
Abstract
E-cadherin is a major component of tubular adherent proteins that maintain intercellular contacts and cell polarity in epithelial tissue. It is involved in pathological processes of renal cell carcinoma and fibrotic diseases via epithelial–mesenchymal transition. Although studies have shown E-cadherin is significantly downregulated in acute kidney injury (AKI), its function in AKI is unknown. Here, we evaluated cell damage and inflammation in cisplatin-stimulated tubular epithelial cell lines after disrupting E-cadherin and restoring it with PPBICA, a small molecule identified by high-throughput screening. We also determined the therapeutic potential of restoring E-cadherin in vivo. Results show cisplatin reduced E-cadherin expression both in mouse kidney and proximal tubular epithelial cell lines (mTECs). PPBICA restored E-cadherin levels, which increased cell viability while attenuating programmed cell death. This may be mediated via deactivation of the RIPK1/RIPK3 axis and decreased caspase3 cleavage. In addition, PPBICA suppressed inflammatory response in cisplatin-treated mTECs, which correlated with suppressed NF-κB phosphorylation and promoter activity. In contrast, disruption of E-cadherin promoted cell damage and inflammation. PPBICA failed to further attenuate kidney damage in E-cadherin knockdown cells, indicating that PPBICA protects against mTECs through E-cadherin restoration. We also found that peritoneal injection of PPBICA in mice prevented loss of renal function and tubular damage by suppressing NF-κB-driven renal inflammation and RIPK-regulated programmed cell death. This was driven by restoration of E-cadherin in cisplatin nephropathy. Additionally, PPBICA attenuated cisplatin-induced kidney damage in an established AKI model, indicating its therapeutic potential in the treatment of AKI. In conclusion, E-cadherin plays functional roles in tubule integrity, programmed cell death, and renal inflammation. Our results underscore the potential of E-cadherin restoration as a novel therapeutic strategy for AKI.
Similar content being viewed by others


Introduction
Acute kidney injury (AKI) is a significant clinical problem and correlated with high morbidity and mortality rates. Recent evidence has shown severe AKI causes abnormal cellular repair and fibrosis, leading to chronic diseases and end stage renal disease (ESRD). AKI is characterized by sudden decline in renal function over a short time period, and it is always induced by hemodynamic, hypoxic, mechanical, and toxic insults [1, 2]. Although programmed cell death and inflammation are believed to play roles in the pathophysiological process of AKI, a detailed mechanism and effective therapy are yet to be determined [3, 4].
Kidney epithelial cells communicate with each other via tight junctions, including major transmembrane proteins and zonular occludins, and adhesion junctions consisting of adherent proteins like E-cadherin [5]. Loss of integrity of renal parenchyma plays critical roles in damaged kidneys of AKI. Previous findings show pharmacological inhibition of Src kinase protects against AKI in a murine model of renal ischemia/reperfusion [6]. In response to this injury, dissociation of cellular junctions and loss of cell-to-cell interplay leads to back-leak of tubular fluid and inflammatory response. This further damages tubular epithelial and endothelial cell integrity in a toxic feedback loop [7]. Therefore, restoring tubule integrity should be considered as a therapeutic tool in AKI treatment.
E-cadherin is a major component of tubular adherent proteins and maintains intercellular contacts and cell polarity in epithelial tissue by regulating actin cytoskeleton [8]. As a calcium-dependent cell adhesion molecule, E-cadherin is a transmembrane protein belonging to the classical cadherin family and widely investigated in renal cell carcinoma (RCC) and renal fibrotic diseases [9, 10]. However, these lines of evidence focused on the importance of E-cadherin in epithelial–mesenchymal transition (EMT) in the progression of renal diseases. Our preliminary studies and recent data from other groups have consistently shown that E-cadherin levels are reduced in response to cisplatin in the acute phase of injury. Although we know pharmacological inhibition of Src kinase protects against renal ischemia/reperfusion-induced injury, partly through enhancing E-cadherin and claudins [6], the exact function and detailed mechanism of E-cadherin in AKI are unknown. Thus, we used PPBICA, a small molecule identified by high-throughput screening, to enhance E-cadherin expression via histone deacetylase (HDAC)-independent mechanism. This technique is superior to HDAC inhibitor-mediated enhancement of E-cadherin, given its non-specific side effects [8]. We disrupted E-cadherin and/or restored E-cadherin in cisplatin-treated tubular epithelial cells and an animal model of cisplatin nephropathy to measure the function and therapeutic potential in AKI.
Materials And methods
Model of Cisplatin-Induced AKI and Drug Treatment
Male C57/BL mice (Laboratory Animal Center, Anhui, China) were housed in constant temperature and humidity environment with a 12:12 h light-dark cycle. All animal procedures were approved by the Animal Experimentation Ethics Committee from Anhui Medical University, Anhui, China. Mice were divided into three groups randomly at 8 weeks age. A volume of 20 mg/kg Cisplatin (Sigma, Shanghai, China) or saline was given by a single intraperitoneal injection as previously described [2]. Our pilot study indicated that 25 mg/kg PPBICA effectively attenuated renal damage without showing nephrotoxicity in vivo, so this dosage of PPBICA was injected intraperitoneally once daily for four times from one day before cisplatin treatment. Mice were killed under anesthesia while kidney tissues and blood samples were collected for further experiments, including renal function, histology, and molecular analysis.
Reagents and materials
Antibodies RIPK1, RIPK3, β-actin, and OCT2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-E-cadherin, anti-P-P65 and anti-P65 were purchased from Cell Signaling Technology (CST, Danvers, MA, USA). Rabbit anti-cleaved-caspase3 was provided by Beijing Biosynthesis Biotechnology (Bioss, Beijing, China). Lipofectamine 2000 was bought from Science Biotechnology (Invitrogen, Beijing, China). Necrostatin-1(Nec-1) and z-VAD were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Human recombinant TNF-α was purchased from R&D systems (R&D Systems Inc, MN, USA). Protein Assay Kit was from Beyotime Institute of Biotechnology (Jiangsu, China). Creatinine and blood urea nitrogen (BUN) kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). ITC AnnexinV/propidiuiodide was purchased from Bestbio (Shanghai, China). Dual-Luciferase® Reporter Assay System was from Promega Corporation.
Cell culture
MTECs (a mouse kidney proximal tubular epithelial cell line) was provided by prof. Hui Yao Lan from The Chinese University of Hong Kong as described [11]. E-cadherin was disrupted by transfection with sequence-specific shRNA or scramble control plasmid (GenePharma, Shanghai, China) by lipofectamine 2000 according to the instructions of manufacture. Cells with shRNA plasmid screened by puromycin were cultured in DMEM/F12 (HyCloneTM, USA), supplemented with 5% fetal bovine serum at 37 °C in humidified 5% CO2. Then, E-cadherin was detected by western blot and real-time PCR to evaluate the knockdown efficiency. Cisplatin (20 µM), TNF-α, TNF-α + z-VAD, and/or PPBICA (1 µM) treated cells were harvested after twenty-four hours for further analysis, including cell viability, the indexes of kidney injury, programmed cell death, inflammatory response through western blot, real-time PCR, and flow cytometry (FCM) etc. Three to four in vitro experiments were performed independently.
Cell viability assay
Cell viability was tested by using Cell Counting MTT assays. MTECs were treated with a set of concentrations of PPBICA and/or cisplatin for 24 h before being exposed to MTT (5 mg/ml) solution for 4 h. At 492 nm wavelength, optical density (OD) detection was performed by microplate reader (Multiskan MK3, Thermo, USA).
RNA extraction and real-time PCR
Total RNA was extracted from kidney homogenate or mTECs using the RNeasy Isolation Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. NanoDrop 2000 Spectrophotometer (Thermo Scientific, USA) was applied for the quantitative RNA concentration and reverse-transcribed cDNA with RealMasterMix (TOYOBO, Japan). Then target gene expression was performed using THUNDERBIRDTM SYBR®Qpcr Mix (TOYOBO, Japan) and quantified using Real-Time Polymerase Chain Reaction (RT-PCR) Detection System (TOYOBO, Japan). The sequences of primers were previously reported or as follows [12, 13]. Mouse E-cadherin, forward 5′- CGACCGGAAGTGACTCGAAAT -3′, reverse 5′- TCAGAACCACTGCCCTCGTAAT -3′. The real-time PCR data were analyzed using the relative gene expression (2-∆∆Ct) method.
Western blot analysis
Proteins were separated from pulverized tissue or cells in ice-cold RIPA-Buffer (Beyotime, Jiangsu, China), whose concentration was quantified by BCA protein kit (Beyotime, Jiangsu, China). Proteins were Supplemented with 10% SDS-PAGE and transferred to nitrocellulose membranes. After blocking, membranes were incubated overnight at 4 °C with the appropriate antibodies against KIM1, E-cadherin, P-P65, P65, RIPK1, RIPK3, Cleaved-caspase3, OCT2, and β-actin, and then incubated with IRDye 800-conjugated secondary antibody for 1.5 h at room temperature (1:10000, Rockland immunochemicals, Gilbertsville, PA). Membranes were detected by LiCor/Odyssey infrared image system (LI-COR Biosciences, Lincoln, NE) and quantified using the Image J software (NIH, Bethesda, MD, USA).
Immunofluorescence assay
For staining of E-cadherin, single-layer mTECs on cover slips were fixed by 4% parafomaldehyde for 10 min. After washing three times with PBS, the cells were blocked with 5% normal serum for 60 min at room temperature, and then labeled with appropriate antibody at 4 °C overnight. Secondary antibodies (Bioss, Beijing, China) were added for 1 h in the dark at room temperature. The nucleus was marked with DAPI. After a final wash, sections were evaluated by Inverted Fluorescence Microscope (Olympus, Japan)
Flow cytometry
For assessing programmed cell death, attached and supernatant cells were stained with 5 µL Annexin V-FITC and 5 µL PI in the dark, performed on BD FACSVerse flow cytometer machine, quantified by FlowJo 7.6 software.
Kidney histology
Kidney tissues were fixed (4% PFA), dehydrated, and embedded in paraffin. Tissue slides (4 µm thick) were stained with periodic acid–Schiff (PAS) to assess the degree of tubulointerstitial damage and subjected to relevant antibody to detect protein expression. PAS sections was scored using a previously described [2]. IHC sections were performed by a microwave-based antigen retrieval technique [14], incubated with rabbit anti-KIM1, anti-TNF-α, anti-E-cadherin, and rabbit anti-F4/80 antibody for 24 h at 4 °C and secondary antibodies for 30 min at 37 °C. Followed by labeled with chromagen liquid DAB (3, 30-diaminobenzidine tetrahydrochloride), sections were photographed using a microscope (Olympus, Japan) at ×200 magnification. We took 10 consecutive pictures in each mouse, and analyzed the average percentage of stained area. Results were analyzed by Image Analysis System (AxioVision 4, Carl Zeiss,Jena, Germany).
Statistical analysis
Data are expressed as the mean ± SE of the mean. Statistical significance was analyzed by two-tailed unpaired t-test or one-way analysis of variance (ANOVA), followed by Tukey post hoc tests using GraphPad Prism 5 software.
Results
PPBICA inhibited cisplatin-induced death of mTECs
We determine the impact of PPBICA on cell viability in a mice tubular epithelial cell line (mTEC) by MTT assay. Results show PPBICA concentrations no higher than 2 µM had limited inhibitory effect on the viability of normal TECs (Fig. 1b). And, PPBICA restored cell viability in cisplatin-treated mTECs at a concentration of 1 µM (Fig. 1c).

PPBICA inhibited cisplatin-induced death of mTECs. a Compound structure of PPBICA. b–c MTT assay results in mTECs show PPBICA concentrations no higher than 2 µM had no inhibitory effect on the viability of normal TECs. 1 µM PPBICA restored cell viability in cisplatin-treated mTECs. Data represent mean ± SEM of 3–4 independent experiments. ∗∗∗p < 0.001 compared to control. ###p < 0.001 compared with cisplatin-treated group. Cis cisplatin
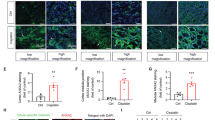
PPBICA restored E-cadherin expression and attenuated cisplatin-induced cell injury and inflammation
Immunofluorescence assay and western blot results show E-cadherin highly expressed in normal mTECs and significantly reduced after cisplatin treatment. Levels of E-cadherin were restored after PPBICA treatment (Fig. 2a, b). We then measured expression of kidney injury molecule-1 (KIM1) and found PPBICA suppressed cisplatin-induced KIM1 protein and mRNA (Fig. 2c, d). To measure the impact on inflammatory response, we analyzed common pathway cytokines. Results show PPBICA downregulated inflammatory cytokines TNF-α, IL-1β, IL-6, and chemokine MCP-1 (Fig. 2e). These findings are supported by western blot and promoter assay results showing NF-κB activation (Fig. 2f, g).

PPBICA restored E-cadherin expression and attenuated cisplatin-induced cell injury and inflammation in mTECs. a Immunofluorescence of E-cadherin in mTECs. b Western blot of E-cadherin in cisplatin-treated mTECs. PPBICA increased E-cadherin in cisplatin-stimulated mTECs. c, d Western blot and real-time PCR of KIM1 in mTECs. PPBICA significantly reduced protein and mRNA levels of KIM1. e Real-time PCR shows PPBICA decreased cisplatin-upregulated mRNA level of TNF-α, IL-1β, IL-6, and MCP-1 in mTECs. f, g Western blot and promoter assay of NF-κB. Data represent the mean ± SEM for 3–4 independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 compared with control. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with cisplatin-treated group. Cis cisplatin
PPBICA attenuated cisplatin-induced programmed cell death in mTECs
We then determine PPBICA’s effect on programmed cell death by using flow cytometric analysis. Results show PPBICA reduced the percentage of necroptotic and apoptotic cells in response to cisplatin (Fig. 3a). We then measured necroptosis using NEC-1, a necroptosis inhibitor that blocks RIPK1. After NEC-1 treatment, PPCIBA failed to further downregulate RIPK signaling and KIM1 expression in response to cisplatin (Fig. 3b). Finally, PPBICA decreased cleaved-caspase3 in cisplatin-treated cells (Fig. 3c). To further confirm the inhibitory effect of PPBICA, we established another in vitro model of necroptosis and apoptosis with TNF-α + z-VAD and TNF-α, respectively, western blot result indicates that PPBICA directly suppressed the RIPK signaling and caspase3 cleavage (Fig. 4).

PPBICA attenuated cisplatin-induced programmed cell death in mTECs. a Flow cytometry of PI/AnnexinV. PPBICA reduced percentage of apoptotic and necrotic cells in cisplatin-treated mTECs. b, c Western blot and quantitative analysis of KIM1 and programmed cell death-correlated signaling in mTECs show PPBICA substantially suppressed KIM1 and necroptosis-related indexes, including RIPK1, RIPK3, and apoptosis-related protein cleaved-caspase3. PPBICA fails to further downregulate RIPK signaling and KIM1 expression in Nec-1-treated mTECs in response to cisplatin. Data represent the mean ± SEM for 3–4 independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 compared with control. #p < 0.05, ##p < 0.01, compared with cisplatin-treated group. Cis cisplatin

PPBICA suppressed programmed cell death-related signaling pathway in mTECs. a Western blot in mTECs treated with TNF-α and z-VAD. b Western blot in mTECs treated with TNF-α. PPBICA decreased RIPK1 and RIPK3 in the in vitro necroptosis model and reduced the caspase3 cleavage in TNF-α-treated mTECs. Data represent the mean ± SEM for 4 independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 compared with control. ##p < 0.01, ###p < 0.001 compared with model group
PPBICA suppressed cisplatin-induced damage via E-cadherin-dependent mechanism
To determine the role of PPBICA in mediating protective effects, we knocked down E-cadherin in mTEC cells and measured the outcome. We found that E-cadherin mRNA and protein significantly reduced in cisplatin-stimulated mTECs (Fig. 5a, b). In addition, PPBICA had no significant effect on restoring E-cadherin (Fig. 5c). Furthermore, PPBICA failed to further reduce cisplatin-induced KIM1 and NF-κB-mediated inflammatory response in E-cadherin deficient cells (Fig. 5d–h). These findings indicate E-cadherin plays an important role in mediating the therapeutic effect of PPBICA.

PPBICA suppressed cisplatin-induced damage via E-cadherin-dependent mechanism a E-cadherin knockdown mTECs. Western blot and real-time PCR show E-cadherin downregulated after transfection of E-cadherin shRNA plasmid. c Western blot analysis of E-cadherin in cisplatin-treated mTECs. PPBICA had no significant impact on restoring E-cadherin expression in E-cadherin knockdown mTECs. d, e Western blot and real-time PCR analysis of KIM1 in E-cadherin knockdown mTECs. f Western blot analysis of P-P65 NF-κB. PPBICA failed to attenuate KIM1 expression and P65 NF-κB phosphorylation when E-cadherin was disrupted in cisplatin-treated mTECs. g Real-time PCR of inflammatory indexes. Knockdown of E-cadherin failed to further decrease inflammatory cytokine IL-1β and chemokine monocyte chemotactic protein (MCP-1) in cisplatin-treated mTECs. h. Promoter assay of NF-κB. Data represent the mean ± SEM for 3–4 independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with control. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with E-cadherin EV group. $p < 0.05, $$p < 0.01 compared with cisplatin-treated group. Cis cisplatin, Control empty vector, E-cad-KD Knockdown of E-cadherin
PPBICA restored E-cadherin level and prevented cisplatin-induced acute kidney injury in mice
To better understand how PPBICA affects E-cadherin in cisplatin-treated mice, we measured E-cadherin levels using immunohistochemistry and western blot. Results showed E-cadherin level decreased in response to cisplatin injection, but were restored by PPBICA (Fig. 6a, b). Additionally, PAS staining showed that cell swelling, cytoplasmic vacuolization, detached cell necrotic tubules, and cast formation were significantly reduced after PPBICA treatment (Fig. 6c). PPBICA also improved renal function as measured by serum creatinine and BUN (Fig. 6d, e). Western blot and IHC results show PPBICA reduced KIM1 protein level, which was supported by real-time PCR results showing lower mRNA levels (Fig. 6f–h). Collectively, these data show PPBICA had a therapeutic effect on cisplatin nephropathy.

PPBICA restored E-cadherin level and prevented cisplatin-induced acute kidney injury in mice. a, b Immunohistochemistry and western blot analysis of E-cadherin in mice. PPBICA upregulated E-cadherin in cisplatin-treated mice. c PAS staining. Histological analysis shows PPBICA reduced tubular necrosis, dilation and cast formation in cisplatin nephropathy. d, e Serum creatinine and blood urea nitrogen. PPBICA improved renal function in cisplatin nephropathy. f–h Western blot, real-time PCR, and immunohistochemistry analysis of KIM1. PPBICA attenuated cisplatin-induced upregulation of KIM1 protein and mRNA. Data represent the mean ± SEM for 6–8 mice. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with control. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with model. IHC immunohistochemistry, Cis cisplatin
PPBICA ameliorated cisplatin-induced inflammatory response and programmed cell death in mice
Results from IHC staining show PPBICA significantly suppressed macrophage infiltration (Fig. 7a). IHC staining and real-time PCR show PPBICA reduced TNF-α protein and mRNA levels, which align with the findings that PPBICA decreased mRNA levels of IL-6, IL-1β, and MCP-1 in cisplatin nephropathy (Fig. 7b, c). Western blot data show PPBICA significantly reduced phosphorylation of p65 NF-κB (Fig. 8a). And, PPBICA suppressed the necroptosis-correlated RIPK1/RIPK3 signaling and apoptosis-correlated cleavage of caspase3 (Fig. 8b). These findings highlight the protective effects of PPBICA on cisplatin-induced renal inflammation and programmed cell death in vivo.

PPBICA ameliorated cisplatin-induced inflammatory response in mice a Immunohistochemistry of F4/80 + macrophages. b Immunohistochemistry of TNF-α. PPBICA attenuated inflammatory response, including recruitment of macrophages and secretion of TNF-α. c Real-time PCR analysis of proinflammatory cytokines and chemokines. Inflammatory factors, including TNF-α, IL-6, IL-1β, and MCP-1, were decreased by PPBICA treatment. Data represent the mean ± SEM for 6–8 mice. ∗p < 0.05,∗∗p < 0.01, ∗∗∗p < 0.001 compared with control. ##p < 0.01, ###p < 0.001 compared with model. IHC immunohistochemistry, Cis cisplatin

PPBICA inhibited inflammation and programmed cell death-related pathways in cisplatin nephropathy. a Western blot and quantification of P-P65. PPBICA prevented NF-κB-driven renal inflammation in cisplatin nephropathy. b Western blot analysis of programmed cell death-related molecules. PPBICA suppressed the activation of RIPK1/RIPK3 axis and cleavage of cleaved-caspase3. Data represent the mean ± SEM for 6–8 mice. ∗∗p < 0.01, ∗∗∗p < 0.001 compared with control. ##p < 0.01, ###p < 0.001 compared with model. IHC immunohistochemistry, Cis cisplatin
PPBICA attenuated cisplatin-induced kidney injury in established AKI mouse model
To determine the therapeutic potential of PPBICA in an established AKI mouse model, we injected PPBICA one day after cisplatin injection. PAS staining shows PPBICA attenuated kidney damage (Fig. 9a). This was further supported by results from serum creatinine and BUN assays and immunostaining of KIM1 (Fig. 9b–d).

PPBICA attenuated cisplatin-induced kidney injury in established AKI mouse model. a PAS staining. b, c. Serum creatinine and blood urea nitrogen assays. d IHC analysis of KIM1. PPBICA attenuated kidney damage in established acute kidney injury model. Data represent the mean ± SEM for 6–8 mice. ∗p < 0.05, ∗∗∗p < 0.001 compared with control. #p < 0.05, ##p < 0.01, ###p < 0.001 compared with model. IHC immunohistochemistry, Cis cisplatin
Discussion
In the current study, we provided novel evidence showing E-cadherin was significantly reduced in cells or kidney tissues challenged by cisplatin. Disruption of E-cadherin increased apoptosis and necroptosis while activating inflammatory response in cisplatin-treated tubular epithelial cells. Restoration of E-cadherin by administration of PPBICA protected against cisplatin-induced injury both in vivo and in vitro via inhibiting renal inflammation and programmed cell death (Shown in Fig. 10).

Restoration of E-cadherin by PPBICA attenuated cisplatin-induced AKI by attenuating inflammation and programmed cell death
E-cadherin is a key adherent protein in the formation of adhesion junctions, which are critical to tubule integrity in normal kidney. E-cadherin has been extensively investigated in regulating phenotype of tubular epithelial cells via EMT in the kidney field. Not surprisingly, E-cadherin deficiency has been found in several types of cancers [15]. In fact, evidence shows that reduced E-cadherin is correlated with β-catenin expression and poor prognosis of renal RCC; and, E-cadherin prevents β-catenin translocation from membrane to cytoplasm, suppressing wnt/β-catenin signaling [16]. A recent study showed that inhibition of E-cadherin-α/E-catenin complex by miR-720 promoted RCC [17], indicating a preventive role of E-cadherin in RCC. In the fibrotic kidney, loss of E-cadherin, which is a marker for epithelial cells, and gain of α-SMA expression, the marker for myofibroblasts, is a key process in myofibroblast transdifferentiation from epithelial origin [18]. Although the concept that activated myofibroblasts are mainly derived from epithelial cells is challenged, new data show that dedifferentiated tubular epithelial cells engage a profibrotic gene expression. This drives interstitial fibrosis without completely transiting to α-SMA + myofibroblasts [19, 20]. This data support the important role of E-cadherin in renal fibrosis. However, the detailed function of E-cadherin in acute injury of kidney cells has not been fully understood. We found that E-cadherin expression was substantially reduced by cisplatin in proximal tubular cells in vivo and in vitro. Knockdown of E-cadherin enhanced the production of kidney injury marker KIM1, but restoration of E-cadherin by PPBICA effectively suppressed KIM1 expression, and reduced cisplatin-induced programmed cell death. These results are consistent with recent finding that pharmacological inhibition of Src kinase prevented the degradation of several adherens and tight junction proteins, which includes E-cadherin, ZO-1 and claudins, in the I/R kidney, and oxidative stress-challenged renal tubular epithelial cells [6]. Additionally, we found that PPBICA did not further protect against renal injury in absence of E-cadherin, indicating PPBICA functions specifically via an E-cadherin-dependent mode.
The mechanisms by which PPBICA prevents cisplatin-induced nephrotoxicity were then evaluated. PPBICA not only limited programmed cell death induced by TNF-α and TNF-α plus z-VAD, but also prevented cisplatin-induced necroptosis and apoptosis. Our previous study showed that cisplatin induced the activation of caspase3-dependent apoptosis and RIPK-mediated necroptosis of tubular epithelial cells [2, 21, 22]. This may lead to loss of tubular epithelial cells and tubule integrity, where adherent junctions are further damaged, back-leaking of tubular fluid and inflammatory responses accelerated, which causes renal dysfunction. However, when the E-cadherin was restored, tubular epithelial cells showed resistance to cisplatin and the cell viability was upregulated. RIPK1/RIPK3 axis activation and cleavage of caspase3 were also inhibited. Additionally, PPBICA attenuated cisplatin-induced inflammatory response, which was evidenced by reduced production of proinflammatory cytokines including TNF-α, IL-1β, IL-6, MCP-1, and NF-κB. Regarding underlying mechanisms, we offer the following explanations. First, PPBICA-mediated restoration of E-cadherin may recover the tubule integrity while limiting back-leak of tubular fluid and subsequent inflammatory response. Second, proximal tubular cells succumb to necroptosis release damage-associated molecular patterns (DAMPs) including high-mobility group box 1 (HMGB1), heat-shock proteins, uric acid, and IL-33. These may activate inflammation-related signaling pathways, such as Toll-like receptors, and form feedback loops of inflammatory response called necroinflammation [23, 24]. In this setting, the anti-necroptotic effects of PPBICA may also contribute to the attenuation of renal inflammation. Third, we know overexpression of E-cadherin, which is highly induced in alternatively activated macrophages (M2), suppresses inflammatory response in macrophages, indicating the novel function of E-cadherin in immune cells [25]. In this setting, the regulative role of E-cadherin on macrophage infiltration and ratio of M2/M1 cells may be also responsible for anti-inflammatory effect of PPBICA in treatment of AKI. Additionally, we detected whether PPBICA limited kidney damage by affecting the cellular uptake of cisplatin. Organic cation transporters-2 (OCT2) is well accepted as a critical transporter in mediating the absorption of cisplatin. In the current study, we found PPBICA did not alter OCT2 expression, indicating PPBICA may function through an OCT-independent mechanism [26].
Importantly, by designing both prevention and treatment studies, we evaluated the E-cadherin-targeted therapy in cisplatin nephropathy by peritoneal injection of PPBICA. Results showed that PPBICA restored E-cadherin expression in injured kidney. Consequently, it prevented the decline of renal function and cell damage, which may be correlated with its inhibitory roles on NF-κB-driven renal inflammation and RIPK-mediated programmed cell death. Finally, it is important to note that PPBICA also has protective effects in kidney injury in established AKI models.
In conclusion, E-cadherin has fundamental and functional roles in controlling tubule integrity, programmed cell death, and renal inflammation. In this regard, restoration of E-cadherin by PPBICA should be considered as a novel therapeutic strategy for acute kidney injury.
References
Sharp CN, Doll MA, Dupre TV, et al. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am J Physiol Ren Physiol. 2016;310:F560–68.
Gao L, Wu WF, Dong L, et al. Protocatechuic aldehyde attenuates cisplatin-induced acute kidney injury by suppressing nox-mediated oxidative stress and renal inflammation. Front Pharmacol. 2016;7:479.
Sancho-Martinez SM, Lopez-Novoa JM, Lopez-Hernandez FJ. Pathophysiological role of different tubular epithelial cell death modes in acute kidney injury. Clin Kidney J. 2015;8:548–59.
Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. 2016;67:293–307.
Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–21.
Xiong C, Zang X, Zhou X, et al. Pharmacological inhibition of Src kinase protects against acute kidney injury in a murine model of renal ischemia/reperfusion. Oncotarget. 2017;8:31238–53.
Jang HR, Rabb H. The innate immune response in ischemic acute kidney injury. Clin Immunol. 2009;130:41–50.
Stoops SL, Pearson AS, Weaver C, et al. Identification and optimization of small molecules that restore E-cadherin expression and reduce invasion in colorectal carcinoma cells. ACS Chem Biol. 2011;6:452–65.
Onder TT, Gupta PB, Mani SA, et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–54.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–38.
Meng XM, Ren GL, Gao L, et al. Anti-fibrotic effect of wogonin in renal tubular epithelial cells via Smad3-dependent mechanisms. Eur J Pharmacol. 2016;789:134–43.
Meng XM, Huang XR, Xiao J, et al. Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J Pathol. 2012;227:175–88.
Meng XM, Huang XR, Xiao J, et al. Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012;81:266–79.
Meng XM, Huang XR, Chung AC, et al. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol. 2010;21:1477–87.
Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796:75–90.
Zhang X, Yang M, Shi H, et al. Reduced E-cadherin facilitates renal cell carcinoma progression by WNT/beta-catenin signaling activation. Oncotarget. 2017;8:19566–76.
Bhat NS, Colden M, Dar AA, et al. MicroRNA-720 regulates E-cadherin-alphaE-catenin complex and promotes renal cell carcinoma. Mol Cancer Ther. 2017;16:2840–8.
Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol. 2014;10:493–503.
Grande MT, Sanchez-Laorden B, Lopez-Blau C, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21:989–97.
Lovisa S, LeBleu VS, Tampe B, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998–1009.
Meng XM, Ren GL, Gao L, et al. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab Invest. 2018;98:63–78.
Meng XM, Li HD, Wu WF, et al. Wogonin protects against cisplatin-induced acute kidney injury by targeting RIPK1-mediated necroptosis. Lab Invest. 2018;98:79–94.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5.
He S, Liang Y, Shao F, et al. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 2011;108:20054–9.
Van den Bossche J, Laoui D, Naessens T, et al. E-cadherin expression in macrophages dampens their inflammatory responsiveness in vitro, but does not modulate M2-regulated pathologies in vivo. Sci Rep. 2015;5:12599.
Ciarimboli G, Deuster D, Knief A, et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am J Pathol. 2010;176:1169–80.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81570623), Science and Technological Fund of Anhui Province for Outstanding Youth of China (Grant number: 1608085J07), and by Anhui Provincial Natural Science Foundation (No. 1508085QH196).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
These authors contributed equally: Li Gao, Ming-Ming Liu and Hong-mei Zang.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Gao, L., Liu, MM., Zang, Hm. et al. Restoration of E-cadherin by PPBICA protects against cisplatin-induced acute kidney injury by attenuating inflammation and programmed cell death. Lab Invest 98, 911–923 (2018). https://doi.org/10.1038/s41374-018-0052-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0052-5
This article is cited by
-
Histone methyltransferase MLL1 drives renal tubular cell apoptosis by p53-dependent repression of E-cadherin during cisplatin-induced acute kidney injury
Cell Death & Disease (2022)
-
LncRNA TUG1 regulates the development of ischemia-reperfusion mediated acute kidney injury through miR-494-3p/E-cadherin axis
Journal of Inflammation (2021)
-
Blocking Connexin-43 mediated hemichannel activity protects against early tubular injury in experimental chronic kidney disease
Cell Communication and Signaling (2020)
-
3-deazaneplanocin A protects against cisplatin-induced renal tubular cell apoptosis and acute kidney injury by restoration of E-cadherin expression
Cell Death & Disease (2019)
