
Abstract
The non-medical use of opioids has become a national crisis in the USA. Developing non-opioid pharmacotherapies for controlling this opioid epidemic is urgent. Dopamine D3 receptor (D3R) antagonists and low efficacy partial agonists have shown promising profiles in animal models of opioid use disorders (OUD). However, to date, advancement to human studies has been limited. Here we report the effects of (S)- and (R)-enantiomers of (±)-ABS01-113, structural analogs of the D3R partial agonist, (±)-VK4-40, in which the 3-OH in the linking chain is replaced by 3-F group. (S)- and (R)-ABS01-113 are identical in chemical structure but with opposite chirality. In vitro receptor binding and functional assays indicate that (S)-ABS01-113 is an efficacious (55%) and potent (EC50 = 7.6 ± 3.9 nM) D3R partial agonist, while the (R)-enantiomer is a potent D3R antagonist (IC50 = 11.4 nM). Both (S)- and (R)-ABS01-113 bind with high affinity to D3R (Ki = 0.84 ± 0.16 and 0.37 ± 0.06 nM, respectively); however, the (S)-enantiomer is more D3/D2-selective (>1000-fold). Pharmacokinetic analyses indicate that both enantiomers display excellent oral bioavailability and high brain penetration. Systemic administration of (S)- or (R)-ABS01-113 alone failed to alter open-field locomotion in male rats and mice. Interestingly, pretreatment with (S)- or (R)-ABS01-113 attenuated heroin-enhanced hyperactivity, heroin self-administration, and (heroin + cue)-induced reinstatement of drug-seeking behavior. Together, these findings reveal that both enantiomers, particularly the highly selective and efficacious D3R partial agonist (S)-ABS01-113, demonstrate promising translational potential for the treatment of OUD.
Similar content being viewed by others

Introduction
In the midst of the COVID-19 pandemic opioid overdose fatalities soared to a record 101,263 in 2021 [1]. This estimate translates to an average of 277 deaths/day. According to the CDC data, more than 74% of the overdose deaths last year involved fentanyl and other opioids. Indeed, the rampant abuse of opioids continues to be a major public health problem. In addition, lockdowns and social distancing made access to opioid-based treatments such as methadone, that require in person administration, much more difficult. Although the United States Food and Drug Administration (FDA) has approved methadone and buprenorphine for the treatment of opioid use disorders (OUD), there is a high rate of relapse after cessation of treatment. Further concerns for these opioid agonist therapies include their dependence liability and severe withdrawal symptoms after cessation of use [2]. Hence, developing non-opioid-based medications, without their own dependence liability for the treatment of OUD, would provide a valuable alternative or addition to current pharmacotherapeutic options.
Over the past 2 decades, the dopamine D3 receptor (D3R) has become a target for medication development to treat substance use disorders and many D3R-selective ligands with high affinity have been discovered [3,4,5,6]. In particular, selective D3R partial agonists and antagonists have been investigated extensively and have demonstrated promising results in experimental animals [6,7,8,9,10,11]. However, to date, advancement to human studies has been limited to a very few selective D3R antagonists such as GSK598,809 [12, 13]. We previously focused on developing such compounds as therapeutics to treat psychostimulant use disorders [3, 14, 15]. However, some concerns from the reports that D3R antagonists might exacerbate cardiotoxicity produced by cocaine [16] led us to shift our attention to OUD, as the opioid crisis was pervasive and typically opioids do not produce cardiovascular toxicity [10, 17, 18]. Toward this goal, we used the D3R crystal structure and extensive structure activity relationships (SAR) to develop bitopic D3R ligands [15, 19,20,21], which has led to the development of a number of highly selective D3R antagonists or low efficacy partial agonists, exemplified by (±)-VK4-116 and (±)-VK4-40 [15, 22,23,24]. As these lead compounds have a chiral center in their linking chain (3-OH), the synthesis and evaluation of the (S)- and (R)-enantiomers has continued [10, 23,24,25]. Extensive metabolism, pharmacokinetic (PK) and behavioral evaluation in numerous animal models of OUD indicate both these lead molecules are effective in reducing opioid reward and relapse [6, 8, 10, 11] and further development toward human trials is underway.
We previously reported that replacing the 3-OH with a 3-F in the linking chain in these bitopic ligands resulted in some of the most selective D3R partial agonists/antagonists reported [26, 27]. Here, we report another novel pair of enantiomers — (S)- and (R)-ABS01-113, in which the 3-OH in the linking chain of (S)- or (R)-VK4-40 was replaced with a 3-F group [23]. Unlike the low efficacy partial agonist (±)-VK4-40, (±)-ABS01-113 is a selective D3R antagonist, as is its (R)-enantiomer. However, we discovered that (S)-ABS01-113 is an efficacious D3R partial agonist. As the only difference between these enantiomers is the linking chain chirality at the 3-position, rendering them mirror images of one another, we were curious to compare their behavioral profiles and drug-like properties. We hypothesized that the (S)-enantiomer might not be as effective in these models as an antagonist or low efficacy partial agonist. Nevertheless, we reasoned that if (S)-ABS01-113 produced a behavioral profile in our rodent models of OUD, it may have advantages over a D3R antagonist. Indeed, partial agonists have been discovered to be more favorable pharmacotherapeutics for treatment of a variety of neuropsychiatric disorders ranging from OUD (e.g., buprenorphine) to schizophrenia (e.g., aripiprazole or cariprazine) [28,29,30].
We had previously reported the synthesis and binding affinities at D3R and D2R of (S)- and (R)-ABS01-113 [20]. Herein we investigated their functional activity at D3R in vitro as well as binding affinities at a few off-target sites with known homology to D3R. We also evaluated their brain: plasma ratios after intragastric drug administration. Lastly, we examined their pharmacological efficacy in antagonizing opioid actions, including opioid-induced hyperactivity, self-administration, and reinstatement of drug-seeking behavior to predict their translational potential for the treatment of OUD in humans.
Materials and methods
Exp. 1: (S)- and (R)-ABS1-113 off-target and functional assays
(S)- and (R)-ABS01-113 off-target and functional activity assays were performed in Dr. Aaron Janowsky’s laboratory at the VA Portland Health Care System, Portland OR.
[3H]-SCH-23390 (D1R) receptor binding assays
Mouse fibroblast cells expressing the human D1 receptor at high density (LhD1 cells) were used. The cells were grown to confluence in Dulbecco’s Minimal Essential Medium (DMEM) containing 10% FetalClone1 serum (FCS, HyClone), 0.05% penicillin-streptomycin (pen-strep), and 200 µg/ml of Geneticin (G418). One confluent 150 mm plate yielded enough membranes for 1 assay plate with ~10–15 µg protein/well. The cells from 1 150 mm plate were washed with phosphate-buffered saline (PBS), scraped into 50 mM Tris buffer (pH 7.4), and centrifuged at 27 K × g for 20 min. The cell pellet was homogenized in 50 mM Tris (pH 7.4) with a polytron and centrifuged at 27 K × g for another 20 min. The pellet was overlaid with 2 ml of 50 mM Tris buffer (pH 7.4) and frozen at −70 °C. On the day of experiment, each pellet was homogenized in 10 ml assay buffer (50 mM Tris-HCl, pH 7.4, containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2) with a polytron. Cell homogenate (100 µl) was added to wells containing 800 µl of test drug (blinded to the investigator) or buffer. After 10 min preincubation, 100 µl of [3H]-SCH-23390 (0.18 nM final concentration) was added. The plates were incubated at 25 °C for 60 min. The reaction was terminated by filtration through polyethylenimine-soaked (0.05%) filters using a Tomtec 96-well harvester and radioactivity on the filters was counted using a Perkin Elmer microbetaplate 1405 liquid scintillation counter. Nonspecific binding was determined with 1 µM SCH-23390.
[3H]-Spiperone (D4R) receptor binding assays
Human embryonic kidney cells coexpressing the human D4.4 receptor and adenylate cyclase type I (HEK-D4.4-AC1) were grown in DMEM supplemented with 5% FetalClone (HyClone), 5% bovine calf serum (BCS), 0.05% pen-strep, 2 µg/ml of puromycin and 200 µg/ml of hygromycin. Membranes were prepared according to the procedures described for D1 cells, using 50 mM Tris (pH 7.4 at 4 °C). Assay buffer contained 120 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 4 mM MgCl2, 1 mM ascorbic acid and 1 mM EDTA (pH 7.4 at 37 °C). One confluent 150 mm plate of D4.4 cells, yielded enough membranes for 1 assay plate with ~14–25 µg protein/well. Proteins were determined using a BCA assay (Pierce). Cell homogenate (100 µl) was added to wells containing 800 µl of test drug (blinded to the investigator) or buffer. After 10 min, 100 µl of [3H]-spiperone (specific activity 80.2, 0.2–0.3 nM final concentration) was added. The plates were incubated at 37 °C for 60 min. The reaction was terminated by filtration through polyethylenimine-soaked (0.05%) filters using a Tomtec 96-well harvester. Radioactivity on the filters was counted using a Perkin Elmer microbeta counter. Nonspecific binding was determined with 1 µM haloperidol.
[3H]8-OH-DPAT (5-HT1A) receptor binding assays
Human embryonic kidney cells expressing the human 5HT1A receptor (HEK-h5HT1A) were used. The cells were grown to confluence in DMEM containing 5% FetalClone (FC, HyClone), 5% bovine calf serum, 0.05% penicillin-streptomycin (pen-strep), and 200 µg/mL of Geneticin (G418). The cells were scraped from 150 mm plates into phosphate-buffered saline and centrifuged at 1000 × g, for 10 min. The cell pellet was homogenized in 50 mM Tris-HCl (pH 7.4) with a polytron, and centrifuged at 27,000 × g. The homogenization and centrifugation were repeated twice to wash any remaining 5-HT from the growth media. The final pellet was covered with 2 ml Tris buffer and stored at −80 °C until needed. The assay was performed in duplicate in a 96-well plate. Serial dilutions of test compounds were made using the Biomek 4000 robotics system. The reaction mixture contained compound whose identity was not known to the experimenter, 100 µl of cell homogenate (0.05 mg protein/well) and 100 µl of [3H]8-OH-DPAT (0.5 nM final concentration, 170 Ci/mmol, Perkin Elmer) and assay buffer (25 mM Tris-HCl, pH 7.4, containing 100 µM ascorbic acid and 10 µM pargyline) in a final volume of 1 ml. Nonspecific binding was determined with 1.0 µM dihydroergotamine. The plates were incubated at room temperature for 60 min and then filtered through polyethylenimine-soaked (0.05%) “A” filtermats on a Tomtec cell harvester. The filters were washed with cold 50 mM Tris buffer (pH 7.4) for 6 s, dried, spotted with scintillation cocktail, and counted for 2 min, after a 4 h delay, on a Perkin Elmer microbetaplate counter. IC50 values were calculated with GraphPad Prism, and IC50 values were converted to Ki values using the Cheng–Prusoff correction [31].
[3H]5-HT (5-HT2A and 5-HT2C) receptor binding assays
The method was adapted from AR Knight et al. [32]. Human embryonic kidney cells expressing the human 5HT2A receptor (HEK-h5HT2A) or human 5HT2C receptor (HEK-h5HT2C) were used. The cells were grown until confluent on 15 cm plates. Media was removed, cells were washed with phosphate-buffered saline (PBS), scraped into 10 ml PBS and centrifuged at 1000 × g, for 10 min. The pellet was resusupended in 10 ml, and polytronned at setting 6 for 5 s. The homogenate was centrifuged at 27,000 × g for 20 min. To minimize the residual 5-HT concentration, the pellet was resuspended in buffer, polytronned, and centrifuged as above. The final pellet was resuspended in 2 ml buffer/plate of cells.
The binding assay included 50 µl drug, 5-HT or buffer, 50 µl cell homogenate, 50 µl [3H]5-HT (~4 nM) and assay buffer (50 mM Tris, pH 7.4 at 37 °C, with 0.1% ascorbic acid and 5 mM CaCl2) buffer in a final volume of 250 µl. Specific binding was defined as the difference between total binding and binding in the presence of 10 µM 5-HT. The reaction was incubated for 1 h at 37 °C, and terminated by filtration through Wallac A filtermats presoaked in 0.05% polyethylenimine using a Tomtec 96-well harvester. Radioactivity remaining on filters was counted in a Perkin Elmer microbetaplate counter. IC50 values were calculated using GraphPad Prism. IC50 values were converted to Ki values using the Cheng–Prusoff equation [31].
Exp. 2: D3R mitogenesis functional assay
CHOp-D3 cells were maintained in alpha-MEM with 10% fetal bovine serum (FBS, Atlas Biologicals), 0.05% pen-strep, and 200 µg/ml of G418. To measure D3 stimulation of mitogenesis (agonist assay) or inhibition of quinpirole stimulation of mitogenesis (antagonist assay), CHOp-D3 cells were seeded in a 96-well plate at a concentration of 5000 cells/well. The cells were incubated at 37 °C in alpha-MEM with 10% FBS, pen-strep, and G418. After 48–72 h, the cells were rinsed twice with serum-free alpha-MEM and incubated for 24 h at 37 °C. Serial dilutions of test compounds were made by the Biomek robotics system in serum-free alpha-MEM. In the functional assay for agonists, the medium was removed and replaced with 100 µl of test compound in serum-free alpha-MEM. In the antagonist assay, the serial dilution of the putative antagonist test compound was added in 90 μl (1.1× of final concentration) and 300 nM quinpirole (30 nM final) was added in 10 μl. After another 16-h incubation at 37 °C, 0.3 µCi of [3H]-thymidine in alpha-MEM supplemented with 10% FBS was added to each well and the plates were further incubated for 2 h at 37 °C. The cells were trypsinized by addition of 10× trypsin solution (1% trypsin in calcium–magnesium-free phosphate-buffered saline) and the plates were filtered and counted as usual. Quinpirole was run each day as an internal control and dopamine was included for comparative purposes.
Exp. 3: (S)- and (R)-ABS1-113 pharmacokinetic studies
To determine whether the enantiomers were orally available and brain penetrable, we evaluated plasma vs. brain drug concentrations after intragastric administration of (R)-ABS01-113 or (S)-ABS01-113 in male Long–Evans rats (Charles River Laboratories, Frederick, MD) (initially weighing 250–300 g). The experimental procedures are the same as we reported before [10]. The experimental methods in detail are provided in the Supplementary Materials.
Exp. 4: Open-field locomotion
Increased DA release produces an increase in open-field locomotion by activation of DA receptors [33, 34]. However, as D3R are not highly expressed in brain regions associated with motor activity such as the dorsal striatum [3], we speculated that neither the D3R partial agonist (S)-ABS01-113 nor the D3R antagonist (R)-ABS01-113 would produce significant locomotor activity by itself. Hence, we tested (S)- and (R)-ABS01-113 on locomotor activity in wild-type mice. The experimental methods in detail are provided in the Supplementary Materials.
Exp. 5: Heroin self-administration
Subjects
Male Long–Evans rats (Charles River Laboratories, Frederick, MD) (initially weighing 250–300 g, n = 48) were used for heroin self-administration and reinstatement tests. Male wild-type (C57/BL6J, 25–30 g) mice were used for open-field locomotion and sucrose self-administration tests. We chose males in this initial proof-of-concept study. However, females will be used in our follow-up studies. Both rats and mice were chosen based on the availability of the test compounds, experimental animals, and equipment systems in the laboratory. To the best of our knowledge, there have been no reports of sex differences in D3R antagonist or partial agonist behavioral effects. During heroin self-administration experimentation animals were housed individually and mice used in the locomotor experiment were housed in groups of 4/cage in climate-controlled animal colony rooms on a reversed 12-h light–dark cycle (lights on at 7 a.m.) with free access to food and water. All experiments were conducted during the animals’ active period (reversed dark cycle). The housing conditions and care of the animals were consistent with the Guide for the Care and Use of Laboratory Animals. The protocols used in the present experiments were approved by the National Institute on Drug Abuse Animal Care and Use Committee.
Surgery
To determine whether (S)- and (R)-ABS01-113 attenuated heroin intake, rats underwent intravenous catheter surgeries and intravenous self-administration procedures as described previously [9] (see details in the Supplementary Materials).
Heroin self-administration procedures
The i.v. heroin self-administration procedures were the same as reported previously [10, 11]. Heroin self-administration training was conducted in an operant conditioning chamber equipped with two response levers (Med Associates Inc., Georgia, VT, USA). Each rat was allowed to press the active lever for heroin (0.0125 or 0.05 mg/kg/infusion) under a FR1 schedule of reinforcement during daily 3-h sessions. We used two heroin doses in order to determine whether the pharmacological efficacy of both the compounds is heroin dose dependent. In addition, both the heroin doses are located on the descending limb of the heroin self-administration dose-response curve [34,35,36]. Using two different doses of heroin would help us to determine whether the test compounds produce an increase or a decrease in heroin’s rewarding effects (or efficacy) based on the negative correlation between the number of heroin infusions and heroin dose — higher numbers of heroin self-administration (infusions) at lower doses, while lower numbers of heroin self-administration at higher heroin doses. The experimental methods in detail are provided in the Supplementary materials.
Exp. 6: Oral sucrose self-administration in mice
Procedures for oral sucrose self-administration in mice were the same as we reported previously [37] (see details in the Supplementary materials). After the active lever response was stabilized, we observed the effects of the same doses of (R)- or (S)-ABS01-113 on oral sucrose self-administration.
Exp. 7: (Heroin + cue)-induced reinstatement of drug seeking
We then examined whether (S)- or (R)-ABS01-113 can also reduce (heroin + cue)-induced reinstatement of drug seeking. A total of 57 rats with a history of heroin self-administration were used in this experiment. After stable heroin self-administration was achieved, the animals underwent extinction training for 3 weeks. Each extinction session lasted for 3 h and responding on either lever produced no consequences — no heroin or heroin-related cues. This phase continued until extinction criteria were met, defined as <20 lever presses for 3 consecutive sessions. Subsequently, the animals were divided into 6 drug dose groups (n = 8–10 per group) and received either vehicle or one of two doses of (S)- or (R)-ABS01-113 (10 or 30 mg/kg, i.p.). Thirty min later, all animals received a non-contingent injection of heroin (1 mg/kg, i.p.) and then were placed into the same operant chambers for a drug-induced reinstatement test session. Responses on the active and inactive levers were recorded. Each response on the active lever was reinforced with the drug cues (but no heroin). Responding on the inactive lever produced no consequences.
Drugs
(S)- and (R)-ABS1-113 were synthesized at the NIDA-IRP by A.B. Shaik and E.S. Gorgarnoiu according to a modification of the published procedure [21] and dissolved in 25% 2-hydroxypropyl beta-cyclodextrin in distilled H2O. Heroin hydrochloride (provided by NIDA pharmacy) was dissolved in 0.9% saline to achieve final doses of 0.05 and 0.0125 mg/kg/infusion (self-administration experiment). For the reinstatement test and locomotor activity experiment, heroin was given intraperitoneally (1 mg/kg, i.p.).
Data analysis
For the binding data, data were normalized to the binding in the absence of drug. Three or more independent competition experiments were conducted with duplicate determinations. GraphPAD Prism was used to analyze the ensuing data, with IC50 values converted to Ki values using the Cheng-Prusoff equation (Ki = IC50/(1 + ([drug*]/Kd drug*))), where drug* is the labeled ligand used in the binding assays. The Kd values used in the equations are 0.62 nM for [3H]-SCH-23390 at D1 receptors, 0.262 nM for [3H] spiperone at D4.4 receptors, 5.02 nM for [3H]8-OH-DPAT at 5-HT1A receptors and 27.37 nM and 10.02 nM for [3H]5-HT at 5-HT2A and 5-HT2C receptors, respectively. For functional assays, GraphPAD Prism was used to calculate either EC50 (agonists) or IC50 (antagonists) values using data expressed as % quinpirole stimulation for mitogenesis.
Plasma and brain concentrations of (S)- and (R)-ABS01-113 were analyzed using a non-compartmental method as implemented in the computer software program Phoenix® WinNonlin® version 7.0 (Certara USA, Inc., Princeton, NJ). The maximum plasma and tissue concentration (Cmax) and time to Cmax (Tmax) were the observed values. The area under the plasma and tissue concentration time curve (AUC) value was calculated up to 8 h (AUC0−t) by use of the log-linear trapezoidal rule. Brain to plasma ratio is calculated from mean total AUCbrain versus AUCplasma.
All behavioral data are presented as means ± SEM. One-way or two-way analyses of variance (ANOVA) with repeated measures over time or drug dose was used to analyze data across experiments. Post-hoc multiple group comparisons were carried out using the Tukey test. p < 0.05 was considered to indicate statistical significance.
Results
(S)- and (R)-ABS01-113 are potent and selective D3R ligands with differing functional efficacies
Supplementary Table 1 (Table S1) shows the off-target receptor binding profiles of (S)- and (R)-ABS01-113 in in vitro mouse fibroblast cells expressing DA D1, and HEK cells expressing DA D4 or 5-HT (5-HT1A, 5-HT2A and 5-HT2C) receptors, compared to previously reported binding affinities at D2R and D3R, illustrating that (S)- and (R)-ABS01-113 are highly selective D3R ligands.
(S)-ABS01-113 is an efficacious D3R partial agonist and (R)-ABS01-113 is a D3R antagonist
We next examined functional activity of (S)- and (R)-ABS01-113 at D3R using the mitogenesis functional assay. Figure 1A shows the chemical structures of (S)- and (R)-ABS01-113. Figure 1B shows that both DA and quinpirole (a D2-like receptor agonist) produced a robust increase in mitogenesis in a concentration-dependent manner, as indicated by increased [3H]-thymidine incorporation in cultured CHO cells expressed human D3R, while (S)-ABS01-113 produced a potent increase (EC50 = 7.6 ± 3.9 nM) in mitogenesis with ~55% efficacy, demonstrating that (S)-ABS01-113 is an efficacious partial D3R agonist, relative to DA and quinpirole. In contrast, (R)-ABS01-113 had no effect on mitogenesis in CHO cells expressing human D3R (Fig. 1C), demonstrating that (R)-ABS01-113 has no intrinsic functional activity at D3R.

A Chemical structures of (S)- and (R)-ABS01-113. B (S)-ABS01-113, DA, or quinpirole alone all stimulated mitogenesis, as assessed by increased [3H]thymidine incorporation in CHO cells. (S)-ABS01-113 is less efficacious than DA or quinpirole, suggesting that it acts as a partial D3R agonist. C In contrast to the (S)-enantiomer, (R)-ABS01-113 had no effect on mitogenesis in the same functional assay; D (S)-ABS01-113 inhibited quinpirole-stimulated mitogenesis in a way similar to butaclamol or NGB2904 with the IC50 value of 213 nM; E (R)-ABS01-113 is ~20-fold more potent than its (S)-enantiomer in attenuating quinpirole-stimulated mitogenesis with an IC50 of 11.4 nM.
We then examined the effects of (S)- and (R)-ABS01-113 on quinpirole-stimulated mitogenesis in D3R-expressing CHO cells. We found that butaclamol (a typical antipsychotic and non-selective DA receptor antagonist), NGB2904 (a selective D3R antagonist) and (S)- and (R)-ABS01-113 all attenuated quinpirole-induced mitogenesis in a concentration-dependent manner (IC50 = 213 ± 57 nM for (S)-ABS01-113 (Fig. 1D) and 11.4 ± 3.2 nM for (R)-ABS01-113 (Fig. 1E), demonstrating that in the presence of the D3R agonist (quinpirole), both the ABS enantiomers function as D3R antagonists. However, the (R)-enantiomer is ~20-fold more potent than the (S)-enantiomer in attenuating quinpirole-induced mitogenesis. In contrast, (S)-ABS1-113 was ~28-fold more potent as a partial agonist than as an antagonist.
(S)- and (R)-ABS01-113 show high oral availability and blood-brain barrier penetration
Figure 2 shows the pharmacokinetic (PK) study results conducted in rats following intragastric administration of 10 mg/kg (S)- or (R)-ABS01-113. Compared to its parent compound (R)-VK4-40 [22], (R)-ABS01-113 displayed quicker absorption and higher oral availability, as assessed by the maximum drug levels in plasma (Cmax) and the time to reach the maximum levels (Tmax). (R)-ABS01-113 is absorbed four-fold faster (Tmax, 0.5 h vs. 2 h) achieving the maximum plasma/brain concentrations, which are approximately seven-fold higher than those of (R)-VK4-40 (plasma Cmax 2.13 ± 0.35 vs. 0.31 ± 0.11 nmol/mL). (R)-ABS01-113 also displayed excellent brain penetration ability, as assessed by four-fold higher AUC of drug concentration in brain tissue than in plasma after 10 mg/kg (R)-ABS01-113 administration. Importantly, (S)-ABS01-113 (Fig. 2A) displayed similarly improved oral bioavailability as its (R)-enantiomer and high brain penetration ability (Fig. 2B). (R)- and (S)-ABS01-113 also displayed very high selectivity for D3R over D2R (217-fold vs. 1060-fold, respectively) (Table S1).

A Plasma and brain concentrations of (S)-ABS01-113 over time; B Plasma and brain concentrations of (R)-ABS01-113 over time; data are expressed as mean ± SD, n = 3, per time point. C Plasma and brain pharmacokinetic parameters of (S)- and (R)-ABS01-113.
(S)- and (R)-ABS01-113 do not alter open-field locomotion
To further determine the functional activity of both ABS enantiomers in vivo, we observed their effects on open-field locomotion. Figure 3A shows that (S)-ABS01-113 alone had no effect on locomotor activity. A two-way ANOVA failed to reveal either a (S)-ABS01-113 treatment main effect (F2,14 = 2.75, p > 0.05) or time × dose interaction (F22,154 = 1.58, p > 0.05) although it revealed a significant time main effect (F11,77 = 16.83, p < 0.001). Post-hoc Tukey’s tests for multiple group comparisons did not reveal significant differences in locomotion after either 10 mg/kg or 30 mg/kg (S)-ABS01-113 administration as compared to the vehicle control group (p > 0.05). Given that (S)-ABS01-113, at 30 mg/kg, appeared to produce an increase in open-field locomotion beginning at 45 min after (S)-ABS01-113 administration (Fig. 3A), we repeated this experiment. We found that (S)-ABS01-113, at the same drug doses, failed to alter locomotion within 2 h after the drug administration (Fig. S1).

A, B (S)- and (R)-ABS01-113 alone failed to alter open-field locomotion; C, D Pretreatment with (S)- or (R)-ABS01-113 decreased heroin-enhanced locomotor activity at lower doses (10 mg/kg). Within-subjects design, n = 8 in each treatment. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with vehicle control.
Figure 3B shows spontaneous locomotor activity in mice treated with the same doses of (R)-ABS01-113. A two-way ANOVA also failed to reveal a (R)-ABS01-113 treatment main effect (F2,14 = 0.33, p > 0.05), although it revealed a significant time main effect (F11,77 = 17.07, p < 0.001) and time × dose interaction (F22,154 = 1.82, p < 0.05). However, post-hoc Tukey’s tests for multiple group comparisons did not detect a significant reduction after 10 mg/kg or 30 mg/kg (R)-ABS01-113 administration as compared to the vehicle group (p > 0.05), suggesting that this D3R antagonist has no effect on locomotion.
(S)- and (R)-ABS01-113 inhibit heroin-induced hyperactivity
Next, we examined whether pretreatment with (S)- or (R)-ABS01-113 alters heroin-induced increase in locomotor activity. We found that both compounds produced a significant reduction in heroin-induced hyperactivity at lower doses (10 mg/kg), but not higher doses (30 mg/kg). A two-way ANOVA for the data shown in Fig. 3C revealed a significant time main effect (F11,77 = 45.29, p < 0.001) and time × dose interaction (F33,231 = 6.81, p < 0.001), but not (S)-ABS01-113 treatment main effect (F3,21 = 2.25, p > 0.05). Post-hoc Tukey’s test for multiple group comparisons revealed a significant reduction in heroin-enhanced locomotion at 10–20 min after 10 mg/kg (S)-ABS01-113 pretreatment compared to the vehicle control group (p < 0.001).
Similarly, a two-way ANOVA for the data shown in Fig. 3D also revealed a significant (R)-ABS01-113 treatment main effect (F3,21 = 3.58, p < 0.05), time main effect (F11,77 = 44.32, p < 0.001), and time × dose interaction (F33,231 = 4.74, p < 0.001). Post-hoc Tukey’s test revealed a significant reduction in heroin-enhanced locomotion at 15 min time point after 10 mg/kg (R)-ABS01-113, administration (p < 0.05).
(S)- and (R)-ABS01-113 inhibit heroin self-administration
We then examined the effects of both ABS enantiomers on intravenous heroin self-administration. Figure 4 shows that pretreatment with (S)-ABS01-113 dose-dependently decreased heroin self-administration maintained by a lower dose (0.0125 mg/kg/infusion) (Fig. 4A), but not by a higher dose (0.05 mg/kg/infusion) (Fig. 4B), of heroin. A two-way ANOVA for repeated measures over (S)-ABS01-113 treatment and drug dose for the data shown in the Fig. 4A indicated a significant (S)-ABS01-113 main effect (F1,7 = 15.49, p < 0.001), dose main effect (F3,21 = 6.89, p < 0.01) and treatment × dose interaction (F3,21 = 3.98, p < 0.05). Post-hoc test for multiple group comparisons revealed a significant reduction after 10 mg/kg and 30 mg/kg (S)-ABS01-113 administration (p < 0.05, p < 0.001). However, a two-way ANOVA for the data shown in Fig. 4B did not reveal significant (S)-ABS01-113 treatment main effect (F1,6 = 0.26, p = 0.63), dose effect (F3,18 = 0.79, p = 0.51) and treatment × dose interaction (F3,18 = 0.94, p = 0.44), suggesting that (S)-ABS01-113 is not effective in attenuating self-administration maintained by a higher heroin dose.

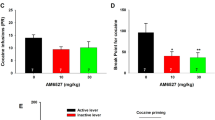
Pretreatment with (S)-ABS01-113 dose-dependently inhibited heroin self-administration maintained by a lower dose (A), but not by a higher dose (B) of heroin; Pretreatment with (R)-ABS01-113 dose-dependently inhibited heroin self-administration maintained by both lower (C) and higher (D) doses of heroin. E, F Original heroin self-administration records, illustrating the patterns of heroin self-administration in the presence or absence of (S)- or (R)-ABS01-113. (n = 7–8 per group) *p < 0.05, **p < 0.01, ***p < 0.001, compared to the vehicle group.
We also examined the effects of (R)-ABS01-113 on heroin self-administration under the same experimental conditions in a separate group of rats. We found that (R)-ABS01-113 is similarly effective in attenuating heroin self-administration maintained by the two doses of heroin (Fig. 4C, D). For a low dose of heroin (0.0125 mg/kg/infusion, Fig. 4C), a two-way ANOVA revealed a significant (R)-ABS01-113 treatment main effect (F1,7 = 15.00, p < 0.01), dose main effect (F3,21 = 5.29, p < 0.01) and treatment × dose interaction (F3,21 = 10.28, p < 0.001). Post-hoc Tukey’s test for multiple groups comparisons indicated a significant reduction in heroin self-administration after 10 or 30 mg/kg (R)-ABS01-113 treatment. Similarly, a two-way ANOVA for the data shown in Fig. 4D revealed a significant dose × treatment interaction (F3,21 = 3.49, p < 0.05). Post-hoc Tukey’s test indicated that (R)-ABS01-113, at 30 mg/kg, significantly inhibited heroin self-administration maintained by 0.05 mg/kg/infusion heroin as compared to vehicle group (Fig. 4D). Figure 4E, F shows the original self-administration records in the presence or absence of the test drugs, illustrating a classical extinction-like pattern of drug seeking, i.e., an initial increase in drug intake, followed by cessation of drug-taking and drug-seeking, suggesting a reduction in heroin’s rewarding effects after D3R blockade.
(S)- and (R)-ABS01-113 do not reduce oral sucrose self-administration
To determine whether the actions observed above are drug- or heroin-specific, we observed the effects of (R)- or (S)-ABS01-113 on oral sucrose self-administration in mice. (R)- or (S)-ABS01-113, at 10 and 30 mg/kg. We found that systemic administration of the same doses that inhibit heroin self-administration failed to alter oral sucrose self-administration (Fig. S2). A one-way RM ANOVA did not reveal a significant (S)-ABS01-113 treatment main effect (Fig. S2A, F2,10 = 1.54, p > 0.05). Similarly, a one-way RM ANOVA failed to reveal a significant (R)-ABS01-113 treatment main effect (Fig. S2B, F2, 14 = 1.48, p > 0.05).
(S)- and (R)-ABS01-113 inhibit heroin-induced reinstatement of drug seeking
Lastly, we examined whether both ABS01-113 enantiomers can block heroin-induced reinstatement of drug-seeking behavior. In this experiment, we first trained rats to self-administer heroin (0.05 mg/kg/infusion) until stable self-administration was achieved as described above. Then the rats underwent extinction training until the heroin-seeking behavior was extinguished (Fig. 5A). On the reinstatement test day, rats received a non-contingent injection of heroin (1 mg/kg, i.p.), which caused a robust reinstatement of responding on the active but not inactive lever in the absence of (S)-ABS01-113 (Fig. 5B). A three-way ANOVA on the active and inactive lever pressing revealed a significant treatment × dose × lever interaction (F2,23 = 4.65, p < 0.05). Interaction comparisons at the level of active lever (Fig. 5B) revealed a significant treatment × dose interaction (F2,23 = 5.56, p < 0.05). Post-hoc multiple group comparisons showed that 30 mg/kg of (S)-ABS01-113 caused a significant reduction in active lever pressing during the reinstatement test as compared to the vehicle pretreatment group (p < 0.05). Interaction comparison for inactive lever pressing (Fig. 5B) showed no significant treatment effect (F1,23 = 3.33, p = 0.08), dose effect (F2,23 = 1.72, p = 0.20), and phase × dose interaction (F2,23 = 2.28; p = 0.12), suggesting that (S)-ABS01-113 selectively reduced drug-associated active, but not inactive, lever pressing.

A General experimental procedures of drug + cue-induced reinstatement of drug seeking. B, C Pretreatment with (S)- or (R)-ABS01-113 dose-dependently inhibited (heroin + cue)-induced reinstatement of heroin-seeking behavior. *p < 0.05, **p < 0.01, ***p < 0.001, compared to the vehicle group.
Figure 5C shows that pretreatment with (R)-ABS01-113 also caused a robust reduction in heroin-triggered reinstatement of drug seeking in a dose dependent manner. A three-way ANOVA on active and inactive lever pressing revealed a significant treatment × dose × lever interaction (F2,23 = 8.07, p < 0.01). Interaction comparison at the level of active lever revealed a significant treatment × dose interaction (F2,23 = 9.55, p = 0.001). A post-hoc comparisons showed that 10 and 30 mg/kg (R)-ABS01-113 caused a significant reduction in active lever pressing during the reinstatement test (p < 0.001). Interaction comparison for inactive lever pressing showed a significant treatment main effect (F1,23 = 4.59; p = 0.043), but did not show dose main effect (F2,23 = 1.81; p = 1.81) and treatment × dose interaction (F2,23 = 1.78; p = 0.19), suggesting that (R)-ABS01-113 selectively reduced heroin-associated active, but not inactive, lever pressing.
Discussion
The major purpose of this study was to further develop novel bitopic D3R ligands with translational potential for the treatment of OUD in humans. Moreover, we were intrigued by the significant enantiomeric differences in functional efficacies at D3R with ABS01-113, which provided the opportunity to compare side-by-side compounds with identical chemical and physical properties, but with a single chiral center in the linking chain. Once thought to only function to optimally separate the primary pharmacophore from the secondary pharmacophore [38], the demonstration that chirality matters and that it effects D3R/D2R selectivity [23, 24, 39] as well as efficacy at D3R is abundantly evident, with ABS01-113 being a prime example. Here we systematically evaluated (S)- and (R)-ABS01-113 both in vitro and in vivo. We identified that (S)-ABS01-113 is a highly potent and selective D3R partial agonist while its (R)-enantiomer is a highly potent and selective D3R antagonist. Both enantiomers displayed excellent brain penetration in rats and both compounds significantly attenuated actions produced by heroin, including heroin-induced hyperactivity, heroin self-administration and (heroin + cue)-induced reinstatement of drug-seeking behavior. The reduction in heroin taking and seeking is unlikely due to nonspecific sedation or locomotor impairment as (S)- or (R)-ABS01-113 neither altered oral sucrose self-administration nor inhibited locomotor behavior. This is consistent with our previous findings with other D3R antagonists/partial agonists (±)-VK4-40 and BAK4-54 [9, 18, but see 10]. This is also consistent with previous reports that the D3R antagonists/partial agonists (BAK4-54 and CAB2-015) significantly inhibited heroin self-administration in wild-type mice, but not in D3-knockout mice [17], and other D3R antagonists (SR-21502 and VK4-116) inhibited heroin-induced CPP, oxycodone self-administration, and oxycodone- or cue-triggered reinstatement of drug-seeking behavior in rats [7, 18]. Together, these findings suggest that both enantiomers deserve further investigation as novel non-opioid pharmacotherapies for OUD.
As stated above, (S)- and (R)-ABS01-113 are the analogs of (S)- and (R)-VK4-40. Both (S)- and (R)-ABS01-113 showed similar pharmacodynamic and pharmacokinetic profiles compared to their parent compounds. For example, both (S)- and (R)-ABS01-113 displayed very high selectivity for D3R over D2R (1060- and 217-fold, respectively), and excellent oral bioavailability and brain penetration, as indicated by the Cmax and Tmax values in blood and brain after intragastric administration. We have previously reported that the (±)-VK4-40 is a low efficacy D3R partial agonist [9, 23, 24], which dose-dependently reduced cocaine reward and relapse to drug-seeking behavior without producing significant unwanted side effects such as abuse potential and locomotor impairment [9]. Further separation of enantiomers indicates that (R)-VK4-40 is a selective D3R antagonist [10] and (S)-VK4-40 is a low efficacy partial D3R agonist [15]. Systemic administration of (R)-VK4-40 dose-dependently inhibited oxycodone self-administration, lowered break-points for oxycodone under a PR schedule, and blocked oxycodone-enhanced brain-stimulation reward [10]. (R)- or (S)-VK4-40 alone also significantly inhibited brain-stimulation reward maintained by optical stimulation of VTA DA neurons [15].
Herein, we show that both (R)- and (S)-ABS01-113 are effective in attenuating heroin self-administration and (heroin + cue)-triggered reinstatement of drug-seeking behavior in rats. Together, these data support further development of both compounds and highlight the consideration of further pursuing efficacious partial D3R agonists, such as (S)-ABS01-113. As partial agonists have proven effective in treating neuropsychiatric disorders such as schizophrenia and bipolar disorder, and substance use disorders are highly prevalent in these patients, we propose the possibility that a D3R partial agonist may be helpful in treating these dual disorders [30, 40] that have historically been most difficult to manage medically.
The mechanisms underlying D3R modulation of opioid reward have been discussed extensively in previous reports [6, 10, 41, 42]. Briefly, opioid reward has been shown to be mediated by activation of mu-opioid receptors located on GABAergic interneurons or afferents in the VTA and substantia nigra pars reticulata (SNr), which subsequently disinhibits (activates) DA neurons in the VTA and substantia nigra pars compacta (SNc) [6, 42, 43], causing an increase in DA release in the striatum. Since D3R has the highest affinity for DA among all 5 subtypes of DA receptors, D3R blockade by selective D3R antagonists or partial agonists would attenuate DA transmission, producing a reduction in opioid reward, without concomitant and untoward D2R antagonist side effects.
In addition, repeated administration of heroin or re-exposure to heroin-related cues also increases DA neuron firing in the VTA and DA release in the NAc and prefrontal cortex [44,45,46,47], suggesting that the mesolimbic DA system may also be involved in reinstatement of drug seeking. This is supported by the finding that opioids or opioid-conditioned cues increase expression of c-fos and other immediate-early genes in the mesolimbic system [48,49,50] and blockade of DA (D1-like) receptors reduces context- and cue-induced reinstatement of heroin seeking [51, 52]. Given that D3R are mostly located on postsynaptic cells [3] and colocalized with D1-receptor-expressing medium-spiny neurons in the striatum [3, 42], it is plausible to hypothesize that blockade of postsynaptic D3R may attenuate opioid- or cue-induced reinstatement of drug-seeking behavior.
We note that both ABS01-113 enantiomers inhibited heroin-induced hyperactivity only at lower doses. The mechanisms underlying this observation are unclear. One possibility is that both enantiomers, at high doses, may bind to unknown off targets, which compromises their actions against heroin-induced hyperactivity.
In summary, in this report, we systematically evaluated two novel D3R-selective enantiomers, (S)- and (R)-ABS01-113 with differing efficacies for their therapeutic potential in the treatment of OUD. We found that (S)-ABS01-113 is a potent, highly selective and efficacious D3R partial agonist. In contrast, (R)-ABS01-113 is a potent and highly selective D3R antagonist. Both enantiomers displayed excellent oral bioavailability and PK profiles, suggesting drug-like properties suitable for further development. Importantly, both enantiomers displayed pharmacological efficacy in attenuating opioid reward and relapse to drug-seeking behavior, despite significant differences in D3R intrinsic activity. These findings suggest that both (S)- and (R)-ABS01-113 deserve further investigation in translational studies for the treatment of OUD. Further, a D3R partial agonist, exemplified by (S)-ABS01-113, might be more effective in patients with dual diagnoses of substance use disorder and other neuropsychiatric disorders such as schizophrenia or bipolar disorder, where dopaminergic receptor antagonists have proven ineffective.
References
Ahmad FB, Cisewski JA, Rossen LM, Sutton P. Provisional drug overdose death counts. National Center for Health Statistics. 2022. https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm.
Novick DM, Salsitz EA, Joseph H, Kreek MJ. Methadone medical maintenance: an early 21st-century perspective. J Addict Dis. 2015;34:226–37.
Newman AH, Xi ZX, Heidbreder C. Current perspectives on selective dopamine D3 receptor antagonists/partial agonists as pharmacotherapeutics for opioid and psychostimulant use disorders. Curr Top Behav Neurosci. 2022.
Newman AH, Grundt P, Nader MA. Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J Med Chem. 2005;48:3663–79.
Keck TM, John WS, Czoty PW, Nader MA, Newman AH. Identifying medication targets for psychostimulant addiction: unraveling the dopamine D3 receptor hypothesis. J Med Chem. 2015;58:5361–80.
Galaj E, Newman AH, Xi ZX. Dopamine D3 receptor-based medication development for the treatment of opioid use disorder: rationale, progress, and challenges. Neurosci Biobehav Rev. 2020;114:38–52.
Galaj E, Manuszak M, Babic S, Ananthan S, Ranaldi R. The selective dopamine D3 receptor antagonist, SR 21502, reduces cue-induced reinstatement of heroin seeking and heroin conditioned place preference in rats. Drug Alcohol Depend. 2015;156:228–33.
de Guglielmo G, Kallupi M, Sedighim S, Newman AH, George O. Dopamine D(3) receptor antagonism reverses the escalation of oxycodone self-administration and decreases withdrawal-induced hyperalgesia and irritability-like behavior in oxycodone-dependent heterogeneous stock rats. Front Behav Neurosci. 2019;13:292.
Jordan CJ, He Y, Bi GH, You ZB, Cao J, Xi ZX, et al. (±)VK4-40, a Novel D(3) R partial agonist, attenuates cocaine reward and relapse in rodents. Br J Pharmacol. 2020.
Jordan CJ, Humburg B, Rice M, Bi GH, You ZB, Shaik AB, et al. The highly selective dopamine D(3)R antagonist, R-VK4-40 attenuates oxycodone reward and augments analgesia in rodents. Neuropharmacology. 2019;158:107597.
You ZB, Bi GH, Galaj E, Kumar V, Cao J, Gadiano A, et al. Dopamine D(3)R antagonist VK4-116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology. 2019;44:1415–24.
Dodds CM, O’Neill B, Beaver J, Makwana A, Bani M, Merlo-Pich E, et al. Effect of the dopamine D3 receptor antagonist GSK598809 on brain responses to rewarding food images in overweight and obese binge eaters. Appetite. 2012;59:27–33.
Murphy A, Nestor LJ, McGonigle J, Paterson L, Boyapati V, Ersche KD, et al. Acute D3 antagonist GSK598809 selectively enhances neural response during monetary reward anticipation in drug and alcohol dependence. Neuropsychopharmacology. 2017;42:1925–6.
Heidbreder CA, Newman AH. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann N Y Acad Sci. 2010;1187:4–34.
Newman AH, Ku T, Jordan CJ, Bonifazi A, Xi ZX. New drugs, old targets: tweaking the dopamine system to treat psychostimulant use disorders. Annu Rev Pharmacol Toxicol. 2021;61:609–28.
Appel NM, Li SH, Holmes TH, Acri JB. Dopamine D3 receptor antagonist (GSK598809) potentiates the hypertensive effects of cocaine in conscious, freely-moving dogs. J Pharmacol Exp Ther. 2015;354:484–92.
Boateng CA, Bakare OM, Zhan J, Banala AK, Burzynski C, Pommier E, et al. High affinity dopamine D3 receptor (D3R)-selective antagonists attenuate heroin self-administration in wild-type but not D3R knockout mice. J Med Chem. 2015;58:6195–213.
You ZB, Gao JT, Bi GH, He Y, Boateng C, Cao J, et al. The novel dopamine D3 receptor antagonists/partial agonists CAB2-015 and BAK4-54 inhibit oxycodone-taking and oxycodone-seeking behavior in rats. Neuropharmacology. 2017;126:190–9.
Newman AH, Kumar V, Shaik, AB. Dopamine D3 receptor selective antagonists/partial agonists; method of making and uses thereof. International Patent Application 2017;E-053-2016/0-PCT-02, NIH0107PCT.
Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–5.
Keck TM, Burzynski C, Shi L, Newman AH. Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv Pharmacol. 2014;69:267–300.
Newman AH, Blaylock BL, Nader MA, Bergman J, Sibley DR, Skolnick P. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol. 2012;84:882–90.
Kumar V, Bonifazi A, Ellenberger MP, Keck TM, Pommier E, Rais R, et al. Highly selective dopamine D3 receptor (D3R) antagonists and partial agonists based on eticlopride and the D3R crystal structure: new leads for opioid dependence treatment. J Med Chem. 2016;59:7634–50.
Shaik AB, Kumar V, Bonifazi A, Guerrero AM, Cemaj SL, Gadiano A, et al. Investigation of novel primary and secondary pharmacophores and 3-substitution in the linking chain of a series of highly selective and bitopic dopamine D(3) receptor antagonists and partial agonists. J Med Chem. 2019;62:9061–77.
Jordan CJ, Humburg BA, Thorndike EB, Shaik AB, Xi ZX, Baumann MH, et al. Newly developed dopamine D(3) receptor antagonists, R-VK4-40 and R-VK4-116, do not potentiate cardiovascular effects of cocaine or oxycodone in rats. J Pharmacol Exp Ther. 2019;371:602–14.
Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, et al. N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J Med Chem. 2011;54:3581–94.
Kumar V, Banala AK, Garcia EG, Cao J, Keck TM, Bonifazi A, et al. Chiral resolution and serendipitous fluorination reaction for the selective dopamine D3 receptor antagonist BAK2-66. ACS Med Chem Lett. 2014;5:647–51.
Ohlsen RI, Pilowsky LS. The place of partial agonism in psychiatry: recent developments. J Psychopharmacol. 2005;19:408–13.
Grunze H, Csehi R, Born C, Barabassy A. Reducing addiction in bipolar disorder via hacking the dopaminergic system. Front Psychiatry. 2021;12:803208.
Peris L, Szerman N. Partial agonists and dual disorders: focus on dual schizophrenia. Front Psychiatry. 2021;12:769623.
Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108.
Knight AR, Misra A, Quirk K, Benwell K, Revell D, Kennett G, et al. Pharmacological characterisation of the agonist radioligand binding site of 5-HT(2A), 5-HT(2B) and 5-HT(2C) receptors. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:114–23.
Han X, He Y, Bi GH, Zhang HY, Song R, Liu QR, et al. CB1 receptor activation on VgluT2-expressing glutamatergic neurons underlies delta(9)-Tetrahydrocannabinol (delta(9)-THC)-induced aversive effects in mice. Sci Rep. 2017;7:12315.
Galaj E, Han X, Shen H, Jordan CJ, He Y, Humburg B, et al. Dissecting the role of GABA neurons in the VTA versus SNr in opioid reward. J Neurosci. 2021;40:8853–69.
Wang XF, Barbier E, Chiu YT, He Y, Zhan J, Bi GH, et al. T394A mutation at the mu opioid receptor blocks opioid tolerance and increases vulnerability to heroin self-administration in mice. J Neurosci. 2016;36:10392–403.
Gao JT, Jordan CJ, Bi GH, He Y, Yang HJ, Gardner EL, et al. Deletion of the type 2 metabotropic glutamate receptor increases heroin abuse vulnerability in transgenic rats. Neuropsychopharmacology. 2018;43:2615–26.
Bi GH, Galaj E, He Y, Xi ZX. Cannabidiol inhibits sucrose self-administration by CB1 and CB2 receptor mechanisms in rodents. Addict Biol. 2020;25:e12783.
Newman AH, Battiti FO, Bonifazi A. 2016 Philip S. Portoghese medicinal chemistry lectureship: designing bivalent or bitopic molecules for G-protein coupled receptors. The whole is greater than the sum of its parts. J Med Chem. 2020;63:1779–97.
Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, et al. N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J Med Chem. 2009;52:2559–70
Laszlovszky I, Barabassy A, Nemeth G. Cariprazine, A Broad-Spectrum Antipsychotic for the Treatment of Schizophrenia: Pharmacology, Efficacy, and Safety. Adv Ther. 2021;38:3652–73.
Jordan CJ, Cao J, Newman AH, Xi ZX. Progress in agonist therapy for substance use disorders: Lessons learned from methadone and buprenorphine. Neuropharmacology. 2019;158:107609.
Galaj E, Xi ZX. Progress in opioid reward research: from a canonical two-neuron hypothesis to two neural circuits. Pharmacol Biochem Behav. 2021;200:173072.
Fields HL, Margolis EB. Understanding opioid reward. Trends Neurosci. 2015;38:217–25.
Wise RA, Leone P, Rivest R, Leeb K. Elevations of nucleus accumbens dopamine and DOPAC levels during intravenous heroin self-administration. Synapse. 1995;21:140–8.
Kiyatkin EA, Rebec GV. Impulse activity of ventral tegmental area neurons during heroin self-administration in rats. Neuroscience. 2001;102:565–80.
Bassareo V, De Luca MA, Di Chiara G. Differential impact of pavlovian drug conditioned stimuli on in vivo dopamine transmission in the rat accumbens shell and core and in the prefrontal cortex. Psychopharmacology. 2007;191:689–703.
Bassareo V, Musio P, Di, Chiara G. Reciprocal responsiveness of nucleus accumbens shell and core dopamine to food- and drug-conditioned stimuli. Psychopharmacology. 2011;214:687–97.
Bontempi B, Sharp FR. Systemic morphine-induced Fos protein in the rat striatum and nucleus accumbens is regulated by mu opioid receptors in the substantia nigra and ventral tegmental area. J Neurosci. 1997;17:8596–612.
Koya E, Spijker S, Voorn P, Binnekade R, Schmidt ED, Schoffelmeer AN, et al. Enhanced cortical and accumbal molecular reactivity associated with conditioned heroin, but not sucrose-seeking behaviour. J Neurochem. 2006;98:905–15.
Liu J, Nickolenko J, Sharp FR. Morphine induces c-fos and junB in striatum and nucleus accumbens via D1 and N-methyl-D-aspartate receptors. Proc Natl Acad Sci USA. 1994;91:8537–41.
Bossert JM, Poles GC, Wihbey KA, Koya E, Shaham Y. Differential effects of blockade of dopamine D1-family receptors in nucleus accumbens core or shell on reinstatement of heroin seeking induced by contextual and discrete cues. J Neurosci. 2007;27:12655–63.
Bossert JM, Wihbey KA, Pickens CL, Nair SG, Shaham Y. Role of dopamine D(1)-family receptors in dorsolateral striatum in context-induced reinstatement of heroin seeking in rats. Psychopharmacology. 2009;206:51–60.
Funding
AHN and ABS are inventors on an NIH patent that covers (S)- and (R)-ABS01-113. All rights are reserved by NIH. This research was supported by the National Institute on Drug Abuse Intramural Research Program (Z1A DA000424), U.S. DOJ/DEA [Interagency agreement D-15-OD-0002] (AJ), Department of Veterans Affairs Merit Review [I01BX002758] and Department of Veterans Affairs Award Senior Research Career Scientist programs [1IK6BX005754] (AJ), and NIH/NIDA [Interagency agreement ADA12013] (AJ). The contents do not represent the views of the U.S. Department of Veterans Affairs, U.S. Department of Justice, Drug Enforcement Administration, or the United States Government.
Author information
Authors and Affiliations
Contributions
EG, Z-XX, AHN, AJ, and AJE designed the experiments. ABS, ESG synthesized (S)- and (R)-ABS01-113. JL and JF performed the PK experiments. JFR, SHB, TLS, JLS, and AJE performed the receptor binding and function experiments. EG, G-HB, BK, and BH performed the behavioral experiments. EG, Z-XX., AJ, AJE, RR, and AHN analyzed the data and prepared the figures. EG, Z-XX, AJ, AJE, and AHN wrote or edited the manuscript and all other coauthors contributed to the final draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Galaj, E., Bi, GH., Klein, B. et al. A highly D3R-selective and efficacious partial agonist (S)-ABS01-113 compared to its D3R-selective antagonist enantiomer (R)-ABS01-113 as potential treatments for opioid use disorder. Neuropsychopharmacol. 47, 2309–2318 (2022). https://doi.org/10.1038/s41386-022-01379-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-022-01379-1
This article is cited by
-
The selective D3Receptor antagonist VK4-116 reverses loss of insight caused by self-administration of cocaine in rats
Neuropsychopharmacology (2024)
-
Effects of selective dopamine D3 receptor partial agonist/antagonists on oxycodone self-administration and antinociception in monkeys
Neuropsychopharmacology (2023)
-
Blockade of the Dopamine D3 Receptor Attenuates Opioids-Induced Addictive Behaviours Associated with Inhibiting the Mesolimbic Dopamine System
Neuroscience Bulletin (2023)
