
Abstract
Inflammatory leiomyosarcoma (ILMS), defined as “a malignant neoplasm showing smooth muscle differentiation, a prominent inflammatory infiltrate, and near-haploidization”, is a very rare soft tissue tumor with a generally favorable prognosis. The morphologic features of “histiocyte-rich rhabdomyoblastic tumor” (HRRMT) are similar to those of ILMS, although this lesion shows by definition a skeletal muscle phenotype. Recent gene expression profiling and immunohistochemical studies have also suggested that ILMS and HRRMT may be related. We studied the clinicopathologic, immunohistochemical and genetic features of four cases previously classified as ILMS and nine classified as HRRMT. Tumors from both groups tended to occur in the deep soft tissues of the extremities of young to middle-aged males and exhibited indolent behavior. Morphologically, all were well-circumscribed, often encapsulated, and showed a striking histiocyte-rich inflammatory infiltrate admixed with variably pleomorphic tumor cells showing spindled and epithelioid to rhabdoid morphology, eosinophilic cytoplasm, and prominent nucleoli, but few, if any, mitotic figures. Immunohistochemically, the tumor cells expressed desmin, alpha-smooth muscle actin, and the rhabdomyoblastic markers PAX7, MyoD1, and myogenin. H-caldesmon expression was absent in all cases, using the specific h-CD antibody. Karyotypic study (1 HRRMT) and genome-wide copy number analysis (7 HRRMT, OncoScan SNP assay), revealed near-haploidization in four cases, with subsequent genome doubling in one, an identical phenotype to that seen in ILMS. We propose reclassification of ILMS and HRRMT as “inflammatory rhabdomyoblastic tumor”, a name which accurately describes the salient morphologic and immunohistochemical features of this distinctive tumor, as well as its intermediate (rarely metastasizing) clinical behavior.
Similar content being viewed by others

Introduction
Inflammatory leiomyosarcoma (ILMS), first recognized by the WHO as a distinct entity in 2020, is defined as “… a malignant neoplasm showing smooth muscle differentiation, a prominent inflammatory infiltrate, and near-haploidization” [1]. ILMS was initially described by Merchant et al. in a series of 12 consultation cases previously coded as “leiomyosarcoma with inflammation” or “leiomyosarcoma simulating inflammatory ‘malignant fibrous histiocytoma’ (undifferentiated pleomorphic sarcoma)” [2]. By definition, these tumors showed morphologic and immunohistochemical evidence of smooth muscle differentiation (e.g., fascicles of eosinophilic spindled cells with cigar shaped nuclei showing strong desmin and/or smooth muscle actin expression), and prominent inflammation, in the form of aggregates of xanthoma cells, lymphocytes, and occasionally neutrophils.
ILMS is quite rare, with fewer than 30 reported cases, and typically presents as a deeply situated mass in the extremities of young to middle-aged adults, most often males [2,3,4,5,6,7,8,9]. The prognosis for patients with ILMS is favorable, with distant spread in only three reported patients [2, 4]. Perhaps the most distinctive feature of ILMS is its karyotype, with most examined cases showing “near-haploidization”, with loss of one copy of nearly all chromosomes [3,4,5, 7]. Less often, inactivating NF1 mutations are present [3, 6].
In 2019, Martinez et al. reported as “histiocyte-rich rhabdomyoblastic tumor” (HRRMT) a series of ten unusual lesions displaying some morphologic overlap with ILMS, including a striking xanthomatous chronic inflammatory cell infiltrate, but showing by definition skeletal muscle differentiation in the form of desmin, myogenin and MyoD1 expression [10]. To date, only two additional cases of cases of HRRMT have been reported [11]. Similar to ILMS, HRRMT most often presents as a deep soft tissue mass in a young to middle-aged male, and appears to have an excellent prognosis, without documented metastatic potential to date [10, 11]. Very little is known about the genetic events underlying the pathogenesis of HRRMT, although two cases have harbored pathogenic inactivating mutations in NF1 [10].
Two very recent studies have also suggested a possible link between ILMS and HRRMT. Using gene expression profiling, Arbajian et al. demonstrated upregulation of several skeletal muscle-specific genes in seven ILMS, all showing a typical near-haploid genetic profile [3]. Immunohistochemical confirmation of skeletal muscle marker expression was not, however, performed. Conversely, Michal et al. noted expression of skeletal muscle markers in nine cases previously classified as ILMS, although testing for near-haploidization was not performed [6].
Prompted by our own recent finding of near-haploid genetic features, both with traditional karyotyping and the OncoScan single-nucleotide polymorphism (SNP) assay, in a tumor showing morphologic and immunohistochemical features of HRRMT, we studied the morphologic, immunophenotypic, and molecular genetic features of 13 tumors previously classified as either ILMS or HRRMT, with the goal of better understanding the relationship or lack thereof between these 2 very rare tumors.
Materials and methods
Case selection
Approval for this study was granted by the Institutional Review Boards of Mayo Clinic and Stanford University. All available slides and blocks from 13 cases previously classified as ILMS or HRRMT were retrieved. Cases 1, 2, 4, and 13 of our cohort are previously unpublished cases from the Mayo Clinic institutional and consultation archives, classified as HRRMT. Some details of case 4 have recently been published elsewhere [11]. Case 5, previously classified as ILMS, was identified in the surgical pathology archives of Stanford University School of Medicine. Cases 6–9 were previously reported as examples of HRRMT by Martinez et al. (corresponding to cases 1–3 and 6 from this series, respectively) [10]. Cases 10–12 were previously reported as ILMS by Nord et al. [7] and Arbajian et al. [3] (corresponding to cases 1–3 in both studies, respectively). Genetic findings for cases 10–12 have been reported in detail in these two publications: case 10 is known to have a hyperdiploid karyotype with loss of heterozygosity (LOH) for chromosomes 1–4, 6–17, and 19, with retained heterozygosity of chromosomes 5, 18, and 20–22; cases 11 and 12 have near-haploid karyotypes with LOH of chromosomes 1–4, 6–19, and 21 and retained heterozygosity of 5, 20, and 22 (case 11), and LOH of 1–4 and 6–19 with retained heterozygosity of 5 and 20–22 (case 12).
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed paraffin-embedded (FFPE) tissue sections using antibody-specific epitope retrieval with the Dako Envision (Dako, Carpinteria, CA, USA) automated system for detection of the following primary antigens: desmin (DE-R-11, 1:50–1:100; Leica, Newcastle Upon Tyne, UK), alpha-smooth muscle actin (BS66, 1:100–1:400; Nordic Biosite), h-caldesmon (h-CD, 1:50–1:100; Dako), MyoD1 (EP212, 1:25–1:100; Cell Marque, Rocklin CA), myogenin (F5D, 1:25–1:50; Dako, Santa Clara, CA), and CD163 (10D6, 1:200; Leica, Newcastle Upon Tyne, UK). Anti-PAX7 antibody (1:200; Developmental Studies Hybridoma Bank, Iowa City, IA) was processed using a Leica Bond II platform (Buffalo Grove, IL) with Leica ER2 solution (pH 9.0) for heat-induced epitope retrieval. Only nuclear immunoreactivity was scored as positive for PAX7, MyoD1, and myogenin. Staining was graded semiquantitatively as negative (no cells positive), 1 + (1–10% of cells positive), 2 + (10–50% of cells positive), or 3 + (>50% of cells positive).
Genetic analysis
During the routine clinical work-up of one case (Case 2), traditional cytogenetic karyotyping was performed on cultured tumor cells, using routine laboratory protocol. Case 5 was evaluated for rearrangements of the EWSR1 (22q12.2) and FUS (16p11.2) genes, also using standard laboratory protocol.
Affymetrix OncoScan assay was used for genomic characterization of tumors diagnosed as HRRMT (cases 1–4, 6, 7, and 13). This is a SNP-based microarray platform designed to assess genome-wide copy number alternations and LOH. It utilizes molecular inversion probe technology to capture the alleles of over 220,000 SNPs distributed across the genome. Samples were prepared from FFPE tumor tissue collected from several consecutive, unstained, 5-μm-thick sections placed on positively charged slides. DNA was extracted using QIAamp DNA FFPE Tissue Kit (QIAGEN, Germantown, MD), the DNA was quantified using a Qubit 2.0 Fluorometer (Life Technologies/Thermo Fisher Scientific, Waltham, MA), and the OncoScan assay was performed per the manufacturer’s protocol. Array fluorescence intensity data (CEL files), generated by Affymetrix GeneChip Command Console Software version 4.0, were processed using OncoScan Console software version 1.1.034 to produce OSCHP files. Chromosome Analysis Suite software version 3.1 was used for analysis of CN and LOH events from OSCHP files. Autosomal probes were considered “copy number loss” if the genomic interval deviating from normal contained a minimum of 25 probes and were considered “copy number gain” if the genomic interval deviating from normal contained a minimum of 50 probes.
Results
Clinical features including follow-up
The clinicopathological features of cases previously classified as ILMS and HRRMT were strikingly similar (Table 1). Both showed a marked male predilection (ILMS: 4 males, 0 females; HRRMT: 7 males, 2 female), with a similar median age at diagnosis (ILMS: 41.5 years; HRRMT: 38.5 years). The tumors occurred in the deep soft tissues of the extremities (7 cases; 4 ILMS and 3 HRRMT), trunk (5 cases; all HRRMT) and parapharyngeal soft tissue (1 HRRMT). One patient (case 3) had a clinical diagnosis of neurofibromatosis type-1 (NF1) with multiple neurofibromas.
Clinical follow-up was available for three of the five previously unreported patients (cases 2, 3 and 5), of 7, 5, and 56 months duration, respectively. These tumors were treated with wide local excision, resulting in negative surgical margins, and the three patients are currently disease free, without evidence of local recurrence or distant metastases. As indicated in Table 1, published follow-up information from four patients previously reported by Martinez et al. [10] and two patients previously reported by Arbajian et al. [3] indicated all to be recurrence and/or metastasis free. We did not attempt to obtain further follow-up on these six patients. Case 13 is too recent for meaningful follow-up.
Morphologic features
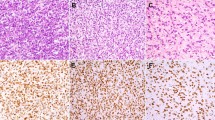
The morphologic features of tumors previously diagnosed as ILMS and HRRMT were strikingly similar, although some morphologic variation was seen from case to case (ILMS, case 5: Fig. 1; HRRMT, cases 4 and 13: Figs. 2 and 3). All tumors were on the whole circumscribed, often demonstrating a well-defined fibrous capsule which surrounded most of the mass. Close inspection, however, typically showed small foci of infiltration into the surrounding soft tissues. Small, peripherally located lymphoid aggregates and intralesional calcifications were present in many cases. All tumors displayed a prominent inflammatory infiltrate comprised predominately of histiocytes, small lymphocytes and plasma cells. Neutrophils were not prominent. The histiocytes showed a range of morphologies from small spindle-shaped cells to large foamy cells; Touton-type multinucleated giant cells were also often present.

At low-power, this lesion demonstrated the fibrous pseudocapsule, calcifications, and lymphoid aggregates typically seen in these tumors (A). The eosinophilic tumor cells grew in vague fascicles (B), and contained enlarged, irregular nuclei with visible nucleoli (C). In addition to desmin (not shown), many neoplastic cells expressed MyoD1 (D). Strong smooth muscle actin expression was also seen (E), but caldesmon was entirely negative (F). Traditional karyotyping of this lesion was performed; of 20 metaphases, 17 metaphases were hyper-haploid with gain of chromosomes 5, 20, 22 (G), and 3 metaphases represented a tetraploid subclone (H).


This encapsulated tumor consisted of a sheetlike proliferation of large, eosinophilic cells, with a smaller number of nonlipidized histiocytes (A). Higher power magnification showing large, markedly pleomorphic tumor cells with abundant eosinophilic cytoplasm, vesicular nuclei, and small nucleoli (B). Immunohistochemistry for CD163 (C) highlights the numerous histiocytes admixed with the desmin-positive (D) tumor cells. The tumor was variably positive for smooth muscle actin (E), but caldesmon-negative (F). Expression of myogenin and MyoD1 was extremely limited, confined to scattered, isolated MyoD1-positive cells (G). In contrast, PAX7 was diffusely positive, confirming the skeletal muscle phenotype of this lesion (H). The final (I) illustrates a whole-genome single-nucleotide polymorphism array plot, depicting copy number changes and loss of heterozygosity in this case. Weighted Log2 Ratio represents relative copy number and Allele Difference and B-allele frequency (BAF) represent ratios of polymorphic SNPs. In contrast to case 4 (illustrated in Fig. 3), which showed relative gains and loss of heterozygosity for most autosomes, this case shows relative loss of most autosomes, with retention of chromosomes 5, 12, 18, 20, 21, and 22, consistent with a near-haploid karyotype.

Case 4, also originally classified as a “histiocyte-rich rhabdomyoblastic tumor”, showed predominantly spindle cell features, both in the larger, eosinophilic tumor cells and in the smaller histiocytes (A). Immunohistochemically, this tumor also showed a skeletal muscle phenotype, with expression of desmin, myogenin and PAX7 (not shown). Whole-genome single-nucleotide polymorphism array plot depicting copy number changes and loss of heterozygosity, showing relative gain of chromosomes 5, 20, and 22 and “normal” copy state for all other autosomes (B). Allele Difference and BAF represent the ratios of polymorphic SNPs across the genome, and show loss of heterozygosity for all autosomes at the “normal” copy state. This pattern suggests a doubling of a near-haploid karyotype.
The neoplastic cells were often largely obscured by this histiocytic and lymphoid infiltrate, and consisted of individual cells, small aggregates and short fascicles of spindled, epithelioid and often pleomorphic cells with abundant, deeply eosinophilic, “glassy” cytoplasm. Cross striations were not seen. Many of the tumor cells had markedly enlarged, irregular, hyperchromatic nuclei, but mitotic activity was extremely low (<1 mitotic figure/10 400× microscopic fields). Necrosis was absent.
Immunohistochemical features
The immunohistochemical results are summarized in Table 1 and illustrated in Figs. 1c–f, 2d–f, and 3d–g. The immunophenotype of cases previously classified as ILMS and HRRMT was essentially identical. As expected, the overwhelming majority of cells within all of the masses represented reactive CD163-positive histiocytes. The neoplastic cells consistently showed a skeletal muscle phenotype, with diffuse expression of desmin (ILMS: 4 of 4; HRRMT: 9 of 9) and more variable expression of MyoD1 (ILMS: 3 of 4; HRRMT: 9 of 9), myogenin (ILMS: 3 of 4; HRRMT: 8 of 9), and PAX7 (ILMS: 4 of 4; HRRMT: 4 of 4). In general, PAX7 was expressed by the majority (>50%) of tumor cells, whereas expression of MyoD1 and myogenin was more limited (1–50% and 1–10% of cells, respectively). Evidence for smooth muscle differentiation was considerably more limited, with variable expression of smooth muscle actin in all tested ILMS and in just over half of HRRMT (4 of 7); caldesmon was negative in all tumors (4 ILMS; 7 HRRMT).
Genetic findings
Karyotypic study of Case 2 showed 17 of 20 metaphases to be hyper-haploid with gain of chromosomes 5, 20, 22, and 3 of 20 to demonstrate a tetraploid subclone (Fig. 3).
Table 1 summarizes the results of SNP array analysis for genome-wide copy number and LOH in seven studied cases of HRRMT (cases 1–4, 6, 7, and 13). As expected, Case 2 displayed LOH for all autosomes except for 5, 20, and 22, which were disomic, consistent with a near-haploid karyotype. Case 3 had a similar near-haploid karyotype with LOH for all autosomes except for 5, 7, 20, 21, and 22, which were all present in a normal diploid state. Case 4 showed relative gain of chromosomes 5, 20, and 22 and complete LOH for the remaining autosomes, but in a near-diploid clone (Fig. 4). Case 7 showed a diploid karyotype with complex chromosomal gains and losses involving portions of chromosomes 19 and X. Case 13 showed a near-haploid phenotype, with retained disomy for chromosomes 5, 18, 20, 21, and 22. Two cases (cases 1 and 6) did not show LOH or copy number changes, most likely reflecting the small number of neoplastic cells and the large number of nonneoplastic histiocytes present in the submitted tissue sections.

At low-power magnification, this tumor was notable for sheets of foamy macrophages and smaller, nonlipidized histiocytes, which largely obscured the underlying neoplasm (A). Closer inspection, however, demonstrated scattered tumor cells with brightly eosinophilic cytoplasm and peripherally placed nuclei (B). By immunohistochemistry, the tumor showed a skeletal muscle phenotype, with expression of desmin (C), MyoD1 (D), PAX7 (E), and myogenin (not shown). Although limited smooth muscle actin expression was present (not shown), caldesmon expression was entirely absent (F). Fluorescence in situ hybridization studies for the EWSR1 (G) and FUS genes (H), located at 22q12.2 and 16p11.2, respectively, showed the great majority of studied cells to contain only 1 copy of FUS, with 2 copies of EWSR1, suggestive of loss of one copy of chromosome 16. In this morphologic and immunohistochemical context, this finding suggests that this may also have been a near-haploid tumor, although this cannot be confirmed.
As noted above, three previously reported ILMS (cases 10–12) were known to have essentially identical LOH findings [3, 7].
In case 5, FISH for EWSR1 and FUS showed the great majority of studied cells to contain only 1 copy of FUS, with 2 copies of EWSR1, suggestive of chromosome 16 loss in the context of a tumor showing otherwise classical morphologic and immunohistochemical features of ILMS (Fig. 4g, h). Regrettably, further genetic study was not possible on this case.
Discussion
The results of the present study show the clinical, morphologic, immunohistochemical, and genetic features of cases previously classified as ILMS and HRRMT to be essentially identical, strongly suggesting that these tumors represent a single entity. In particular, we have shown immunohistochemical evidence of skeletal muscle marker expression in genetically typical (i.e., near-haploid) ILMS, and conversely have demonstrated identical genetic findings in cases previously classified as HRRMT on morphologic and immunohistochemical grounds.
Although ILMS is currently defined as a tumor showing smooth muscle differentiation [1], the preponderance of evidence from this and prior studies points instead toward skeletal muscle lineage. Gene expression profiling data, published by Arbajian et al., have shown upregulation of several genes considered to be highly specific for skeletal muscle differentiation in genetically typical ILMS, including MYF5, MYF6, MYOD1, MYOG, and PAX7; high-level expression of smooth muscle-related genes, such as ACTA2, SMTN, or CALD1 was not seen [3]. These findings were first corroborated at the protein level by Michal et al., who demonstrated consistent expression of MyoD1, myogenin and PAX7 in cases previously classified on morphologic grounds as ILMS [6]. Our results are essentially identical, with near-uniform expression of these skeletal muscle-specific markers in tumors previously classified as ILMS and HRRMT. Co-expression of these transcription factors is considered to be highly specific for skeletal muscle differentiation [12]. Mammalian myogenesis occurs through a highly coordinated pathway in which PAX7 controls specification of early progenitors and maintenance of skeletal muscle stem cells, while MyoD1 determines lineage commitment and myogenin regulates terminal differentiation [13]. The observation of increased PAX7 and MyoD1 expression relative to myogenin in ILMS/HRRMT suggests a primitive skeletal muscle phenotype, an impression mirrored by the morphology of these lesions, without well-differentiated rhabdomyoblasts or visible cross striations.
In contrast, evidence for smooth muscle differentiation in ILMS is rather limited, or even arguably absent. As the initial description of ILMS by Merchant et al. predated the widespread availability of antibodies to sensitive and specific skeletal muscle markers (e.g., MyoD1, myogenin, PAX7), the cases from this study were classified as “leiomyosarcoma” on the basis of immunoreactivity with antibodies to pan-muscle actins (HHF35), alpha-smooth muscle actin (1A4) and desmin. However, expression of these markers is not specific for smooth muscle differentiation either in isolation or in combination. Desmin is an intermediate filament protein that is widely expressed in muscle of all types, and in numerous nonmyogenous neoplasms [14]. Alpha-smooth muscle actin has a higher specificity for smooth muscle, but it is also expressed in myofibroblasts and up to 18% of rhabdomyosarcomas, in particular those of spindle cell and embryonal subtypes [15,16,17]. Notably, alpha-smooth muscle actin is known to be transiently produced in myogenic progenitor cells during normal skeletal muscle differentiation, where it is thought to play a role in regulating sarcomeric actin assembly and morphologic changes relating to cell fusion [18,19,20]. Furthermore, expression of smooth muscle actin is not always seen in ILMS/HRRMT, with absent expression in two of three cases reported by Chang et al. [4], one of two cases reported by Bourgeau and Martinez [11], and three cases from the present series.
The heavy isoform of caldesmon (h-caldesmon) is considered to be the most specific marker for smooth muscle differentiation [21,22,23]. Thus, the presence or absence of caldesmon expression is of particular import in the determination of smooth muscle differentiation in ILMS/HRRMT. Although caldesmon expression was absent in three cases reported by Chang et al. [4], it was reported to be positive in three of four cases examined by Arbajian et al. [3], and five of eight cases reported by Michal et al. [6]. In contrast, we have not been able to demonstrate caldesmon expression in any of the ten cases studied herein, including three cases from the prior Arbajian et al. study. We strongly suspect that these differences are the result of antibody selection, as the present study utilized the highly specific h-CD antibody to h-caldesmon, rather than the strikingly nonspecific E89 clone used by Michal et al. and Arbajian et al. A comparative study of the h-CD and E89 clones, published by Beck et al., showed the specificities of the h-CD and E89 caldesmon clones to be 97% and 8%, respectively, for the distinction of true smooth muscle tumors from morphologic mimics [24]. We are not aware of any cases of ILMS or HRRMT which have been shown to be caldesmon-positive, using the h-CD clone.
Our results also confirm that the genetic hallmark of ILMS/HRRMT is a highly unusual and apparently specific pattern of near-haploidization, with retention of both parental copies of chromosomes 5 and 22, with or without subsequent whole-genome doubling. The presence of a near-haploid genetic profile in ILMS was first described by Dal Cin et al., who reported two cases both showing monosomy for all chromosomes except 5, 18, 20, 21, and 22, which were each present in two copies [5]. Chang et al. reported similar findings in one case, although close evaluation of the published karyotype suggests that this tumor, although near-haploid, had somewhat different abnormalities than are seen in ILMS/HRRMT. However, haploidization is not restricted to ILMS/HRRMT, and may be seen in subsets of pleomorphic sarcomas [25, 26]. The most comprehensive study of the genetics of ILMS/HRRMT to date is that of Arbajian et al., who demonstrated seven cases to show near-genome-wide LOH with retained disomy of chromosomes 5 and 22 [3]. Three cases had near-haploid karyotypes, while the remaining four showed near-diploid or hyperdiploid karyotypes, the consequence of genome doubling of a haploid clone. SNP array analysis in the present study showed identical near-genome-wide LOH and relative gain of chromosomes 5 and 22 in four of seven studied cases, with near-haploid karyotypes in three and a near-diploid karyotype in the fourth. We suspect that Case 5, which showed only a single copy of FUS by FISH, also may have been near-haploid, although this cannot be proven. The mechanism that gives rise to near-haploidization and its pathogenetic link to tumorigenesis remain largely unknown.
Although somatic mutations are rare in ILMS/HRRMT, inactivating mutations in the tumor suppressor gene NF1 have been reported in five cases of ILMS/HRRMT [3, 6, 10]. Although the present series did not include NF1 mutational analysis, it is of interest that one patient in this series did have a documented clinical history of NF1, indicative of a germline mutation in NF1. Recurrent mutations in NF1 have also been reported in other tumors with near-haploidization [27,28,29], although haploidization is quite rare in the most common NF1-related tumors (e.g., neurofibroma, malignant peripheral nerve sheath tumor).
Although it is difficult to be entirely certain, we suspect that not all previously reported cases of “inflammatory leiomyosarcoma” represent the same entity. For example, the large, aggressive retroperitoneal tumor reported by Morovic et al. likely represented a dedifferentiated liposarcoma with inflammatory features and myogenous marker expression [8], and the ankle tumor reported by Efstathopoulos et al. might be better considered simply a pleomorphic sarcoma showing limited myogenous differentiation [9]. It is considerably more difficult to individually re-evaluate the cases reported in the seminal description of ILMS by Merchant et al., as the morphologic features of each individual case are not provided in detail. It is highly likely that most (but perhaps not all) of the cases in this initial series represented the same entity as that reported herein as ILMS/HRRT [2]. A subset of cases reported by Merchant et al., however, showed morphologic features which would be quite unusual in ILMS/HRRMT, such as a prominent fascicular or storiform growth pattern, high mitotic activity, vascular space invasion and an inflammatory infiltrate composed predominantly of eosinophils and neutrophils. Thus, it is possible that some of the tumors from this series may have represented dedifferentiated liposarcoma with “inflammatory malignant fibrous histiocytoma”-like histology [30] or more conventional types of leiomyosarcoma/myofibrosarcoma with prominent inflammatory elements. One of us (ALF) has illustrated a relatively conventional leiomyosarcoma with a prominent inflammatory cell infiltrate in a textbook [31]. Similarly, the cases reported by Chang et al. may represent something other than ILMS/HRRMT, as they occurred in atypical locations such as the ovary and lung, presented with unusual systemic symptoms, showed elevated mitotic activity with atypical forms, and resulted in distant metastases and death from disease [4].
If we restrict our analysis of ILMS/HRRMT to those cases previously reported by Nord et al. [7], Martinez et al. [10], Bourgeau and Martinez [11], Michal et al. [6], and the those of the present series, all of which show essentially identical morphologic, immunohistochemical and genetic features, a clearer picture of this tumor type emerges. ILMS/HRRMT almost always presents as a slowly growing mass of the deep soft tissues, often present for years prior to diagnosis. Most occur in young to middle-aged adults and are more common in males. Histologically, these lesions are characterized by circumscribed growth (frequently with a well-formed fibrous pseudocapsule), peripheral lymphoid aggregates, scattered intralesional calcifications, a striking infiltrate of xanthomatous histiocytes (often obscuring the underlying neoplastic cells), a variable component of lymphocytes, plasma cells and other inflammatory cells, and short fascicles, small aggregates, and individual pleomorphic tumor cells with eosinophilic cytoplasm and few if any mitotic figures. Expression of skeletal muscle-specific markers is the rule, smooth muscle actin expression is less common, and caldesmon expression is not present. A near-haploid genotype with retained disomy of chromosomes 5 and 22 is present in most, but not all tumors conforming to the above description. The natural history of these lesions appears to be quite favorable, with available clinical follow-up on 26 previously reported cases and 3 new cases from the present series showing only a single patient with intra-abdominal spread of disease; all 28 patients are reported to be alive without disease at the time of last follow-up.
We would argue that the slow growth, long preclinical duration and indolent clinical behavior of these tumors is that of a mesenchymal tumor having intermediate (rarely metastasizing) malignant potential, rather than a fully malignant sarcoma. This obviously raises the question of whether “inflammatory leiomyosarcoma” is the best name for this group of lesions, whether “histiocyte-rich rhabdomyoblastic tumor” is more suitable, or whether a new consensus term might be preferable. Michal et al. have very recently suggested the name “low-grade inflammatory myogenic tumor” to describe these tumors [6], a suggestion with some merit, but one that implies a mixed or undifferentiated myogenic phenotype, which we do not believe these lesions truly demonstrate. As detailed above, there is strong evidence that these lesions show skeletal muscle differentiation at the gene and protein level, but little if anything to support smooth muscle differentiation. Furthermore, current classification systems require myogenous tumors to be clearly assigned to either smooth or skeletal muscle lineage. We propose reclassifying these distinctive tumors as “inflammatory rhabdomyoblastic tumors”, a name which (1) preserves at least part of the original name given these lesions by Merchant et al., (2) accurately describes the line of differentiation that they exhibit, and (3) replaces the term “sarcoma” with “tumor”, emphasizing their less than fully malignant, borderline behavior. From a classification perspective, these lesions are likely best thought of as skeletal muscle tumors of intermediate malignancy, a new category between benign rhabdomyomas and fully malignant rhabdomyosarcomas.
In summary, the results of the present study and careful review of the published literature strongly suggest that ILMS and HRRMT represent the same entity, which we propose to rename “inflammatory rhabdomyoblastic tumor”. At the present time, we believe the diagnosis of this entity should rest chiefly on demonstration of its classic clinicopathological and immunohistochemical features, rather than demonstration of near-haploidy with retained disomy of chromosomes 5 and 22, as the frequency of this genetic event in inflammatory rhabdomyoblastic tumor is not yet fully established, and the natural history of otherwise-typical tumors with and without these typical genetic findings is the same. Study of additional, well-characterized cases is necessary to fully understand the clinicopathologic and genetic features of these very rare, fascinating neoplasms.
References
WHO Classification of Tumours Editorial Board. Soft tissue and bone tumours. Lyon: International Agency for Research on Cancer; 2020.
Merchant W, Calonje E, Fletcher CDM. Inflammatory leiomyosarcoma: a morphological subgroup within the heterogeneous family of so-called inflammatory malignant fibrous histiocytoma. Histopathol. 1995;27:525–32.
Arbajian E, Köster J, Vult von Steyern F, Mertens F. Inflammatory leiomyosarcoma is a distinct tumor characterized by near-haploidization, few somatic mutations, and a primitive myogenic gene expression signature. Mod Pathol. 2018;31:93–100.
Chang A, Schuetze SM, Conrad EU 3rd, Swisshelm KL, Norwood TH, Rubin BP. So-called “inflammatory leiomyosarcoma”: a series of 3 cases providing additional insights into a rare entity. Int J Surg Pathol. 2005;13:185–95.
Dal Cin P, Sciot R, Fletcher CD, Samson I, De Vos R, Mandahl N. et al. Inflammatory leiomyosarcoma may be characterized by specific near-haploid chromosome changes. J Pathol. 1998;185:112–5.
Michal M, Rubin BP, Kazakov DV, Michalová K, Šteiner P, Grossmann P. et al. Inflammatory leiomyosarcoma shows frequent co-expression of smooth and skeletal muscle markers supporting a primitive myogenic phenotype: a report of 9 cases with a proposal for reclassification as low-grade inflammatory myogenic tumor. Virchows Arch. 2020;10:1007/s00428–020-02774-z.
Nord KH, Paulsson K, Veerla S, Wejde J, Brosjö O, Mandahl N. et al. Retained heterodisomy is associated with high gene expression in hyperhaploid inflammatory leiomyosarcoma. Neoplasia. 2012;14:807–12.
Morovic A, Delcore R, Damjanov I. Inflammatory leiomyosarcoma of the retroperitoneum. Pathol Res Pract. 2003;199:41–3.
Efstathopoulos N, Lazarettos J, Nikolaou V, Chronopoulos E. Inflammatory leiomyosarcoma of the ankle: a case report and review of the literature. J Foot Ankle Surg. 2006;45:127–130.
Martinez AP, Fritchie KJ, Weiss SW, Agaimy A, Haller F, Huang H-Y. et al. Histiocyte-rich rhabdomyoblastic tumor: rhabdomyosarcoma, rhabdomyoma, or rhabdomyoblastic tumor of uncertain malignant potential? A histologically distinctive rhabdomyoblastic tumor in search of a place in the classification of skeletal muscle neoplasms. Mod Pathol. 2019;32:446–57.
Bourgeau M, Martinez AP. Histiocyte-rich rhabdomyoblastic tumor: a report of two cases and a review of the differential diagnoses. Virchows Arch. 2020;10:1007/s00428–020-02857-x.
Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. https://doi.org/10.1126/science.aan2507.
Bentzinger CF, Wang YX, Rudnicki MA. Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol. 2012;4:a008342.
Folpe AL. Chapter 6: Immunohistochemistry for Analysis of Soft Tissue Tumors. In: Enzinger and Weiss’s Soft Tissue Tumors. 7th Ed. Philadephia PA USA: Elsevier; 2019.
Mentzel T, Kuhnen C. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch. 2006;449:554–60.
Rekhi B, Gupta C, Chinnaswamy G, Qureshi S, Vora T, Khanna N. et al. Clinicopathologic features of 300 rhabdomyosarcomas with emphasis upon differential expression of skeletal muscle specific markers in the various subtypes: a single institutional experience. Ann Diagn Pathol. 2018;36:50–60.
Rubin BP, Hasserjian RP, Singer S, Janecka I, Fletcher JA, Fletcher CD. Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol. 1998;22:459–64.
Babai F, Musevi-Aghdam J, Schurch W, Royal A, Gabbiani G. Coexpression of α-sarcomeric actin, α-smooth muscle actin and desmin during myogenesis in rat and mouse embryos I. Skeletal muscle. Differentiation. 1990;44:132–42.
Cossu G, Biressi S. Satellite cells, myoblasts and other occasional myogenic progenitors: possible origin, phenotypic features and role in muscle regeneration. Semin Cell Dev Biol. 2005;16:623–31.
Springer ML, Ozawa CR, Blau HM. Transient production of α-smooth muscle actin by skeletal myoblasts during differentiation in culture and following intramuscular implantation. Cell Motil. 2002;51:177–86.
Nucci MR, O’Connell JT, Huettner PC, Cviko A, Sun D, Quade BJ. h-Caldesmon expression effectively distinguishes endometrial stromal tumors from uterine smooth muscle tumors. Am J Surg Pathol. 2001;25:455–63.
Watanabe K, Kusakabe T, Hoshi N, Saito A, Suzuki T. h-Caldesmon in leiomyosarcoma and tumors with smooth muscle cell-like differentiation: its specific expression in the smooth muscle cell tumor. Hum Pathol. 1999;30:392–6.
Watanabe K, Tajino T, Sekiguchi M, Suzuki T. h-Caldesmon as a specific marker for smooth muscle tumors. Comparison with other smooth muscle markers in bone tumors. Am J Clin Pathol. 2000;113:663–8.
Beck EM, Bauman TM, Rosman IS. A tale of two clones: caldesmon staining in the differentiation of cutaneous spindle cell neoplasms. J Cutan Pathol. 2018;45:581–7.
Mandahl N, Johansson B, Mertens F, Mitelman F. Disease-associated patterns of disomic chromosomes in hyperhaploid neoplasms. Genes Chromosomes Cancer. 2012;51:536–44.
Steele CD, Tarabichi M, Oukrif D, Webster AP, Ye H, Fittall M. et al. Undifferentiated sarcomas develop through distinct evolutionary pathways. Cancer Cell. 2019;35:441–56.e8.
Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding. et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–52.
Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA. et al. Comprehensive genomic characterization of adrenocortical carcinoma. Cancer Cell. 2016;30:363
Walther C, Mayrhofer M, Nilsson J, Hofvander J, Jonson T, Mandahl N. et al. Genetic heterogeneity in rhabdomyosarcoma revealed by SNP array analysis. Genes Chromosomes Cancer. 2016;55:3–15.
Coindre JM, Hostein I, Maire G, Derre J, Guillou L, Leroux A. et al. Inflammatory malignant fibrous histiocytomas and dedifferentiated liposarcomas: histological review, genomic profile, and MDM2 and CDK4 status favour a single entity. J Pathol. 2004;203:822–30.
Goldblum JR, Folpe AL, Weiss SW. Enzinger & weiss’s soft tissue tumors. Philadelphia: Elsevier, 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cloutier, J.M., Charville, G.W., Mertens, F. et al. “Inflammatory Leiomyosarcoma” and “Histiocyte-rich Rhabdomyoblastic Tumor”: a clinicopathological, immunohistochemical and genetic study of 13 cases, with a proposal for reclassification as “Inflammatory Rhabdomyoblastic Tumor”. Mod Pathol 34, 758–769 (2021). https://doi.org/10.1038/s41379-020-00703-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00703-8
This article is cited by
-
Pulmonary inflammatory leiomyosarcoma represents a potential diagnostic pitfall of DNA methylation-based classification of sarcomas: a case report
BMC Pulmonary Medicine (2023)
-
Non-cutaneous syncytial myoepitheliomas are identical to cutaneous counterparts: a clinicopathologic study of 24 tumors occurring at diverse locations
Virchows Archiv (2023)
-
Xanthogranulomatous epithelial tumors and keratin-positive giant cell-rich soft tissue tumors: two aspects of a single entity with frequent HMGA2-NCOR2 fusions
Modern Pathology (2022)
-
Myxoid pleomorphic liposarcoma is distinguished from other liposarcomas by widespread loss of heterozygosity and significantly worse overall survival: a genomic and clinicopathologic study
Modern Pathology (2022)
-
Response to Lee et al: Toward a unifying entity that encompasses most, but perhaps not all, inflammatory leiomyosarcomas and histiocyte-rich rhabdomyoblastic tumors
Modern Pathology (2021)
