
Abstract
Studies in post-mortem human brain tissue have associated major depressive disorder (MDD) with cortical transcriptomic changes, whose potential in vivo impact remains unexplored. To address this translational gap, we recently developed a transcriptome-based polygenic risk score (T-PRS) based on common functional variants capturing ‘depression-like’ shifts in cortical gene expression. Here, we used a non-clinical sample of young adults (n = 482, Duke Neurogenetics Study: 53% women; aged 19.8 ± 1.2 years) to map T-PRS onto brain morphology measures, including Freesurfer-derived subcortical volume, cortical thickness, surface area, and local gyrification index, as well as broad MDD risk, indexed by self-reported family history of depression. We conducted side-by-side comparisons with a PRS independently derived from a Psychiatric Genomics Consortium (PGC) MDD GWAS (PGC-PRS), and sought to link T-PRS with diagnosis and symptom severity directly in PGC-MDD participants (n = 29,340, 59% women; 12,923 MDD cases, 16,417 controls). T-PRS was associated with smaller amygdala volume in women (t = −3.478, p = 0.001) and lower prefrontal gyrification across sexes. In men, T-PRS was associated with hypergyrification in temporal and occipital regions. Prefrontal hypogyrification mediated a male-specific indirect link between T-PRS and familial depression (b = 0.005, p = 0.029). PGC-PRS was similarly associated with lower amygdala volume and cortical gyrification; however, both effects were male-specific and hypogyrification emerged in distinct parietal and temporo-occipital regions, unassociated with familial depression. In PGC-MDD, T-PRS did not predict diagnosis (OR = 1.007, 95% CI = [0.997–1.018]) but correlated with symptom severity in men (rho = 0.175, p = 7.957 × 10−4) in one cohort (N = 762, 48% men). Depression-like shifts in cortical gene expression have sex-specific effects on brain morphology and may contribute to broad depression vulnerability in men.
Similar content being viewed by others

Introduction
Major depressive disorder (MDD or ‘depression’) is a common and debilitating psychiatric illness characterized by low mood and anhedonia. With a lifetime prevalence of up to 17% [1], MDD constitutes the leading cause of disability worldwide [2]. Despite the monumental impact of MDD, its biological bases remain incompletely understood.
Converging data from human neuroimaging studies, preclinical experimental models, and post-mortem human brain tissue research suggest depression may be associated with aberrant functioning of a conserved corticolimbic circuit (CLC) [3], which facilitates perception, emotion, and cognition across species. Canonical CLC nodes include the amygdala, hippocampus, and regulatory prefrontal cortical regions. Gene expression studies in post-mortem tissue derived from these and other regions have begun to shed light on the molecular mechanisms that may contribute to the emergence and maintenance of MDD [4,5,6]. The largest gender-balanced post-mortem transcriptome study of the CLC to date is a meta-analysis [4] of eight legacy datasets, including 51 MDD cases and 50 matched controls. This study used robust statistical approaches [7] designed to control for multiple testing and for study-specific confounds, to identify genes and pathways of circuit-wide relevance in both men and women. A total of 566 ‘metaA-MDD’ genes were identified as consistently altered across the dorsolateral prefrontal cortex (dlPFC), subgenual anterior cingulate cortex (sgACC), and the amygdala. Of these genes, 56% were downregulated in individuals with depression, relative to controls. Subsequent pathway analysis revealed associations with biological functions that are consistent with current hypotheses for biological disturbances in MDD, including dysregulated cell death and survival, cell-to-cell signaling, reduced neurotrophic support and impaired GABA function.
Although transcriptomic findings in post-mortem brain tissue provide important information about the molecular pathways whose dysregulation may underlie MDD pathophysiology, they offer little insight into how these microscale processes may impact larger-scale neural structure and function to ultimately contribute to symptom emergence. To address this important gap, our group developed a novel transcriptome-based polygenic risk score (T-PRS) that makes use of common genetic variants to translate molecular changes observed in post-mortem tissue into an in vivo peripheral proxy measure capturing similarity to the MDD cortical transcriptome. We recently mapped this score onto sex-specific patterns of CLC function in a non-clinical sample of young adults [8]. In this prior study, higher T-PRS was associated with elevated reactivity to emotional faces in men, but blunted reactivity to neutral faces in women. The latter was further predictive of subclinical anhedonia [8]. This effect emerged across a network of CLC regions including the sgACC and portions of the dlPFC. Although the T-PRS was uncorrelated with a PRS computed based on results from a Psychiatric Genomics Consortium Genome-Wide Association Study (GWAS) of MDD (PGC-PRS) [9], higher PGC-PRS was similarly associated with blunted reactivity to neutral faces in women, in partially overlapping regions. These findings suggest that molecular shifts toward a depression-like corticolimbic transcriptome may contribute to a sex-specific functional risk phenotype even in the absence of clinically significant depression, and independently of broader genetic risk for MDD indexed by PGC-PRS. However, the impact of these molecular shifts on clinical MDD risk or more stable trait-like measures of brain morphology remains unknown.
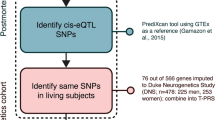
To answer these important questions, we sought to delineate the neuroanatomical signature of T-PRS and its potential link to familial depression risk in a non-clinical sample of young adults participating in the Duke Neurogenetics Study, where we also conducted a comparative analysis examining the neuroanatomical effects of PGC-PRS. To complement and extend these analyses, we further evaluated the association of T-PRS with MDD diagnosis and symptom severity in a large sample of Psychiatric Genomics Consortium MDD (PGC-MDD) participants [9, 10] (see Fig. 1 for study overview).

CT cortical thickness, CSA cortical surface area, DNS Duke Neurogenetics Study, LGI local gyrification index, MDD Major Depressive Disorder, PGC Psychiatric Genomics Consortium, SNPs single nucleotide polymorphisms, T-PRS transcriptome-based polygenic risk score.
To obtain a thorough characterization of the neuroanatomical signature of T-PRS, we examined the effects of T-PRS on volume in subcortical regions, as well as on cortical thickness and surface area. We also examined its effects on local gyrification, a less-studied cortical phenotype that develops in early life and has been linked to cortical complexity, across the cortex. By examining each of these brain-based phenotypes, which have unique genetic origins [11] and developmental and aging-related trajectories [12], we sought to capture links between depression-associated changes in gene expression and distinct variations in brain structure that could indicate atypical neurodevelopment (i.e., variations in cortical surface area or local gyrification) or accelerated aging (i.e., variations in cortical thickness or subcortical volume), both of which have been implicated in the pathophysiology of MDD [13, 14]. Given consistent evidence of sex differences in depression [15, 16] and sex-specificity of T-PRS effects in our prior work [8], we tested associations between T-PRS and brain morphology separately in men and women.
Although we aimed to maintain a discovery component to this study, we held several predictions when testing our main hypotheses that T-PRS would be associated with brain morphology, MDD diagnosis, and symptom severity. Above all, we expected to observe associations between T-PRS and morphology in corticolimbic regions, including those from which T-PRS was originally derived [4]. Second, we expected to observe particularly strong negative associations between T-PRS and cortical surface area, a highly heritable morphological phenotype [11] that has been genetically linked to depression [17, 18].
Materials and methods
Neuroimaging sample
This study used archival data from 482 university students (226 men, 256 women; aged 19.78 ± 1.23 years) who participated in the Duke Neurogenetics Study (DNS). All participants provided informed consent in accordance with Duke University guidelines, and all were in good general health (for full exclusionary criteria, see [19]). Participants were screened for Diagnostic and Statistical Manual of Mental Disorders IV (DSM-IV) Axis I disorders plus select DSM-IV Axis II disorders (Antisocial Personality Disorder, Borderline Personality Disorder) using the electronic Mini International Neuropsychiatric Interview (eMINI) [20], but a current or lifetime diagnosis was not necessarily exclusionary. A total of 114 participants met criteria for at least one lifetime diagnosis (Supplementary Table 1). To examine the potential cross-diagnostic relevance of T-PRS, these participants were included in all initial analyses. All emerging associations were then confirmed in follow-up statistical models controlling for lifetime diagnosis and in a restricted sample excluding those individuals. The Duke University Institutional Review Board approved all study procedures.
We restricted our analyses to participants of non-Hispanic Caucasian descent to match the ethnic background of the post-mortem cohorts used to develop T-PRS. Our sample was reduced to 478 subjects after assessing the presence of relatedness and population stratification, as previously described [8] (see also Supplementary Methods). To account for residual population substructure we used 20 genetic components derived from a multidimensional scaling (MDS) analysis [21] as covariates in all analyses.
Calculation of the polygenic risk scores
Methods for DNA extraction and genotyping are described in [22]. T-PRS was calculated as in our previous study [8] (see also Supplementary Methods, Supplementary Tables 2, 3). Briefly, we used PrediXcan [23] and Genotype-Tissue Expression (GTEx) ‘cortex’ tissue as a reference transcriptome to impute at the individual participant level the relative cortical expression of 76 out of the 566 genes identified in the original post-mortem meta-analysis. Expression levels were not imputed for the remaining genes due to limited statistical power in the reference transcriptome and/or lower expression heritability [23]. Imputed expression values were weighted by direction of effect in the original post-mortem meta-analysis [4], and summed into a single score (T-PRS). To allow side-by-side comparison, we calculated a PRS based on the latest MDD GWAS results with complete summary statistics available publicly (PGC-PRS) [9]. The score was calculated as previously described [8]. We used a variant selection threshold of p < 0.001, as it has been shown to be most predictive of diagnosis [9] and CLC function [8] in prior work.
Acquisition and preprocessing of MRI data
Each participant was scanned using one of the two identical research-dedicated GE MR750 3T scanners at the Duke-UNC Brain Imaging and Analysis Center. This scanner is equipped with high-power high-duty-cycle 50-mT/m gradients at 200 T/m/s slew rate and an eight-channel head coil for parallel imaging at high bandwidth up to 1 MHz. High-resolution 3D T1-weighted structural images were obtained using a 3D Ax FSPGR BRAVO sequence with the following parameters: TE = 3.22 ms, TR = 8.148 ms, FOV = 240 mm, flip angle = 12°, 162 sagittal slices, matrix = 256 × 256, slice thickness = 1 mm with no gap. Magnetic resonance imaging (MRI) data were processed using Freesurfer (http://surfer.nmr.mgh.harvard.edu, version 6.0). Pipelines and parameters are described in detail in Supplemental Methods.
Volume- and surface-based analyses
Sex-specific main effects of T-PRS on subcortical volume were tested with separate linear regressions including age, estimated total intracranial volume (eTIV), and 20 genetic principal components (PCs) as nuisance variables. To account for testing in multiple regions (n = 7), we determined significance using a false discovery rate (FDR) correction threshold of pFDR < 0.05.
Sex-specific main effects of T-PRS on vertex-wise cortical thickness (CT), cortical surface area (CSA), and local gyrification index (LGI) were tested with separate linear regressions including age, eTIV (for CSA only), and 20 genetic PCs as nuisance variables. Regions of interest (ROIs) were identified using a modified cluster-size exclusion method for multiple comparisons correction whereby cluster-wise probability was estimated using a Monte Carlo simulation with a vertex-wise threshold, p < 0.05, and 10,000 repetitions [24]. To account for testing of multiple cortical phenotypes (n = 3), a Bonferroni-adjusted threshold, p ≤ 0.0167 (0.05/3), was used to determine cluster-wise significance for each PRS. All genetic effects on brain structure were confirmed in follow-up analyses additionally partialling out PC-related variability from both PRS.
Links to familial depression
Sex-specific linear regressions, also fitted in R, were used to test associations between self-reported family history of depression (‘familial depression’) and covariate-adjusted morphology in each of the T-PRS-associated regions in the DNS sample. Family history of depression was assessed using the following item, to which participants responded ‘yes’ or ‘no’: “Has anyone in your family ever felt sad, blue, or depressed for most of the time for 2 weeks or more?” For this question, family was defined as ‘immediate, biological family only (biological mother, biological father, biological brothers, or sisters).’ A follow-up question sought to control for the potentially confounding effects of physical illness or grief with the added phrase “excluding times of physical illness or mourning after a death […]”. Participant responses to the first question were used in our main analysis, while responses to the second were used as a covariate in follow-up analyses. Participants were additionally asked to indicate whether they were ‘not confident at all’, ‘reasonably confident’, or ‘very confident’ in their responses (“How confident do you generally feel about the information you have just given about your family members?”). A causal mediation analysis, performed in R with nonparametric bootstrapping, was used to test relationships among T-PRS, T-PRS-related brain structural phenotypes, and familial depression.
PGC analyses
To evaluate a potential link between T-PRS and depression in a clinical sample, we computed T-PRS in 29,340 European individuals (59% women; 12,923 MDD cases and 16,417 controls) from 21 cohorts included in the PGC. Twenty genetic PCs were derived for each individual and used to account for residual population substructure in each cohort. Calculation of the T-PRS was performed separately for each cohort. Logistic regressions, including sex and PCs as covariates, were used to test associations between T-PRS and MDD diagnosis (case/control) in each of the 21 PGC cohorts. Initial analyses only included PCs significantly associated with diagnosis as covariates (as in [9]); however, all results were confirmed when controlling for all 20 PCs. One-sided p-values were obtained for each regression. Results from cohort-specific logistic regressions were used to perform a generic inverse variance meta-analysis using the META package in R. Three meta-analyses were performed: (1) not stratified by sex and including sex as a covariate; (2) stratified by sex (women only); (3) stratified by sex (men only).
Dimensional measures of depressive symptom severity with sufficient sample size (see Supplementary Methods) were available in three PGC cohorts—the Munich Antidepressant Response Signature (MARS; (n = 762, 48% male), the Netherlands Study of Depression and Anxiety (NESDA; n = 1359 cases and 290 controls; 34% male), and the Sequenced Treatment Alternatives to Relieve Depression (STAR*D; n = 932, 40% male). The clinician-administered Hamilton Depression Rating Scale (HDRS, 21-item version) was only available in the MARS cohort, while the other two studies used self-report measures: the Inventory of Depressive Symptoms (IDS) and the Quick Inventory of Depressive Symptoms (QIDS), respectively. General linear models in R were used to test the main effect of T-PRS on depressive symptom severity separately in men and women.
Results
Sample demographics
There was a trend-level association between sex and T-PRS in our neuroimaging sample, where men had slightly higher T-PRS than women (p = 0.068, adjusted for 20 PCs). Interestingly, the opposite pattern was observed in a meta-analysis across all PGC cohorts, where T-PRS was higher in women (SMD = 0.032, 95% CI = [0.009–0.056]; Supplementary Fig. 1). Notably, any potentially confounding effects of these sex differences are precluded by our analytic approach, wherein all analyses are conducted and reported separately in men and women.
Neuroanatomical signature of T-PRS vs. PGC-PRS
We identified partially convergent and highly sex-specific neuroanatomical signatures of T-PRS and PGC-PRS on both subcortical and cortical brain morphology. Subcortical volume analyses revealed that higher T-PRS was strongly associated with lower amygdala volume in women (t = −3.478, p = 0.001, pFDR = 0.014, corrected across all subcortical regions). No significant effects emerged in other subcortical regions in either sex (Table 1). Intriguingly, PGC-PRS was also strongly and specifically associated with lower amygdala volume, however, this effect emerged only in men (t = −2.832, p = 0.005, pFDR = 0.035).
Cortical morphology analyses revealed that T-PRS was not associated with cortical thickness and cortical surface area in either sex. However, it was associated with sex-specific patterns of vertex-wise local gyrification (Fig. 2, Supplementary Table 4). In women, higher T-PRS was associated with lower gyrification (max ≤ −2.797, cluster-wise p ≤ 0.005) across several frontoparietal regions, including clusters with peak vertices in the left rostral middle frontal cortex (cluster #1w), bilateral paracentral cortex (clusters #2w and #4w), and right caudal middle frontal cortex (cluster #5w), as well as one cluster in the left fusiform cortex (cluster #3w). In men, higher T-PRS was associated with lower gyrification in two clusters (max ≤ −2.430, cluster-wise p ≤ 0.001), with peak vertices in the left rostral middle frontal cortex (cluster #1m) and right lingual cortex (cluster #3m). T-PRS was also associated with higher gyrification in three clusters (max ≥ 2.310, cluster-wise p ≤ 0.016), with peak vertices in the right supramarginal cortex (cluster #2m), the right middle temporal cortex (cluster #4m), and the bank of the superior temporal sulcus (cluster #5m). Intriguingly, there was substantial overlap between cluster #1 in men and cluster #1 in women, both of which had peak vertices in the left rostral middle frontal cortex and were negatively associated with T-PRS. There was no additional overlap between T-PRS-associated clusters in men and women.

T-PRS was associated with gyrification in 5 clusters in men (labeled 1m–5m, shown in red) and 5 clusters in women (labeled 1w–5w, shown in yellow). In men, these associations were negative in two clusters (clusters 1m, 3m) and positive in the remaining three (clusters 2m, 4m, 5m). Associations were negative in all clusters in women (1w, 2w, 3w, 4w, 5w). LGI local gyrification index, lh left hemisphere, rh right hemisphere.
Similarly to the T-PRS, PGC-PRS was most strongly associated with gyrification in men, however, this effect emerged in three clusters that did not overlap with those associated with T-PRS (Fig. 3, Supplementary Table 4). Specifically, higher PGC-PRS was associated with lower gyrification in the left lateral occipital (cluster #1), left superior parietal (cluster #2), and right fusiform cortex (cluster #3). Unlike the T-PRS, PGC-PRS was not associated with gyrification in women; however, it did show a female-specific association with higher cortical thickness in a single cluster near the temporal pole (Fig. 3, cluster #1w, Supplementary Table 4). No effects of PGC-PRS on surface area were identified in either sex.

A Clusters in which PGC-PRS was negatively associated with LGI in men (clusters 1–3, shown in green): |max| = 2.071–3.489, cluster-wise p ≤ 0.001. Peak vertices were located in the left lateral occipital cortex (1), left superior parietal cortex (2), and right fusiform cortex (3). B Cluster in which PGC-PRS was positively associated with cortical thickness in women: max = 3.310, cluster-wise p = 0.015, peak vertex in the right temporal pole.
The effects of both PRS on brain morphology remained significant when tested with a follow-up linear regression including psychiatric diagnosis as an additional covariate (p ≤ 0.022, Supplementary Table 5). Almost all effects also remained significant when tested in a restricted sample, excluding participants with a lifetime psychiatric diagnosis (n = 163 men, n = 198 women; p ≤ 0.024, except T-PRS-associated cluster #5 m: p = 0.132; and PGC-PRS-associated cluster #2 in men: p = 0.072), Supplementary Table 5). Highlighting the specificity of the results to each PRS, post hoc analyses showed no association between T-PRS and gyrification/thickness in PGC-PRS-associated clusters, and vice versa (p > 0.21).
Neuroanatomical link between T-PRS and familial depression
Familial depression was associated with morphology in one T-PRS-associated region in men, but not in women (Supplementary Table 6). Specifically, in men, familial depression, like T-PRS, was negatively associated with gyrification in the left rostral middle frontal cortex (cluster #1m: t = −2.688, p = 0.007). This association remained significant when retested in a restricted sample (n = 218), excluding participants reporting low confidence in their family history questionnaire responses (t = −2.598, p = 0.010) and when controlling for potentially confounding effects of physical illness or grief in the family (full sample: t = −2.273, p = 0.024; high-confidence sample: t = −2.251, p = 0.025). Moreover, this cluster formed part of a broader region identified in a follow-up vertex-wise analysis, testing main effects of familial depression on LGI, independent of T-PRS (Fig. 4A, B). This analysis revealed widespread associations between familial depression and hypogyrification in men (max ≤ −2.354, cluster-wise p ≤ 0.028), but none in women. While there was no direct effect of T-PRS on familial depression (p = 0.30), a causal mediation analysis further indicated that hypogyrification in the lateral frontal region in men mediates an indirect link between T-PRS and family history of depression in this otherwise healthy sample (Fig. 4C). PGC-PRS-associated clusters showed no link to familial depression (p > 0.30; Supplementary Table 6).

A Clusters in which familial depression was a significant predictor of vertex-wise local gyrification in men |(max| = 2.354–4.836, cluster-wise p ≤ 0.028). B Overlap with left rostral middle frontal cluster, in which T-PRS was negatively associated with LGI in men. C Indirect effect of T-PRS on broad depression risk, indexed by familial history of depression, in men. Regression coefficients are unstandardized. The 95% confidence interval is in brackets. LGI local gyrification index; p < 0.05 (*), p < 0.01 (**).
T-PRS association with MDD diagnosis and symptom severity
Meta-analyses including results from all 21 PGC cohorts did not reveal links between T-PRS and MDD diagnosis in the entire sample (OR = 1.007, 95% CI = [0.997–1.018]) or in the sex-stratified subsamples (women: OR = 1.009, 95% CI = [0.996–1.023]; men: OR = 1.004, 95% CI = [0.987–1.022]) (Supplementary Figs. 2–4, Supplementary Table 7). However, T-PRS showed a male-specific association with greater symptom severity in the only cohort where the clinician-administered HDRS was available (men: rho = 0.175, p = 7.957 × 10−4, n = 363; women: rho = 0.024, p = 0.638, n = 399; Supplementary Fig. 5). This association remained when controlling for 20 genetic PCs (β = 0.155, 95% CI = [0.017–0.293], p = 0.028). In the same cohort, T-PRS was nominally associated with increased risk for MDD in men (MARS 1: OR = 1.073, p = 0.038), but not women (OR = 0.991, p = 0.607). There were also no associations between T-PRS and symptom severity in the cohorts with self-report measures (p > 0.1; Supplementary Fig. 5).
Discussion
In this study, we set out to identify the neuroanatomical and clinical correlates of a novel polygenic risk score (T-PRS) that captures common variants biasing gene expression toward a depression-like corticolimbic transcriptome. T-PRS had a distinct sex-specific neuroanatomical signature in a well-characterized non-clinical sample, where it was associated with lower amygdala volume and hypogyrification in frontoparietal regions in women. In men, it was associated with lower gyrification in medial occipital and higher gyrification in temporal regions. Hypogyrification in the lateral prefrontal region was shared between men and women and showed a male-specific association with familial depression. These patterns were distinct from those associated with traditional measures of polygenic risk for MDD (PGC-PRS). Although T-PRS was not a significant predictor of depression diagnosis in a large-scale clinical sample of 21 PGC-MDD cohorts, it was associated with clinician-assessed symptom severity in men with MDD in one of these cohorts. Together, these results show for the first time that common functional variants that partially mimic the transcriptomic signature of MDD are associated with structural brain changes detectable in otherwise healthy samples, where they may index novel sex-specific broad depression risk pathways distinct from those associated with genetic variants directly linked to syndromal MDD in vivo.
Consistent with our prior work, T-PRS was uncorrelated with PGC-PRS [8] and the two scores showed both convergent and distinct phenotypic effects. Similarly to T-PRS, PGC-PRS was associated with lower amygdala volume and reduced cortical gyrification. However, unlike the pattern observed in T-PRS analyses, both PGC-PRS effects were male-specific and gyrification effects emerged in distinct regions spanning parietal and temporo-occipital cortices, rather than prefrontal cortex. PGC-PRS also showed a female-specific link to greater cortical thickness in a temporal cluster. These neuroanatomical patterns are partially consistent with our previously reported sex-specific effects of both PRS on brain function during a perceptual processing task in an overlapping sample [8]. In this prior work, we similarly identified stronger effects of T-PRS on lateral prefrontal regions across both sexes, while PGC-PRS more strongly impacted temporal and occipital regions. The broad phenotypic convergence between the two PRS suggests that genetically indexed cortical transcriptomic similarity to MDD may influence brain structure via mechanisms partially shared with those associated with broad genetic vulnerability to MDD (e.g., altered synaptic function). The divergence in sex- and region-specificity may reflect the distinct molecular pathway enrichment patterns and broader transcriptomic contexts of each PRS.
Although we did not identify effects of either PRS on surface area as predicted, consistent with our preliminary hypotheses, neuroanatomical patterns associated with higher T-PRS did show significant spatial overlap with corticolimbic regions, including the amygdala and dlPFC, from which T-PRS is derived. Moreover, these findings are partially consistent with previous imaging studies in depression. For example, reduced local gyrification in mid-frontal cortical regions has been reported in depressed individuals, where it is proportional to the number of prior depressive episodes [25], which in turn has been linked to greater heritability [26]. The latter finding is particularly noteworthy given that we observed links between T-PRS and reduced prefrontal gyrification, including in regions overlapping the dlPFC, in both men and women. Likewise, although the literature on depression-associated changes in amygdala volume is mixed, reduced amygdala volume has been demonstrated in unmedicated patients, relative to healthy controls [27], and smaller amygdala volume has been linked to more depressive symptoms in young adults from a large-scale population-based study [28]. A trend toward a smaller amygdala volume also emerged in individuals with early-onset depression (<21 years old) included in the largest to date case-control meta-analysis of subcortical volumes in MDD [29].
Despite this convergence, the effects of either PRS did not generally recapitulate the neuroanatomical signature of MDD identified in recent ENIGMA-MDD consortium large-scale meta-analyses. These analyses, which did not explicitly assess gyrification, associated the disorder with smaller hippocampal volume [29] and age-dependent reductions in frontotemporal cortical thickness [18] as well as cortical surface area in frontal, visual, somatosensory, and somatomotor regions [18]. Since smaller hippocampal volume was primarily observed in individuals with recurrent MDD [29], we may not have detected any prominent effects on this structure due to the fact that we used a relatively healthy non-clinical sample. Given that there is evidence of genetic correlation between cortical surface area and MDD based on a recent ENIGMA GWAS [17], and that surface area is phenotypically correlated with gyrification [12], it is possible that differences in cortical folding reminiscent of those associated with either or both PRS would emerge if modeled explicitly in ENIGMA’s large-scale case-control meta-analyses. It is also possible that the specific genetic variants captured by T-PRS exert a unique or particularly meaningful influence on cortical folding patterns, while the primarily non-overlapping genetic variants identified in the ENIGMA GWAS [17] specifically shape the degree of cortical expansion independent of gyrification.
Cortical gyrification develops in early life, being nearly complete by the age of two, when differential rates of tissue growth give rise to cortical folding, thereby increasing the cortical surface area, and, by extension, the number of neurons in a limited cranial volume [30, 31]. This patterning is thought to optimize connectivity between adjacent regions [32], and remains relatively stable in adulthood [12]. Thus, cortical gyrification can serve as an index of early brain development, and it can convey information about disruptions to neurodevelopmental processes whose effects may persist into adulthood [33]. The phenotype specificity of our findings suggests a uniquely impactful role of depression-like cortical gene expression in early life, when associated genetic and non-genetic risk factors could contribute to alterations or disruptions in cortical expansion and subsequent cortical folding. Reduced neurotrophic support and/or alterations in biological functions related to cell death and survival as well as cell-to-cell signaling, each of which was associated with genes in T-PRS [4], could contribute to atypical neurodevelopment during this critical period. In turn, these variations in cortical folding could contribute to structural dysconnectivity, subtle but widespread evidence of which has been reported in MDD [34]. By extension, the site-specificity of our findings suggests a uniquely impactful role of depression-like cortical gene expression on structural connectivity within certain regions, namely the left dlPFC, stimulation of which has been shown to modulate resting-state functional connectivity within a meso-cortico-limbic network and has been the primary focus of neuromodulatory treatment of depression [35].
Our PGC analyses indicated that, among men with MDD, higher T-PRS was associated with greater clinician-assessed depressive symptom severity in one large cohort, where T-PRS also predicted diagnosis. Despite this link, we did not identify an association with self-reported depressive symptom severity in two other cohorts or with diagnosis in the full PGC sample. The sparsity of these association signals is readily attributable to the fact that, rather than explicitly modeling overall genetic risk for MDD, our primary aim was to delineate an in vivo neuroanatomical signature associated with depression-related molecular changes identified post-mortem, using peripheral cis-eQTL SNPs only as a proxy measure. Indeed, most depression-associated changes identified in the post-mortem brain transcriptome are likely not the result of common cis-eQTL SNPs, which only capture an average 10% of total gene expression variability [36], but rather stem from environmental risk factors or are themselves a consequence of the disease. Therefore, even though variability in the expression of a particular gene may be associated with depression in the post-mortem brain, this association may not be driven by common genetic variants. Although we hypothesized that more proxy alleles that mimic the depression-associated cortical transcriptome would be associated with elevated MDD risk, our focus was on delineating the impact of these variants on in vivo brain structure to attain an improved mechanistic understanding of depression-related pathophysiological changes in neuroanatomy, which may or may not be genetically driven in the context of the disease.
Further consistent with the differences between our analytic strategy and more conventional genetic associations approaches, the genes included in our T-PRS are also distinct from those emerging from transcriptome-wide association studies (TWAS) of depression [37,38,39]. Unlike our approach, which centers on the MDD cortical transcriptome, TWAS use MDD GWAS results as a starting point and leverage brain tissue expression data to prioritize potentially causal genes, irrespective of whether these genes have been found to be transcriptionally altered in case-control studies of post-mortem brain tissue. The two approaches thus offer distinct and complementary perspectives on molecular pathways of MDD risk and depression pathophysiology. Given disorder heterogeneity and our focus on the CLC, future studies in samples allowing more phenotypic precision (e.g., via the identification of distinct MDD subtypes) may foster further insight and synergy between these approaches.
The sex-specificity of our findings is intriguing and warrants further consideration. Studies suggest MDD may be associated with markedly distinct [5], and even opposite transcriptomic signatures [40] in men and women. The meta-analytic approach that defined the gene list used for T-PRS development was explicitly designed to capture agreement across a gender-balanced sample, which may have limited the initial gene list but may also explain why we detected effects in both men and women. It is still likely that depression-associated genes shared between the sexes are further embedded in a unique sex-specific transcriptomic context, which is not directly measured by the T-PRS (or PGC-PRS) but whose effects are nonetheless reflected in the divergent downstream phenotypic profiles identified in each sex. To provide additional insight into these pathways, future research should seek to explicitly model molecular effects that are sex-specific (i.e., via sex-specific T-PRS) separately from those that may be shared between the sexes.
This study has several limitations. First, we did not observe a link between T-PRS and MDD diagnosis in the full PGC-MDD sample. It is important to recall, however, that the T-PRS was developed as a translational tool to assess the impact of depression-associated cortical transcriptomic changes on in vivo brain structure and function, rather than to capture broad genetic contributions to MDD; hence its primary aim was achieved nonetheless. Second, we were only able to reliably impute cortical gene expression of a subset of the original metaA-MDD genes. This partially reflects a limitation of PrediXcan and other cis-eQTL-based approaches, whose predictive power is limited by the sample size of the reference dataset and the overall heritability of individual gene expression levels. Despite this limitation, however, PrediXcan uses a robust machine learning approach to impute expression based on multiple SNPs, hence we have high confidence in the genes we were able to impute. Third, our initial gene list was based on a study using microarray gene expression technology, which shows good, though not complete, agreement with more contemporary RNA sequencing techniques (RNASeq) [41]. Future studies using RNASeq may help detect additional relevant genes and improve the predictive power of T-PRS-like approaches. Relatedly, since the original case-control comparisons adopted a robust analytic technique specifically designed to account for potential confounds in paired designs [7], it is unlikely but possible that residual confounding effects of medication use in the MDD group remain and may be better addressed in future studies employing larger samples with a variety of medication exposures. Finally, we restricted our analyses to participants of non-Hispanic European ancestry to match the demographic characteristics of the GTEx reference dataset and the original case-control meta-analysis the T-PRS derives from [4]. While this decision likely increased our ability to detect statistically significant effects, it also limits the generalizability of our findings to other ethnic groups. Future work should incorporate larger multi-ethnic samples, including MDD cases and controls covering the full range of symptom severity, in order to improve generalizability across ethnicities and facilitate further assessment of clinical relevance.
Despite these limitations, our results support the translational and partial clinical validity of a recently developed transcriptome-based polygenic risk score (T-PRS) indexing “depression-like” cortical gene expression changes previously only accessible via post-mortem tissue analysis. We provide strong evidence of sex-specific effects of T-PRS on the volume of the amygdala, a central corticolimbic node, and cortical gyrification, variations in which could indicate atypical neurodevelopment and contribute to depression-associated connectivity deficits. At the same time, our results highlight the complexity of depression biology and the relatively limited role that common genetic variation may play in shaping the transcriptomic signature of MDD and any downstream intermediate phenotypes of the disorder. Given the discovery component of this study, replication is critical, as is further examination of the aforementioned sex differences and their possible developmental and clinical implications, along with the possible environmental factors that may more strongly modulate the molecular pathways that lead to MDD. To that end, future work should explore the extent to which effects of T-PRS on brain structure and MDD risk may be developmentally mediated or moderated by experiential factors and therefore possibly amenable to early intervention and prevention efforts crucial for reducing disease burden.
References
Üstün TB, Ayuso-Mateos JL, Chatterji S, Mathers C, Murray CJL. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184:386–92.
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602.
Banasr M, Sanacora G, Esterlis I. Macro- and microscale stress-associated alterations in brain structure: translational link with depression. Biol Psychiatry. 2021;16:2021 https://doi.org/10.1016/j.biopsych.2021.04.004.
Ding Y, Chang L-C, Wang X, Guilloux J-P, Parrish J, Oh H, et al. Molecular and genetic characterization of depression: overlap with other psychiatric disorders and aging. Molecular Neuropsychiatry. 2015;1:1–12.
Labonté B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex-specific transcriptional signatures in human depression. Nat Med. 2017;23:1102–11.
Ramaker RC, Bowling KM, Lasseigne BN, Hagenauer MH, Hardigan AA, Davis NS, et al. Post-mortem molecular profiling of three psychiatric disorders. Genome Med. 2017;9:72.
Wang X, Lin Y, Song C, Sibille E, Tseng GC. Detecting disease-associated genes with confounding variable adjustment and the impact on genomic meta-analysis: with application to major depressive disorder. BMC Bioinformatics. 2012;13:52.
Mareckova K, Hawco C, Dos Santos FC, Bakht A, Calarco N, Miles AE, et al. Novel polygenic risk score as a translational tool linking depression-related changes in the corticolimbic transcriptome with neural face processing and anhedonic symptoms. Transl Psychiatry. 2020;10:1–10.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81.
Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Panizzon MS, Fennema-Notestine C, Eyler LT, Jernigan TL, Prom-Wormley E, Neale M, et al. Distinct genetic influences on cortical surface area and cortical thickness. Cereb Cortex. 2009;19:2728–35.
Hogstrom LJ, Westlye LT, Walhovd KB, Fjell AM. The structure of the cerebral cortex across adult life: age-related patterns of surface area, thickness, and gyrification. Cereb Cortex. 2013;23:2521–30.
Hagan CC, Graham JME, Wilkinson PO, Midgley N, Suckling J, Sahakian BJ, et al. Neurodevelopment and ages of onset in depressive disorders. Lancet Psychiatry. 2015;2:1112–6.
Sibille E. Molecular aging of the brain, neuroplasticity, and vulnerability to depression and other brain-related disorders. Dialogues Clin Neurosci. 2013;15:53.
Kessler RC. Epidemiology of women and depression. J Affect Disord. 2003;74:5–13.
Seney ML, Sibille E. Sex differences in mood disorders: perspectives from humans and rodent models. Biol Sex Differ. 2014;5:1–10.
Grasby KL, Jahanshad N, Painter JN, Colodro-Conde L, Bralten J, Hibar DP, et al. The genetic architecture of the human cerebral cortex. Science. 2020;367.
Schmaal L, Hibar DP, Sämann PG, Hall GB, Baune BT, Jahanshad N, et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol Psychiatry. 2017;22:900–9.
Nikolova YS, Knodt AR, Radtke SR, Hariri AR. Divergent responses of the amygdala and ventral striatum predict stress-related problem drinking in young adults: possible differential markers of affective and impulsive pathways of risk for alcohol use disorder. Mol Psychiatry. 2016;21:348–56.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (MINI): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59:22–33.
Li Q, Yu KAI. Improved correction for population stratification in genome‐wide association studies by identifying hidden population structures. Genetic Epidemiol. 2008;32:215–26.
Nikolova Y, Iruku SP, Lin C-W, Conley ED, Puralewski R, French B, et al. FRAS1-related extracellular matrix 3 (FREM3) single-nucleotide polymorphism effects on gene expression, amygdala reactivity and perceptual processing speed: an accelerated aging pathway of depression risk. Front Psychol. 2015;6:1377.
Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091.
Hagler DJ Jr, Saygin AP, Sereno MI. Smoothing and cluster thresholding for cortical surface-based group analysis of fMRI data. Neuroimage. 2006;33:1093–103.
Depping MS, Thomann PA, Wolf ND, Vasic N, Sosic-Vasic Z, Schmitgen MM, et al. Common and distinct patterns of abnormal cortical gyrification in major depression and borderline personality disorder. Eur Neuropsychopharmacol. 2018;28:1115–25.
Kendler KS, Ohlsson H, Lichtenstein P, Sundquist J, Sundquist K. The genetic epidemiology of treated major depression in Sweden. Am J Psychiatry. 2018;175:1137–44.
Hamilton JP, Siemer M, Gotlib IH. Amygdala volume in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Mol Psychiatry. 2008;13:993–1000.
Daftary S, Van Enkevort E, Kulikova A, Legacy M, Brown ES. Relationship between depressive symptom severity and amygdala volume in a large community-based sample. Psychiatry Res Neuroimaging. 2019;283:77–82.
Schmaal L, Veltman DJ, van Erp TGM, Sämann PG, Frodl T, Jahanshad N, et al. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry. 2016;21:806–12.
Armstrong E, Schleicher A, Omran H, Curtis M, Zilles K. The ontogeny of human gyrification. Cereb Cortex. 1995;5:56–63.
Rakic P. Evolution of the neocortex: a perspective from developmental biology. Nat Rev Neurosci. 2009;10:724–35.
Klyachko VA, Stevens CF. Connectivity optimization and the positioning of cortical areas. Proc Natl Acad Sci USA. 2003;100:7937–41.
Mareckova K, Miles A, Andryskova L, Brazdil M, Nikolova YS. Temporally and sex-specific effects of maternal perinatal stress on offspring cortical gyrification and mood in young adulthood. Hum Brain Mapp. 2020;3:2020 https://doi.org/10.1002/hbm.25163. October
van Velzen LS, Kelly S, Isaev D, Aleman A, Aftanas LI, Bauer J, et al. White matter disturbances in major depressive disorder: a coordinated analysis across 20 international cohorts in the ENIGMA MDD working group. Mol Psychiatry. 2020;25:1511–25.
Tik M, Hoffmann A, Sladky R, Tomova L, Hummer A, de Lara LN, et al. Towards understanding rTMS mechanism of action: stimulation of the DLPFC causes network-specific increase in functional connectivity. Neuroimage. 2017;162:289–96.
Grundberg E, Small KS, Hedman ÅK, Nica AC, Buil A, Keildson S, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–9.
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245–52.
Dall’Aglio L, Lewis CM, Pain O. Delineating the genetic component of gene expression in major depression. Biol Psychiatry. 2021;89:627–36.
Li X, Su X, Liu J, Li H, Li M. 23andMe Research Team, et al. Transcriptome-wide association study identifies new susceptibility genes and pathways for depression. Transl Psychiatry. 2021;11:306.
Seney ML, Huo Z, Cahill K, French L, Puralewski R, Zhang J, et al. Opposite molecular signatures of depression in men and women. Biol Psychiatry. 2018;84:18–27.
Miller JA, Menon V, Goldy J, Kaykas A, Lee C-K, Smith KA, et al. Improving reliability and absolute quantification of human brain microarray data by filtering and scaling probes using RNA-Seq. BMC Genomics. 2014;15:154.
Funding
AEM is supported by a CAMH Discovery Fund Postdoctoral Fellowship. ARH received support from NIH grants R01DA033369 and R01AG049789. YSN is supported by a Koerner New Scientist Award administered by the CAMH Foundation, a NARSAD Young Investigator Grant from the Brain & Behavioral Research Foundation, and a Discovery Grant from the Canadian Natural Sciences and Engineering Research Council (NSERC). The PGC has received major funding from the US National Institute of Mental Health (5 U01MH109528-03).
Author information
Authors and Affiliations
Consortia
Contributions
AEM contributed to study design, conducted all neuroimaging analyses, and wrote the manuscript with guidance from YSN. FCD performed the genetic data processing, conducted the analyses on PGC data, and contributed to manuscript editing. ES contributed to study design and manuscript editing. EMB, MER, AMM, MJA, GP, EC, MP, BTB, KOS, CML, LAJ, IJ, RU, JWS, RHP, DFL, JBP, MMW, JS, GL, BWJHP, DIB, and SPH contributed with data to the PGC-MDD working group and revising the final version of the manuscript. The PGC-MDD working group provided data access and analytic support for the case-control analyses. ARH designed and conducted the parent protocol of the Duke Neurogenetics Study. YSN developed the study concept, oversaw all analyses and all stages of manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Miles, A.E., Dos Santos, F.C., Byrne, E.M. et al. Transcriptome-based polygenic score links depression-related corticolimbic gene expression changes to sex-specific brain morphology and depression risk. Neuropsychopharmacol. 46, 2304–2311 (2021). https://doi.org/10.1038/s41386-021-01189-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01189-x
This article is cited by
-
Transcriptomic risk scores for attention deficit/hyperactivity disorder
Molecular Psychiatry (2023)
-
A Comprehensive Overview of the Role of Visual Cortex Malfunction in Depressive Disorders: Opportunities and Challenges
Neuroscience Bulletin (2023)
