
Abstract
Skeletal muscle tumors are traditionally classified as rhabdomyoma or rhabdomyosarcoma. We have identified an unusual adult rhabdomyoblastic tumor not clearly corresponding to a previously described variant of rhabdomyoma or rhabdomyosarcoma, characterized by a very striking proliferation of non-neoplastic histiocytes, obscuring the underlying tumor. Ten cases were identified in nine males and one female with a median age of 43 years (range 23–69 years). Tumors involved the deep soft tissues of the trunk (N = 4), lower limbs (N = 4), and neck (N = 2). Tumors were well-circumscribed, nodular masses, frequently surrounded by a fibrous capsule containing lymphoid aggregates and sometimes calcifications. Numerous foamy macrophages, multinucleated Touton-type giant cells, and sheets/fascicles of smaller, often spindled macrophages largely obscured the underlying desmin, MyoD1, and myogenin-positive rhabdomyoblastic tumor. Cases were wild type for MYOD1 and no other mutations or rearrangements characteristic of a known subtype of rhabdomyoma or rhabdomyosarcoma were identified. Two of four cases successfully analyzed using a next-generation sequencing panel of 170 common cancer-related genes harbored inactivating NF1 mutations. Next-generation sequencing showed no gene fusions. Clinical follow (nine patients; median 9 months; mean 23 months; range 3–124 months) showed all patients received wide excision; four patients also received adjuvant radiotherapy and none received chemotherapy. At the time of last follow-up, all patients were alive and without disease; no local recurrences or distant metastases occurred. We hypothesize that these unusual tumors represent rhabdomyoblastic tumors of uncertain malignant potential. Possibly over time they should be relegated to a new category of skeletal muscle tumors of intermediate (borderline) malignancy.
Similar content being viewed by others
Introduction
It is now well-recognized that many soft tissue tumors cannot be neatly categorized as simply “benign” or “malignant”. Thus, the current WHO Classification of Tumors of Soft Tissue and Bone recognizes an “intermediate” (borderline) category for several types of soft tissue tumors (e.g., atypical lipomatous tumor in adipocytic tumors and hemangioendothelioma in endothelial tumors) [1]. Some soft tissue tumors are by definition considered to be of intermediate malignancy, either because of their capacity for locally aggressive growth (e.g., desmoid-type fibromatosis and hemosiderotic fibrolipomatous tumor) or because of their low but unpredictable risk of distant metastasis (e.g., angiomatoid fibrous histiocytoma and ossifying fibromyxoid tumor) [1]. In other soft tissue neoplasms, such as glomus tumors [2] and perivascular epithelioid cell tumors [3], classification systems have been developed that not only predict clinically benign and clinically malignant tumors with reasonable sensitivity and specificity, but also classify subsets of such tumors as having “uncertain malignant potential,” usually reflecting limited clinical data. Finally, selected soft tissue tumors, such as gastrointestinal stromal tumors [4] and solitary fibrous tumors [5], are best viewed from a “risk assessment” perspective rather than as benign or malignant.
Tumors showing skeletal muscle differentiation, however, continue to be dichotomized as either benign rhabdomyomas or fully malignant rhabdomyosarcomas (RMSs). Rhabdomyomas are rare tumors typically involving the head/neck [6, 7] or genital region [8], consisting of non-infiltrative proliferations of well-differentiated skeletal muscle with epithelioid (adult rhabdomyoma) or spindled (fetal and genital rhabdomyoma) morphology. RMSs are more common, occurring most often in children, and can be classified as embryonal, alveolar, pleomorphic, and spindle cell/sclerosing subtypes [1, 9]. Skeletal muscle tumors of intermediate malignancy or of uncertain malignant potential are not currently recognized.
Over the past several years, we have seen in consultation a small number of very unusual tumors of skeletal muscle origin in adults, which do not clearly correspond to a previously described variant of rhabdomyoma or RMS, showing distinctive morphological features including a striking histiocytic infiltrate. Although we originally regarded these tumors as variants of RMS, we have come to appreciate that these tumors lack the typical features of RMS, such as diffuse infiltration or mitotic activity, and instead show morphological features suggestive of a slowly growing, indolent tumor. We therefore undertook this study, in order to more fully elucidate the clinicopathological features of these rare tumors, herein provisionally termed “histiocyte-rich rhabdomyoblastic tumors”, and to define their proper place in the classification of skeletal muscle neoplasms.
Materials and methods
Case selection
Approval for the study was granted by the Institutional Review Boards of Mayo Clinic, Emory University, St. Jude Children’s Research Hospital, Erlangen University and Chang Gung Hospital. The initial group of seven cases were identified through search of the Mayo Clinic institutional and consultation archives (January 1990–November 2017) for adult cases previously classified as “rhabdomyosarcoma” and noted to contain large numbers of histiocytes. Previously treated tumors were excluded. All available slides and blocks were retrieved and re-reviewed. Following poster presentation of these seven cases at the 2018 United States and Canadian Academy of Pathology annual meeting (Vancouver, BC, Canada), three similar cases were identified from the consultation archives of Drs Weiss, Agaimy, and Huang.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue sections from each case using antibody-specific epitope retrieval techniques with Dako Envision (Dako, Carpinteria, CA, USA) automated system for detection of the following primary antigens: Desmin (DE-R-11, 1:100, Leica, Newcastle Upon Tyne, UK), MyoD1 (EP212, 1:25–1:100, Cell Marque, Rocklin, CA, USA), Myogenin (F5D, 1:25–1:50, Dako), and CD163 (10D6, 1:200, Leica, Newcastle Upon Tyne, UK).
MyoD1 mutational analysis
Mutational analysis targeting the spindle cell/sclerosing RMS-associated MyoD1 mutations was performed at St. Jude Children’s Research Hospital for cases 1–5. Genomic DNA was extracted from formalin-fixed, paraffin-embedded tissue sections using the Maxwell 16 formalin-fixed, paraffin-embedded Tissue LEV DNA Purification Kit (Promega, Madison, WI), according to the manufacturer’s protocol. A recurrently mutated site in MYOD1 was amplified with GoTaq Long PCR Master Mix (Promega) with primers forward 5′-CAAGCGCAAGACCACCAAC-3′ and reverse 5′-GGTTTGGATTGCTCGACGTG-3′ for 45 cycles at 95 °C for 15 s, at 62 °C for 20 s, and at 68 °C for 30 s. PCR reaction products were purified with ExoSAP-IT (Thermo Fisher Scientific, Waltham, MA) and directly sequenced by Sanger sequencing. Wild-type or mutant sequences were visualized at the location of p.L122 with CLC Main Workbench sequence analysis software version 6.0.2 (CLC bio, Cambridge, MA).
Archer sarcoma FusionPlex next-generation sequencing
Case 8 was tested at Emory University with the Archer Sarcoma FusionPlex next-generation sequencing assay for rearrangements involving the ALK, CAMTA, CCNB, CIC, EPC, EWSR1, FOXO1A, FUS GLI, HMGA, JAZF, MEAF6, MKL2, NCOA2, NTRK2, PDGFB, PLAG1, ROS1, SS18, STAT6, TAF15, TCF12, TFE3, TFG, USP6, and YWHAE genes using standard laboratory methods.
NGS analysis
Initially, molecular analysis (aiming to identify gene fusions) was performed on Cases 8–10 using the TruSight RNA Fusion panel (Illumina, Inc., San Diego, CA, USA). RNA was isolated from formalin-fixed, paraffin-embedded tissue sections using the RNeasy FFPE Kit (Qiagen, Hilden, Germany) and quantified spectrophotometrically using NanoDrop-1000 (Thermo Fisher Scientific, Wilmington, USA). A total of 500 ng RNA was used as a starting material. Specifically, cDNA was first synthesized followed by adaptor ligation, amplification of the total cDNA, hybrid capture of 507 fusion-associated genes, and sequencing on the MiSeq instrument using MiSeq Reagent Kit v3 (150 cycles) according to the protocol of Illumina (San Diego, USA). The resulting raw data (fastq files) were then processed by the Illumina’s Basespace RNA-Seq Alignment workflow with STAR aligner. No fusion event was identified in four cases. Therefore, further molecular testing was initiated on tumor DNA extracted from formalin-fixed, paraffin-embedded tissue using the TST170 cancer panel (Illumina). Briefly, DNA was extracted from formalin-fixed, paraffin-embedded tissue sections with the QIAamp DNA FFPE Tissue Kit (Qiagen) and quantified with NanoDrop-1000 (Thermo Fisher Scientific). Approximately 40 ng DNA was used for library preparation using the TruSight Tumor 170 gene panel (Illumina). Libraries were sequenced on a NextSeq 550 machine (Illumina), and alignment and variant calling was done using the TST170 Basespace app (Illumina). Variants were manually filtered against population databases and non-synonymous variants were visually controlled using the integrative genome viewer.
Results
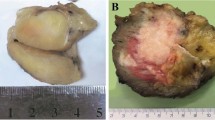
Table 1 summarizes the clinicopathologic features of these ten cases. The tumors occurred in adults ranging from 23 to 69 years of age (median age 43 years), showed a striking male predilection (9M:1F), and involved the deep soft tissues of the trunk (N = 4), lower limbs (N = 4), and neck (N = 2). Patients presented with nonspecific soft tissue masses, slowly growing over a period of 1–2 years in three cases. The tumors ranged from 2.9 to 8.0 cm (median 3.5 cm). The cases were referred in consultation with a range of differential diagnoses, which included RMS in only a minority of cases.
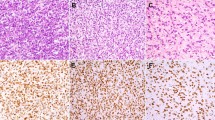
The morphologic and immunohistochemical features of four histiocyte-rich rhabdomyoblastic tumors are illustrated in Fig. 1 (Case 7), Fig. 2 (Case 4), Fig. 3 (Case 1), and Fig. 4 (Case 5). Microscopically, the tumors grew as well-circumscribed, nodular masses, frequently surrounded by a hyalinized fibrous capsule containing lymphoid aggregates and sometimes calcifications. Closer examination of all cases, however, showed at least small areas with infiltrative growth into the surrounding skeletal muscle and fat. Perhaps the most striking feature of these tumors was the presence of numerous large foamy macrophages, multinucleated Touton-type giant cells, and sheets/fascicles of smaller, often spindled macrophages. In several cases, this histiocytic infiltrate largely obscured the underlying neoplastic cell population. However, careful inspection of all cases disclosed distinctly eosinophilic tumor cells, either with epithelioid or spindled morphology, and often having unusual “glassy”-appearing cytoplasm. A variable degree of nuclear pleomorphism was present and cells with large, irregular, hyperchromatic nuclei and prominent nucleoli could be identified in all cases. Cross-striations were not seen in any tumor. Mitotic activity was extremely low, with typically < 1 mitotic figure/50 high-powered fields. Most cases, in fact, contained no more than a single mitotic figure in an entire tissue section. Other microscopic features suggestive of a slowly growing tumor, such as stromal calcification, hyalinized blood vessels, and hemosiderin deposition were commonly present.

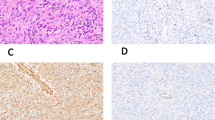
The tumors typically grew in a circumscribed manner, often with a well-formed, hyalinized fibrous capsule (a). Other features suggesting a slowly growing tumor, such as calcifications, were often present (b). Large numbers of foamy macrophages partially obscured the tumor cells (c). Close examination, however, showed atypical rhabdomyoblastic cells with enlarged, hyperchromatic nuclei (d). Desmin (e) and CD163 (f) immunostains highlight the neoplastic cells and macrophages

A well-circumscribed, encapsulated mass with peripheral lymphoid aggregates is seen (a). The tumor consisted of a fascicular proliferation of spindled cells, with occasional cells having nuclear enlargement and hyperchromatism (b). Mitotic activity was extremely low, however (c). Immunohistochemistry showed the overwhelming majority of these spindled cells to represent CD163-positive macrophages (d), with only a very small minority of cells positive for desmin (not shown), MyoD1 (e), and myogenin (f)

Although the tumors tended to grow in a circumscribed manner, as illustrated in Figs. 1 and 2, infiltrative growth into muscle and fat was also present (a). The neoplastic cells were frequently obscured by foamy macrophages and Touton-type giant cells (b). At higher magnification, spindled cells with enlarged, hyperchromatic nuclei were visible. Mitotic activity was extremely low, less than 1/50 HPF in all tumors (c). These spindled cells were highlighted on desmin immunostains (d). The overwhelming majority of the mass represent CD163-positive histiocytes (e). Myogenin (shown) and MyoD1 expression, although present in all cases, was typically very limited in extent (f)


A diffuse proliferation of foamy histiocytes almost entirely obscures the underlying rhabdomyoblastic tumor (a, b). Most lesions tended to demonstrate a low power histiocytic nature (a) with admixed foamy macrophages (b). The tumor itself was composed of variably pleomorphic spindled cells with abundant, often “glassy”-appearing, eosinophilic cytoplasm. Numerous small histiocytes and other chronic inflammatory cells were also typically present (c). Mitotic activity, although present, was extremely low ( < 1/50 HPF (d). Desmin (e) and CD163 (f) immunostains highlighted the rhabdomyoblastic cells and histiocytes, respectively. Next-generation sequencing showed mutation of the NF1 gene (g)
By immunohistochemistry, the overwhelming majority of the cells comprising the masses were strongly positive for CD163, confirming histiocytic lineage. The large, eosinophilic tumor cells were diffusely positive for desmin and showed much more limited expression of MyoD1 and myogenin. Myogenin expression, in particular, was typically confined to a very small number of tumor cells. Immunohistochemistry for other markers, including keratins and S100 protein/SOX10, had been performed in an ad hoc manner by the submitting pathologists; all were negative in the lesional cells.
Sanger sequencing was negative for the known hotspot MYOD1 mutations in RMS in the five cases for which material was available for analysis. Five cases tested for gene fusions (1 case tested with the Archer FusionPlex next-generation sequencing and 4 cases using a 170 and/or 517 gene fusion next-generation sequencing assay) were negative for gene rearrangements, including those involving EWSR1, FUS, and FOXO1A. Molecular genetic testing performed at Erlangen University showed two of four analyzed cases to harbor oncogenic pathogenic inactivating mutations in the NF1 gene. One sample (case 5) harbored a single base pair deletion with a mutated allele frequency of 17% that resulted in a truncating frameshift alteration (c.1398delT; p.Thr467HisfsTer6) (Fig. 4g). Another sample (case 4) had a single base pair missense mutation (c.4600 C > T; p.Arg1534Ter) that resulted in a premature stop codon, which was detected at an allele frequency of 24%. The second case also displayed a likely benign PTCH1 polymorphism (rs115556836; c.2183 C > T; p.Thr728Met) at an allele frequency of 64%. The 3rd and 4th samples (cases 3 and 9) analyzed with the 170 gene panel did not show any pathogenic alterations.
Clinical follow-up was available for 9 patients (median 9 months, mean 23 months, range 3–124 months). Follow-up of > 24 months was available for two patients. All patients were known to have received wide excision showing negative margins. Four patients received adjuvant radiotherapy; none was known to have received chemotherapy. At the time of last follow-up, all patients were alive and without disease; no local recurrences or distant metastases were known to have occurred. No patient was known to have a history of an inherited genetic disorder, in particular neurofibromatosis type-1.
Discussion
We herein have presented the clinicopathological and molecular genetic features of an unusual, hitherto unreported skeletal muscle neoplasm, characterized by circumscribed growth, a diffuse histiocytic infiltrate that largely obscures the underlying tumor cells, extremely low mitotic activity, and microscopic features suggestive of a slowly growing neoplasm, such as encapsulation, calcification, fibrosis, and hemosiderin deposition. As will be discussed below, the morphologic and genetic features of these lesions do not seem to us to correspond to any of the described variants of rhabdomyoma or, more importantly, RMS.
The generally circumscribed growth pattern and the extremely low mitotic activity that characterize histiocyte-rich rhabdomyoblastic tumor raise the possibility that these tumors may be related to rhabdomyoma. However, the clinicopathological features of these lesions are clearly different from those of adult rhabdomyoma, a tumor that usually occurs in the head and neck of older males (median 60 years of age), and is composed of a lobulated proliferation of large polygonal or round cells with abundant eosinophilic cytoplasm, a large nucleus, and a prominent nucleolus, as well as so-called “spider cells” with predominantly clear cytoplasm and thin strands of eosinophilic cytoplasm extending from the nucleus to the cytoplasmic membrane [7, 10,11,12,13,14,15]. Fetal rhabdomyomas, which typically occur as a solitary mass in the subcutaneous tissue of the head and neck region of young boys ( < 5 years of age) are also morphologically quite different from the tumors that comprise the present series, consisting in the “myxoid type” of a variably myxoid growth of bland spindled cells, immature skeletal muscle fibers, and peripherally located mature skeletal muscle, or in the “intermediate type” of differentiated skeletal muscle fibers with little myxoid matrix, ganglion-like rhabdomyoblasts, mature-appearing skeletal muscle cells with easily identified cross-striations, and vacuolated cells [6, 7, 11, 15,16,17,18,19]. Genital rhabdomyomas resemble “intermediate” fetal rhabdomyomas but involve the cervix or vagina in middle-aged women, or rarely the paratesticular region or epididymis of men [20,21,22]. Fetal rhabdomyomas are characterized genetically by PTCH1 mutations [23], events we did not identify in any studied tumor of our present series, although one case contained a likely benign PTCH1 polymorphism.
Histiocyte-rich rhabdomyoblastic tumor also does not seem to correspond to a previously described variant of RMS. Embryonal RMS most often involves the head/neck of young children, and is characterized by primitive mesenchymal cells showing varying degrees of rhabdomyoblastic differentiation, with primitive “small round blue” cells, undifferentiated spindled cells, ganglion-like rhabdomyoblasts, and strap cells with brightly eosinophilic cytoplasm and cross-striations [24,25,26,27]. Mitotic activity is invariably present and necrosis is frequently identified. At the cytogenetic level, embryonal RMS are characterized by complex structural and numerical abnormalities, including trisomies of chromosomes 2, 8, and 13 [28,29,30,31]. Molecular analyses commonly show allelic loss at chromosome 11p15, a site containing a number of tumor suppressor genes, including IGF2, H19, and CDKN1C. RAS pathway alterations, including NF1 mutations, are seen in subsets of embryonal RMS, including highly differentiated ones resembling intermediate forms of fetal rhabdomyoma [32, 33].
The morphological features of histiocyte-rich rhabdomyoblastic tumor are obviously also quite different from those of alveolar RMS, a tumor that usually arises in older children and young adults, and is composed of a solid to pseudoalveolar proliferation of highly malignant-appearing round cells, with scattered rhabdomyoblasts and multinucleated neoplastic giant cells [24, 25, 34]. Genetically, most ARMS are characterized by PAX3/PAX7-FOXO1A fusions, genetic events not present in any of our studied cases.
Although the older patient age, extremity location, and variably pleomorphic eosinophilic tumor cells of histiocyte-rich rhabdomyoblastic tumor might suggest pleomorphic RMS, there are a number of important differences. Pleomorphic RMS typically present as large, destructive, partially necrotic masses, and consist microscopically of a fascicular to sheet-like proliferation of highly malignant-appearing, large, eosinophilic cells with frequent mitotic figures, including atypical forms [35,36,37,38,39,40]. Encapsulation, diffuse histiocytic infiltration, and features of chronicity such as fibrosis and calcification would not be expected in pleomorphic RMS.
Finally, although histiocyte-rich rhabdomyoblastic tumors occur in similar locations and patient ages as spindle cell/sclerosing RMS, their morphological features are obviously quite different. Spindle cell RMS shows a spectrum of appearances, with some well-differentiated tumors consisting of relatively bland-appearing spindled cells arranged in a fascicular or storiform pattern, and other tumors having a much higher grade, fibrosarcoma-like appearance [41, 42]. Rhabdomyoblasts are typically few in number. Sclerosing RMS is characterized by the presence of a strikingly sclerotic, osteochondroid-like stroma, microalveolar and pseudovascular growth patterns, primitive round cells with high nuclear grade, and occasionally small clusters of differentiating rhabdomyoblasts [43,44,45,46]. Spindle cell/sclerosing RMS (in particular tumors showing sclerosing morphology) are known to harbor the MYOD1 L122R mutation [47,48,49,50], or less commonly PIKC3A mutations [47, 48]. Mutations of these genes were not identified in any studied case of histiocyte-rich rhabdomyoblastic tumor.
In addition to rhabdomyoma and RMS, we also considered whether histiocyte-rich rhabdomyoblastic tumor might be related to other desmin-positive mesenchymal tumors, in particular angiomatoid fibrous histiocytoma and diffuse-type tenosynovial giant cell tumor. Angiomatoid fibrous histiocytoma shares some morphologic features with the tumors we have presented, including a fibrous capsule with lymphoid aggregates and intralesional histiocytes [51]. Angiomatoid fibrous histiocytomas may occasionally contain pleomorphic cells, similar to those seen in histiocyte-rich rhabdomyoblastic tumor, although these are essentially always seen alongside more typical areas, composed of bland, histiocytoid cells often arranged in distinctive whorls [52]. Although roughly 50% of angiomatoid fibrous histiocytomas are desmin-positive, they are negative for myogenin and myoD1, distinguishing them from RMSs and the present tumors [51]. Furthermore, angiomatoid fibrous histiocytomas show rearrangements involving the EWSR1 and FUS loci, most often with CREB1 or ATF1 [53], which histiocyte-rich rhabdomyoblastic tumors do not. Although diffuse-type tenosynovial giant cell tumors may contain large numbers of desmin-positive cells and foamy histiocytes, they would not be expected to be encapsulated, contain pleomorphic tumor cells, or express myogenin/MyoD1 [54,55,56]. In addition, the desmin-positive cells present in our tumors lacked dendritic morphology, as is typically seen in the desmin-positive cells of tenosynovial giant cell tumors, and no tumor analyzed with next-generation sequencing techniques showed evidence of CSF1 rearrangements, the molecular hallmark of tenosynovial giant cell tumors. Finally, although NF1 mutations were identified in two cases, the morphological and immunohistochemical features of histiocyte-rich rhabdomyoblastic tumors are quite clearly different from malignant peripheral nerve sheath tumors with rhabdomyoblastic differentiation (malignant Triton tumor).
Our study strongly suggests that histiocyte-rich rhabdomyoblastic tumor is clinically and morphologically distinct from other tumors of skeletal muscle, but it is not clear where in the nosological classification of skeletal muscle tumors they fall. Are they benign, malignant, or possibly rhabdomyoblastic tumors of uncertain malignant potential or even intermediate malignancy? Whereas the long pre-clinical duration, circumscription, partial encapsulation, exceedingly low mitotic activity, and indolent clinical behavior to date suggest they are benign (and thus best classified as “rhabdomyoma”), we are concerned about the cytologic atypia that characterizes these lesions and their lack of well-differentiated skeletal muscle cells (a common feature in rhabdomyomas). In fact, some of us initially thought these were variants of pleomorphic or spindle cell RMS based on these features. We are also uncomfortable labeling these lesions as definitely benign in the absence of larger case numbers and longer clinical follow-up. It is also, however, very difficult to unequivocally label these lesions “rhabdomyosarcoma,” as they lack the morphologic and genetic features of any described RMS subtype, and show instead features suggesting slow growth, including partial encapsulation, extremely low mitotic activity, hyaline fibrosis, and calcifications. Furthermore, none of these tumors have to date presented with metastatic disease, or subsequently resulted in local recurrence or distant metastasis, despite arguably sub-optimal therapy for RMS, with only four patients receiving adjuvant radiotherapy and none chemotherapy.
The molecular pathogenesis of the tumors we are reporting remains to be further characterized. Detection of likely somatic pathogenic mutations in the NF1 gene resulting in protein truncation indicates a likely role for this genetic defect in their pathogenesis, although the absence of clinical stigmata or family history of NF1 in any of these patients suggests that these mutations were likely somatic, given the low allelic frequencies and absence of NF1 phenotype in the patients. The presence of inactivating NF1 mutations in some of our cases might suggest a possible link to embryonal RMS, although the morphology of these tumors would seem to argue strongly against this hypothesis.
In summary, we have reported the clinicopathologic and molecular genetic features of a previously undescribed, distinctive rhabdomyoblastic tumor, characterized by diffuse histiocytic infiltration, pleomorphic, eosinophilic tumor cells lacking cross-striations, morphologic features suggestive of chronicity, and to date favorable outcome. The diffuse histiocytic infiltration and peripheral lymphoid aggregates that characterize these tumors are interesting and unexplained, and it is tempting to speculate that their presence in some way reflects immune modulation of the tumor, as has been described in other mesenchymal tumors. We propose the term “histiocyte-rich rhabdomyoblastic tumor of uncertain malignant potential” for this unusual neoplasm, reflective of its uncertain place in the current nosology of skeletal muscle neoplasms. Study of additional cases with longer follow-up will be necessary to determine whether this tumor is best considered “rhabdomyoma”, “rhabdomyosarcoma”, or perhaps instead a skeletal muscle tumor of intermediate (borderline) malignancy.
References
Fletcher CDM. WHO classification of tumours of soft tissue. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, editors. WHO classification of tumours of soft tissue and bone. 4th ed. Lyons: IARC Press; 2013. p. 10–11.
Folpe AL, Fanburg-Smith JC, Miettinen M, Weiss SW. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1–12.
Folpe AL, Mentzel T, Lehr HA, Fisher C, Balzer BL, Weiss SW. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol. 2005;29:1558–75.
Joensuu H. Gastrointestinal stromal tumors: risk assessment and adjuvant therapy. Hematol Oncol Clin North Am. 2013;27:889–904.
Demicco EG, Wagner MJ, Maki RG, Gupta V, Iofin I, Lazar AJ, et al. Risk assessment in solitary fibrous tumors: validation and refinement of a risk stratification model. Mod Pathol. 2017;30:1433–42.
Kapadia SB, Meis JM, Frisman DM, Ellis GL, Heffner DK. Fetal rhabdomyoma of the head and neck: a clinicopathologic and immunophenotypic study of 24 cases. Hum Pathol. 1993;24:754–65.
Kapadia SB, Meis JM, Frisman DM, Ellis GL, Heffner DK, Hyams VJ. Adult rhabdomyoma of the head and neck: a clinicopathologic and immunophenotypic study. Hum Pathol. 1993;24:608–17.
Lopez Varela C, Lopez de la Riva M, La Cruz, Pelea C. Vaginal rhabdomyomas. Int J Gynaecol Obstet. 1994;47:169–70.
Goldblum JR, Folpe AL, Weiss SW. Rhabdomyosarcoma. In: Goldblum JR, Folpe AL, Weiss SW, Enzinger FM, Weiss SW, editors. Enzinger and Weiss’s soft tissue tumors. 6th ed. Philadelphia, PA: Saunders/Elsevier; 2014. p. 601–38.
Di Sant’Agnese PA, Knowles DM,2nd. Extracardiac rhabdomyoma: a clinicopathologic study and review of the literature. Cancer . 1980;46:780–9.
Eusebi V, Ceccarelli C, Daniele E, Collina G, Viale G, Mancini AM. Extracardiac rhabdomyoma: An immunocytochemical study and review of the literature. Appl Pathol. 1988;6:197–207.
Balatsouras DG, Eliopoulos PN, Economou CN. Adult-type rhabdomyoma of the submandibular region. J Otolaryngol. 1993;22:14–7.
Box JC, Newman CL, Anastasiades KD, Lucas GW, Latouff OM. Adult rhabdomyoma: presentation as a cervicomediastinal mass (case report and review of the literature). Am Surg. 1995;61:271–6.
Sanchez Jimenez J, Dean Ferrer A, Alamillos Granados F, Ruiz Masera JJ, Villar Pastor C, Garcia Lopez A, et al. Adult rhabdomyoma in the masticatory area. New case presentation and review of the literature. Med Oral. 2001;6:64–8.
Hansen T, Katenkamp D. Rhabdomyoma of the head and neck: morphology and differential diagnosis. Virchows Arch. 2005;447:849–54.
Dehner LP, Enzinger FM, Font RL. Fetal rhabdomyoma. Anal nine cases Cancer. 1972;30:160–6.
Seidal T, Kindblom LG, Angervall L. Myoglobin, desmin and vimentin in ultrastructurally proven rhabdomyomas and rhabdomyosarcomas. An immunohistochemical study utilizing a series of monoclonal and polyclonal antibodies. Appl Pathol. 1987;5:201–19.
DiSanto S, Abt AB, Boal DK, Krummel TM. Fetal rhabdomyoma and nevoid basal cell carcinoma syndrome. Pediatr Pathol. 1992;12:441–7.
Crotty PL, Nakhleh RE, Dehner LP. Juvenile rhabdomyoma. An intermediate form of skeletal muscle tumor in children. Arch Pathol Lab Med. 1993;117:43–7.
Lu DY, Chang S, Cook H, Alizadeh Y, Karam AK, Moatamed NA, et al. Genital rhabdomyoma of the urethra in an infant girl. Hum Pathol. 2012;43:597–600.
Hanski W, Hagel-Lewicka E, Daniszewski K. Rhabdomyomas of female genital tract. Report on two cases. Zent Pathol. 1991;137:439–42.
Konrad EA, Meister P, Hubner G. Extracardiac rhabdomyoma: report of different types with light microscopic and ultrastructural studies. Cancer . 1982;49:898–907.
Hettmer S, Teot LA, van Hummelen P, MacConaill L, Bronson RT, Dall’Osso C, et al. Mutations in Hedgehog pathway genes in fetal rhabdomyomas. J Pathol. 2013;231:44–52.
Parham DM. Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod Pathol. 2001;14:506–14.
Qualman SJ, Coffin CM, Newton WA, Hojo H, Triche TJ, Parham DM, et al. Intergroup Rhabdomyosarcoma Study: update for pathologists. Pediatr Dev Pathol. 1998;1:550–61.
Tsokos M. The diagnosis and classification of childhood rhabdomyosarcoma. Sem Diagn Pathol. 1994;11:26–38.
Newton WA Jr, Soule EH, Hamoudi AB, Reiman HM, Shimada H, Beltangady M, et al. Histopathology of childhood sarcomas, Intergroup Rhabdomyosarcoma Studies I and II: clinicopathologic correlation. J Clin Oncol. 1988;6:67–75.
Bridge JA, Liu J, Qualman SJ, Suijkerbuijk R, Wenger G, Zhang J, et al. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosomes Cancer. 2002;33:310–21.
Wang C. Childhood rhabdomyosarcoma: recent advances and prospective views. J Dent Res. 2012;91:341–50.
Pazzaglia L, Chiechi A, Conti A, Gamberi G, Magagnoli G, Novello C, et al. Genetic and molecular alterations in rhabdomyosarcoma: mRNA overexpression of MCL1 and MAP2K4 genes. Histol Histopathol. 2009;24:61–7.
Gallego Melcon S, Sanchez de Toledo Codina J. Molecular biology of rhabdomyosarcoma. Clin Transl Oncol. 2007;9:415–9.
Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–31.
Paulson V, Chandler G, Rakheja D, Galindo RL, Wilson K, Amatruda JF, et al. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosomes Cancer. 2011;50:397–408.
Enzinger FM, Shiraki M. Alveolar rhabdomyosarcoma. Anal 110 cases Cancer. 1969;24:18–31.
Stock N, Chibon F, Binh MB, Terrier P, Michels JJ, Valo I, et al. Adult-type rhabdomyosarcoma: analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am J Surg Pathol. 2009;33:1850–9.
Guillou L, Aurias A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 2010;456:201–17.
Furlong MA, Mentzel T, Fanburg-Smith JC. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
Hollowood K, Fletcher CD. Rhabdomyosarcoma in adults. Sem Diagn Pathol. 1994;11:47–57.
Gaffney EF, Dervan PA, Fletcher CD. Pleomorphic rhabdomyosarcoma in adulthood. Analysis of 11 cases with definition of diagnostic criteria. Am J Surg Pathol. 1993;17:601–9.
Seidal T, Kindblom LG, Angervall L. Rhabdomyosarcoma in middle-aged and elderly individuals. APMIS. 1989;97:236–48.
Cavazzana AO, Schmidt D, Ninfo V, Harms D, Tollot M, Carli M, et al. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol. 1992;16:229–35.
Rubin BP, Hasserjian RP, Singer S, Janecka I, Fletcher JA, Fletcher CD. Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol. 1998;22:459–64.
Chiles MC, Parham DM, Qualman SJ, Teot LA, Bridge JA, Ullrich F, et al. Sclerosing rhabdomyosarcomas in children and adolescents: a clinicopathologic review of 13 cases from the Intergroup Rhabdomyosarcoma Study Group and Children’s Oncology Group. Pediatr Dev Pathol. 2004;7:583–94.
Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26:1175–83.
Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000;436:305–11.
Croes R, Debiec-Rychter M, Cokelaere K, De Vos R, Hagemeijer A, Sciot R. Adult sclerosing rhabdomyosarcoma: cytogenetic link with embryonal rhabdomyosarcoma. Virchows Arch. 2005;446:64–7.
Agaram NP, Chen CL, Zhang L, LaQuaglia MP, Wexler L, Antonescu CR. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer. 2014;53:779–87.
Alaggio R, Zhang L, Sung YS, Huang SC, Chen CL, Bisogno G, et al. A molecular study of pediatric spindle and sclerosing rhabdomyosarcoma: identification of novel and recurrent VGLL2-related fusions in infantile cases. Am J Surg Pathol. 2016;40:224–35.
Szuhai K, de Jong D, Leung WY, Fletcher CD, Hogendoorn PC. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol. 2014;232:300–7.
Rekhi B, Upadhyay P, Ramteke MP, Dutt A. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod Pathol. 2016;29:1532–40.
Fanburg-Smith JC, Miettinen M. Angiomatoid “malignant” fibrous histiocytoma: a clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum Pathol. 1999;30:1336–43.
Smith ME, Costa MJ, Weiss SW. Evaluation of CD68 and other histiocytic antigens in angiomatoid malignant fibrous histiocytoma. Am J Surg Pathol. 1991;15:757–63.
Antonescu CR, Dal Cin P, Nafa K, Teot LA, Surti U, Fletcher CD, et al. EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer. 2007;46:1051–60.
Folpe AL, Weiss SW, Fletcher CD, Gown AM. Tenosynovial giant cell tumors: evidence for a desmin-positive dendritic cell subpopulation. Mod Pathol. 1998;11:939–44.
Boland JM, Folpe AL, Hornick JL, Grogg KL. Clusterin is expressed in normal synoviocytes and in tenosynovial giant cell tumors of localized and diffuse types: diagnostic and histogenetic implications. Am J Surg Pathol. 2009;33:1225–9.
Somerhausen NS, Fletcher CD. Diffuse-type giant cell tumor: clinicopathologic and immunohistochemical analysis of 50 cases with extraarticular disease. Am J Surg Pathol. 2000;24:479–92.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Martinez, A.P., Fritchie, K.J., Weiss, S.W. et al. Histiocyte-rich rhabdomyoblastic tumor: rhabdomyosarcoma, rhabdomyoma, or rhabdomyoblastic tumor of uncertain malignant potential? A histologically distinctive rhabdomyoblastic tumor in search of a place in the classification of skeletal muscle neoplasms. Mod Pathol 32, 446–457 (2019). https://doi.org/10.1038/s41379-018-0145-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0145-0
This article is cited by
-
Pulmonary inflammatory leiomyosarcoma represents a potential diagnostic pitfall of DNA methylation-based classification of sarcomas: a case report
BMC Pulmonary Medicine (2023)
-
Xanthogranulomatous epithelial tumors and keratin-positive giant cell-rich soft tissue tumors: two aspects of a single entity with frequent HMGA2-NCOR2 fusions
Modern Pathology (2022)
-
EWSR1-WT1 gene fusions in neoplasms other than desmoplastic small round cell tumor: a report of three unusual tumors involving the female genital tract and review of the literature
Modern Pathology (2021)
-
“Inflammatory Leiomyosarcoma” and “Histiocyte-rich Rhabdomyoblastic Tumor”: a clinicopathological, immunohistochemical and genetic study of 13 cases, with a proposal for reclassification as “Inflammatory Rhabdomyoblastic Tumor”
Modern Pathology (2021)
-
Toward a unifying entity that encompasses most, but perhaps not all, inflammatory leiomyosarcomas and histiocyte-rich rhabdomyoblastic tumors
Modern Pathology (2021)
