodilator response in the childhood asthma management program. J Allergy Clin. Immunol. 122, 921-928.e4 (2008).
Mak, A. C. Y. et al. Whole-genome sequencing of pharmacogenetic drug response in racially diverse children with asthma. Am. J. Respir. Crit. Care. Med. 197, 1552–1564 (2018).
Blake, K., Madabushi, R., Derendorf, H. & Lima, J. Population pharmacodynamic model of bronchodilator response to inhaled albuterol in children and adults with asthma. Chest 134, 981–989 (2008).
Burchard, E. G. et al. Lower bronchodilator responsiveness in Puerto Rican than in Mexican subjects with asthma. Am J Respir Crit Care Med. 169, 386–392 (2004).
Naqvi, M. et al. Ethnic-specific differences in bronchodilator responsiveness among African Americans, Puerto Ricans, and Mexicans with asthma. J. Asthma. 44, 639–648 (2007).
Martinez, F. D., Graves, P. E., Baldini, M., Solomon, S. & Erickson, R. Association between genetic polymorphisms of the beta2-adrenoceptor and response to albuterol in children with and without a history of wheezing. J. Clin. Invest. 100, 3184–3188 (1997).
Silverman, E. K. et al. Family-based association analysis of β2-adrenergic receptor polymorphisms in the childhood asthma management program. J. Allergy Clin. Immunol. Elsevier 112, 870–876 (2003).
Tantisira, K. G., Small, K. M., Litonjua, A. A., Weiss, S. T. & Liggett, S. B. Molecular properties and pharmacogenetics of a polymorphism of adenylyl cyclase type 9 in asthma: Interaction between beta-agonist and corticosteroid pathways. Hum. Mol. Genet. 14, 1671–1677 (2005).
Brehm, J. M. et al. Stress and bronchodilator response in children with asthma. Am. J. Respir. Crit. Care Med. 192, 47–56 (2015).
Litonjua, A. A. et al. ARG1 is a novel bronchodilator response gene: Screening and replication in four asthma cohorts. Am. J. Respir. Crit. Care Med. 178, 688–694 (2008).
Duan, Q. L. et al. A genome-wide association study of bronchodilator response in asthmatics. Pharmacogenom. J. 14, 41–47 (2014).
Poon, A. H. et al. Association of corticotropin-releasing hormone receptor-2 genetic variants with acute bronchodilator response in asthma. Pharmacogenet. Genom. 18, 373–382 (2008).
Tantisira, K. G. et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N. Engl. J. Med. 365, 1173–1183 (2011).
Moore, P. E. et al. Genetic variants of GSNOR and ADRB2 influence response to albuterol in African-American children with severe asthma. Pediatr. Pulmonol. 44, 649–654 (2009).
Duan, Q. L. et al. A polymorphism in the thyroid hormone receptor gene is associated with bronchodilator response in asthmatics. Pharmacogenom. J. 13, 130–136 (2013).
Himes, B. E. et al. Genome-wide association analysis in asthma subjects identifies SPATS2L as a novel bronchodilator response gene. PLoS Genet. 8, e1002824 (2012).
Padhukasahasram, B. et al. Gene-based association identifies SPATA13-AS1 as a pharmacogenomic predictor of inhaled short-acting beta-agonist response in multiple population groups. Pharmacogenom. J. 14, 365–371 (2014).
Drake, K. A. et al. A genome-wide association study of bronchodilator response in Latinos implicates rare variants. J. Allergy Clin. Immunol. 133, 370–378 (2014).
Nishimura, K. K. et al. Early-life air pollution and asthma risk in minority children: The GALA II and SAGE II studies. Am. J. Respir. Crit. Care Med. 188, 309–318 (2013).
Jin, Y. et al. Dual specificity phosphatase-1 as a pharmacogenetic modifier of inhaled steroid response among asthma patients. J. Allergy Clin. Immunol. 126, 618–625 (2010).
Ong, B. A. et al. Gene network analysis in a pediatric cohort identifies novel lung function genes. PLoS ONE 8, e72899 (2013).
The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Childhood Asthma Management Program Research Group. Control Clin. Trials. 20, 91–120 (1999).
Hunninghake, G. M. et al. Sensitization to Ascaris lumbricoides and severity of childhood asthma in Costa Rica. J. Allergy Clin. Immunol. 119, 654–661 (2007).
Forno, E. et al. Genome-wide interaction study of dust mite allergen on lung function in children with asthma. J. Allergy Clin. Immunol. 140, 996-1003.e7 (2017).
Yan, Q. et al. A meta-analysis of genome-wide association studies of asthma in Puerto Ricans. Eur. Respir. J. 49, 1601505 (2017).
Pellegrino, R. Interpretative strategies for lung function tests. Eur. Respir. J. 26, 948–968 (2005).
Brehm, J. M. et al. African ancestry and lung function in Puerto Rican children. J. Allergy Clin. Immunol. 129, 1484-1490.e6 (2012).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Grnet. 43, 491–498 (2011).
Picard toolkit [Internet]. Broad Inst. GitHub Repos. Broad Institute (2019). https://broadinstitute.github.io/picard/.
Danecek, P. et al. The variant call format and VCFtools. Bioinforma Oxf. Engl. 27, 2156–2158 (2011).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Gogarten, S. M. et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics 35, 5346–5348 (2019).
Han, B. & Eskin, E. Random-Effects Model Aimed at Discovering Associations in Meta-Analysis of Genome-wide Association Studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Joo, J. & Himes, B. Gene-based analysis reveals sex-specific genetic risk factors of COPD. AMIA Annu. Symp. Proc. 2021, 601–610 (2022).
Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. Nucl. Acids Res. 47, D766–D773 (2019).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 526, 68–74 (2015).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Mogil, L. S. et al. Genetic architecture of gene expression traits across diverse populations. PLOS Genet. 14, e1007586 (2018).
Wang, Y. et al. The 3D Genome Browser: A web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19, 151 (2018).
Martin, P. et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nat. Commun. 6, 10069 (2015).
Barrett, T. et al. NCBI GEO: Archive for functional genomics data sets—update. Nucl. Acids Res. 41, D991–D995 (2013).
Kan, M., Shumyatcher, M., Diwadkar, A., Soliman, G. & Himes, B. E. Integration of transcriptomic data identifies global and cell-specific Asthma-related gene expression signatures. AMIA Annu. Symp. Proc. 2018, 1338–1347 (2018).
The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 45, 580–585 (2013).
Kichaev, G. et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. 104, 65–75 (2019).
Lutz, S. M. et al. A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry. BMC Genet. 16, 138 (2015).
Sugier, P.-E. et al. A novel role for ciliary function in atopy: ADGRV1 and DNAH5 interactions. J. Allergy Clin. Immunol. Elsevier 141, 1659-1667.e11 (2018).
Olbrich, H. et al. Axonemal localization of the dynein component DNAH5 is not altered in secondary ciliary dyskinesia. Pediatr. Res. 59, 418–422 (2006).
Olbrich, H. et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left–right asymmetry. Nat. Genet. 30, 143–144 (2002).
Green, K. et al. Ventilation inhomogeneity in children with primary ciliary dyskinesia. Thorax 67, 49–53 (2012).
Srivastava, S., Cai, X., Li, Z., Sun, Y. & Skolnik, E. Y. Phosphatidylinositol-3-kinase C2β and TRIM27 function to positively and negatively regulate IgE receptor activation of mast cells. Mol. Cell Biol. Am. Soc. Microbiol. J. 32, 3132–3139 (2012).
Méndez-Enríquez, E. & Hallgren, J. Mast cells and their progenitors in allergic asthma. Front. Immunol. https://doi.org/10.3389/fimmu.2019.00821/full (2019).
Demenais, F. et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune cell enhancer marks. Nat. Genet. 50, 42–53 (2018).
Zhu, Z. et al. Shared genetics of asthma and mental health disorders: A large-scale genome-wide cross-trait analysis. Eur Respir J. 54, 1901507 (2019).
Coverstone, A. M. et al. Clinical significance of the bronchodilator response in children with severe asthma. Pediatr. Pulmonol. 54, 1694–1703 (2019).
Richardson, T. G. et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 17, e1003062 (2020).
van der Harst, P. & Verweij, N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 122, 433–443 (2018).
Sato, N. et al. Association between neurexin 1 (NRXN1) polymorphisms and the smoking behavior of elderly Japanese. Psychiatr. Genet. 20, 135–136 (2010).
Nussbaum, J. et al. Significant association of the neurexin-1 gene (NRXN1) with nicotine dependence in European- and African-American smokers. Hum. Mol. Genet. 17, 1569–1577 (2008).
Pérez-Rubio, G. et al. SNPs in NRXN1 and CHRNA5 are associated to smoking and regulation of GABAergic and glutamatergic pathways. Pharmacogenomics 17, 1145–1158 (2016).
Polosa, R. & Thomson, N. C. Smoking and asthma: dangerous liaisons. Eur. Respir. J. Eur. Respir. Soc. 41, 716–726 (2013).
Chatkin, J. M. & Dullius, C. R. The management of asthmatic smokers. Asthma Res. Pract. 2, 10 (2016).
Jardim, M. J., Dailey, L., Silbajoris, R. & Diaz-Sanchez, D. Distinct MicroRNA expression in human airway cells of asthmatic donors identifies a novel asthma-associated gene. Am. J. Respir. Cell Mol. Biol. 47, 536–542 (2012).
Krane, C. M. et al. Altered regulation of aquaporin gene expression in allergen and IL-13-induced mouse models of asthma. Cytokine 46, 111–118 (2009).
Israel, E. et al. Genome-wide association study of short-acting β2-agonists: A novel genome-wide significant locus on chromosome 2 near ASB3. Am. J. Respir. Crit. Care Med. 191, 530–537 (2015).
Sullivan, P. F. Spurious genetic associations. Biol Psychiatry. 61, 1121–1126 (2007).
Kan, M., Himes, B.. Genetics and Pharmacogenetics of Asthma (2020), pp 25–37.
Shrine, N. et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir. Med. 7, 20–34 (2019).
Acknowledgements
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Whole genome sequencing (WGS) for “NHLBI TOPMed: Gene-Environment, Admixture and Latino Asthmatics Study” (phs000920) and “NHLBI TOPMed: Study of African Americans, Asthma, Genes and Environments” (phs000921) was performed at the New York Genome Center (3R01 HL-117004-02S3) and the University of Washington Northwest Genomics Center (HHSN268201600032I). WGS for “NHLBI TOPMed: Study of Asthma Phenotypes & Pharmacogenomic Interactions by Race-Ethnicity” (phs001467) and “Genetics of Complex Pediatric Disorders—Asthma” (phs001661) was performed at the University of Washington Northwest Genomics Center (HHSN268201600032I). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Administrative Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. WGS of part of GALA II was performed by New York Genome Center under The Centers for Common Disease Genomics (CCDG) of the Genome Sequencing Program (GSP) grant (UM1 HG-008901). The GSP Coordinating Center (U24 HG-008956) contributed to cross-program scientific initiatives, and provided logistical and general study coordination. GSP is funded by the NHGRI, the NHLBI, and the National Eye Institute.
Funding
The GALA II study, the SAGE II study, and E.G.B. were supported by the Sandler Family Foundation, the American Asthma Foundation, the RWJF Amos Medical Faculty Development Program, the Harry W. and Diana V. Hind Distinguished Professorship in Pharmaceutical Sciences II, the National Heart, Lung, and Blood Institute (NHLBI) (R01 HL-117004, R01 HL-128439, R01 HL-135156, and X01 HL-134589), the National Institute of Environmental Health Sciences (NIEHS) (R01 ES-015794), the National Institute on Minority Health and Health Disparities (P60 MD-006902 and R01 MD-010443), the National Human Genome Research Institute (NHGRI) (U01 HG-009080), and the Tobacco-Related Disease Research Program (24RT-0025). The SAPPHIRE study was supported by the Fund for Henry Ford Hospital, the American Asthma Foundation, the NHLBI (R01 HL-118267, R01 HL-141485, and X01 HL-134589), the National Institute of Allergy and Infectious Diseases (R01 AI-079139), and the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK-113003). The GCPD-A study was supported by an Institutional award from the Children’s Hospital of Philadelphia and by the NHLBI (X01 HL-134589). The HPR study was funded by the NIH (R01 HL-079966). Additional funding was provided by the NHLBI (R01 HL-133433 and R01 HL-141992) and the NIEHS (P30 ES-013508). The funders played no role in the design of the study, the collection, analysis, and interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
J.J., A.C.Y.M., S.X., P.M.S., H.H., L.K.W., E.G.B., and B.E.H. designed and supervised the study. J.J., A.C.Y.M., S.X., P.M.S., D.H., S.H., C.E., M.K., A.R.D., H.H., L.K.W., E.G.B., and B.E.H. performed statistical analysis and interpreted results. J.A.L.-S., S.T.W., J.E.S., A.C.W., M.C., G.C., E.F., J.C.C., and M.A.S. conducted and supervised replication analyses. J.J. and A.C.Y.M. drafted the manuscript with substantial contributions from S.X., P.M.S., E.G.B., and B.E.H. All authors provided feedback on the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Joo, J., Mak, A.C.Y., Xiao, S. et al. Genome-wide association study in minority children with asthma implicates DNAH5 in bronchodilator responsiveness. Sci Rep 12, 12514 (2022). https://doi.org/10.1038/s41598-022-16488-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16488-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
