
Abstract
Hematopoiesis can occur outside of the bone marrow during inflammatory stress to increase the production of primarily myeloid cells at extramedullary sites; this process is known as extramedullary hematopoiesis (EMH). As observed in a broad range of hematologic and nonhematologic diseases, EMH is now recognized for its important contributions to solid tumor pathology and prognosis. To initiate EMH, hematopoietic stem cells (HSCs) are mobilized from the bone marrow into the circulation and to extramedullary sites such as the spleen and liver. At these sites, HSCs primarily produce a pathological subset of myeloid cells that contributes to tumor pathology. The EMH HSC niche, which is distinct from the bone marrow HSC niche, is beginning to be characterized. The important cytokines that likely contribute to initiating and maintaining the EMH niche are KIT ligands, CXCL12, G-CSF, IL-1 family members, LIF, TNFα, and CXCR2. Further study of the role of EMH may offer valuable insights into emergency hematopoiesis and therapeutic approaches against cancer. Exciting future directions for the study of EMH include identifying common and distinct EMH mechanisms in cancer, infectious diseases, and chronic autoimmune diseases to control these conditions.
Similar content being viewed by others

Introduction
Hematopoiesis is the continuous process by which blood and immune cells are produced by the actions of hematopoietic stem cells (HSCs). Thought to be organized similarly to an elevated water source draining into a branching system of rivers, HSCs can continue to self-renew and differentiate to produce mature differentiated cells, including red blood cells, T-cell lineages, B-cell lineages, monocyte lineages, and neutrophils. A critical component of hematopoiesis is the niche that regulates HSC self-renewal and differentiation, which is crucial for hematopoietic output. The niche includes both hematopoietic and nonhematopoietic lineages that perform unique but sometimes overlapping roles. While occurring primarily in the bone marrow of adult animals, hematopoiesis can occur in extramedullary sites during times of organismal stress to increase or sustain hematopoietic output, a phenomenon known as extramedullary hematopoiesis (EMH). In cancer, EMH is increasingly recognized as a mechanism by which cancer cells generate a favorable immune environment for growth. For example, tumor cells can utilize EMH to produce immunosuppressive hematopoietic subsets.
Cancer is a major health concern in the United States of America and globally. In the United States, cancer is the second leading cause of death and costs more to treat than any other disease1. Like other chronic inflammatory pathologies, including arthritis and myocardial infarction, solid tumors enhance the production of myeloid cells, termed myelopoiesis, to further their own growth at the expense of their host2,3,4. In part, this increased myelopoiesis leads to a high ratio of neutrophils to lymphocytes in the peripheral blood, which is correlated with poor survival in breast, colon, pancreatic, and gastric cancer patients and in a systematic review of all cancer types5,6,7,8. As immune-based cancer therapeutics become more widely used, attention has turned to modulating myeloid cells in the tumor microenvironment to improve their efficacy9. One potential avenue is to target their production at the extramedullary hematopoietic site. Here, we review the role of EMH in cancer and other inflammatory conditions and the proteinaceous factors contributing to EMH in adults. Moreover, we discuss the hematopoietic niche in various hematopoietic organs to deepen the understanding of the unique contributions of EMH to physiological and pathological outcomes.
EMH in cancer
EMH refers to the expansion of blood cells in the extramedullary sites of a mature animal in response to an altered organismal state. EMH does not include hematopoiesis in organs such as the thymus at homeostasis and in any organ during development. There are three general processes that induce EMH: (1) trapping of proliferative hematopoietic progenitors in the spleen during hyposplenism; (2) impairment of hematopoietic capacity in the bone marrow due to damage or myelophthisis; and (3) abnormal levels of circulating factors with extramedullary hematopoietic capabilities10. Although the most common organs involved in EMH are the spleen, liver, and lymph nodes, organs as diverse as the skin, pleura, adrenal gland, and pancreas have demonstrated EMH activity11,12,13. For factor-induced EMH, hematopoiesis-active cytokines and pathogen-associated molecular patterns, including G-CSF, GM-CSF, IL-3, and IL-6:sIL-6R complexes, as well as lipopolysaccharide and Pam3CSK4, respectively, have been shown to play roles in stimulating EMH14,15,16,17. Some instances of EMH induced by these factors have marked effects. Subcutaneously injected human IL-3 was reported to induce cutaneous hematopoiesis with trilineage potential at the injection site in cynomolgus monkeys17. One patient was reported to have trilineage cutaneous hematopoiesis following G-CSF therapy to treat myelofibrosis18.
The role of EMH in cancer is a rapidly growing research focus. EMH is associated primarily with hematologic cancers but can also occur in patients with breast, lung, renal, colon, gastric, pancreatic, or prostate cancer4,11,13,19. In particular, splenic EMH has been recognized for more than 30 years in the context of human solid tumors with or without bone marrow metastases20. The presence of EMH is potentially important for cancer treatment because EMH preferentially induces the production of myeloid cells. In the context of cancer, increased myeloid cells are recognized as having a negative impact on survival. A high ratio of neutrophils to lymphocytes in the peripheral blood was correlated with poor prognosis in a systematic review of all cancer types, and in pancreatic, colon, breast, and gastric cancers individually5,6,7,8. In a case series on EMH, Bao et al.11 reported that breast cancer was the most common solid cancer reported with EMH, 7.1% of all patients had confirmed EMH in the spleen, and 24% of patients had splenomegaly, which is clinically associated with splenic EMH. The first mouse model of splenic EMH in the context of solid tumors was created by Cortez-Retamozo et al.21, in which the authors demonstrated that splenic myeloid progenitors contributed directly to the tumor environment and tumor growth and that human spleens similarly expanded myelopoietic progenitors during invasive solid tumor progression. Wu et al.22 suggested that increased circulating myelopoietic progenitors in patients with solid tumors contributed to suppressing myeloid cell generation and the formation of an immunosuppressive tumor microenvironment. Subsequently, Wu et al.4 reported increased human hematopoietic stem and progenitor cells (HSPCs) in the red pulp of the spleen in four separate solid tumors and demonstrated that gastric cancer patients with low levels of EMH in their red pulp have a more favorable prognosis. Additionally, these authors found that the splenic HSPCs generated in their mouse model of hepatocellular carcinoma were myeloid-biased and showed modifications to the splenic niche4. A more recent paper identified HSPCs within the tumor mass of human glioblastomas and revealed that their presence was associated with tumor grade23. We recently evaluated the role of EMH specific, myeloid-expanded HSPCs and their splenic niche in EMH initiation and maintenance24. Together, these data establish a series of novel and important discoveries about the presence and role of EMH in a diverse set of solid tumors. Furthermore, given the correlation between the increased neutrophil to lymphocyte ratio and poor prognosis in patients with solid tumors, targeted, prospective studies to identify EMH sites and EMH-inducing factors in patients with solid tumors and concomitant peripheral myeloid skewing may be justified.
EMH may not mimic all aspects of bone marrow hematopoiesis. EMH seems to occur in humans most commonly because of a loss of hematopoietic capacity in the bone marrow, in agreement with the observation that EMH occurs secondarily to hematologic malignancy13. When eliminating hepatosplenic hematopoiesis, the most common condition associated with EMH was myelofibrosis with myeloid metaplasia, and the most common location was the thoracic vertebral column25. Generally, EMH is associated with myeloid- or erythroid-biased differentiation, although extrathymic T-cell development has been reported in transgenic models26. The mobilization of HSPCs often coincides with the initiation of EMH and is important in seeding cells for this process. However, the extent to which the proliferation of local progenitors contributes to EMH is unknown. Additionally, the various factors that induce EMH begs the question of the degree of diversity that exists within the broad framework of EMH. Is EMH a single unified endpoint for multiple types of inflammation that broadly increase myelopoietic capacity, or do different forms of inflammation produce distinct types of EMH? Answering this question may shed light on the contribution of EMH to various disease states.
EMH and myelopoiesis
Myelopoiesis, the production of myeloid lineage cells from HSPCs, is a process that occurs during homeostasis but is also highly responsive to the organismal state27,28. The development of myeloid cells involves a complex balance of select transcription factors throughout the differentiation process29. The transcription factors that favor myeloid differentiation include PU.1, CCAAT/enhancer binding proteins (C/EBPα, C/EBPβ and C/EBPε), growth factor independent 1 (GFI1), and interferon-regulatory factor 8 (IRF8)30,31,32,33,34,35. PU.1 is a crucial transcriptional regulator of myeloid differentiation, followed by IRF8, which delineates the monocyte lineage, and C/EBPα, which delineates the neutrophil lineage30,31,35. In times of organismal stress, the nature of factors driving myelopoietic differentiation can be altered. For instance, mice lacking Cebpb maintain normal granulocytopoiesis while in homeostasis but fail to induce emergency granulocytopoiesis during challenge36. Cytokine factors, including G-CSF, GM-CSF, M-CSF, IL-1, and IL-27, can stimulate the production of myeloid cells37,38,39,40,41.
The tumor microenvironment (TME) contains a diverse population of myeloid cells similar to that generated during homoeostasis, including neutrophils, macrophages, and dendritic cells, as well as subsets not present at homeostasis, such as myeloid-derived suppressor cells (MDSCs)42,43,44. This review will focus on granulocytic MDSCs (G-MDSCs) due to their association with solid tumors, the ongoing study of their developmental origin, and their presumed effects on treatment outcomes and prognosis. Compared with homeostatic neutrophil subsets, G-MDSCs exhibit positive CD84 expression in mice and humans and positive LOX1 expression in humans in addition to their functional capacity to inhibit T-, B-, and NK-cell activation44,45,46. It is important to note that many historical studies have not separated neutrophils from tumor-associated neutrophils or neutrophils from G-MDSCs due to their similar surface phenotype and the recent discovery of distinctive markers. Because G-MDSCs have been shown to be the most abundant neutrophil subset in the circulation and in the TME during late-stage tumor progression, unless the cited paper specifically distinguishes neutrophils in cancer from G-MDSCs, it is reasonable to assume that these neutrophils are indeed G-MDSCs.
In addition to the expansion of myelopoiesis, changes in the cytokine milieu also impact the cellular products of myelopoiesis, most prominently, the production of G-MDSCs44. Many factors are required for stimulating emergency granulocytopoiesis, and a secondary factor directs the polarization of G-MDSCs toward an immunoregulatory phenotype47. Two particularly important signaling pathways are the NF-κB and STAT1 and STAT3 pathways48. The factors implicated in inducing immunosuppressive activity through the NF-κB pathway include TNFα, IL-1β, and Toll-like receptor ligands, while IFNγ is most commonly linked to STAT1 activation49,50. G-CSF may be important for inducing STAT3 activation in developing G-MSDCs51,52. Interestingly, G-CSF has also been shown to promote the development of immunosuppressive neutrophils at the expense of dendritic cells capable of cancer immunosurveillance53,54. Together, these data implicate an extensive list of cytokines that could contribute to the pathological myelopoiesis identified in cancer. Future research should strive to identify the factors that initiate the cascade of myelopoiesis and the relative contributions of immune-derived and tumor-derived factors to the induction and maintenance of myelopoiesis. Additionally, these studies did not address the contribution of EMH to the induction of pathological myelopoiesis, and emerging data suggest that EMH is an important producer of pathological G-MDSCs in the context of solid tumors.
G-MDSCs are recruited into the tumor environment through the binding of their CXCR2 receptor to CXCL1, CXCL2, and CXCL855. In keeping with its importance in the TME, high systemic CXCL8 levels in tumors increase the number of neutrophils, presumably G-MDSCs, and recruit cells to the tumor microenvironment, reducing the efficacy of anti-PD-L1 therapy56,57. Conversely, CXCR2 deletion or the use of anti-Ly6G antibodies in mouse models slows tumorigenesis58,59,60,61,62. Once in the TME, G-MDSCs maintain immunosuppression through the promotion of angiogenesis and metastasis and reduced responsiveness to immune checkpoint blockade57,63,64. To promote angiogenesis, G-MDSCs have been shown to express MMP9, BV8, and VEGF65,66,67,68. G-MDSCs have also been shown to promote metastasis by enhancing the epithelial–mesenchymal transition of tumor cells through the upregulation of hepatocyte growth factor and TGF-beta69. At the site of metastasis, G-MDSCs can capture circulating tumor cells through neutrophil extracellular traps and surface markers59,70. The presence of granulocytes with altered phenotypes in the presence of chronically inflamed, solid tumors has sparked investigations into the developmental processes that lead to their production and the crucial mechanistic processes that can be targeted for therapeutic intervention.
Bone marrow hematopoietic niche
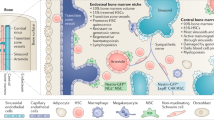
To understand how alterations in hematopoiesis occur during cancer, one must understand the hematopoietic niche of the bone marrow, as this region is the primary site of hematopoiesis during homeostasis. The bone marrow HSC niche exists in a complex microanatomical environment that fosters the differentiation and self-renewal of HSCs while also directing their response to organismal changes71,72. While many cell types have been implicated to play a role in this niche, here, we will focus on perivascular stromal cells, endothelial cells, sympathetic nerves, and macrophages. Prominent within the bone marrow hematopoietic niche are cell types known as perivascular stromal cells, mesenchymal stem cells, or osteoblast precursor cells, which will be collectively treated as the same cell type here, as recent evidence does not support their separation into distinct cell types. The expression of both CXCL12 and KIT ligands distinguishes these cells from other constituents of the bone marrow niche73,74. These mesenchymal stem cells in the bone marrow most commonly distinguished by leptin receptor, nestin, Mx1, Prx1, Osx, PDGFRα, or CD51 expression75,76,77. Initial studies also identified osteoblasts as critical components of the niche78,79. However, when CXCL12 or KIT ligands were deleted from mature osteoblasts, no significant changes in hematopoietic lineages were observed74,77,80. This finding likely reflects the fact that mesenchymal stem cells of the bone appear capable of differentiating into osteoblasts in vitro and were misconstrued as representing mature osteoblasts in early studies81. In fact, under proper culture conditions, mesenchymal stem cells are capable of maintaining HSCs in vitro75,76. Endothelial cells of the bone marrow contribute multiple factors that play a role in the bone marrow niche, including E-selectin, basic FGF, DLL1, IGFBP2, angiopoietin 1, DHH, and EGF82,83,84,85,86,87.
Within the bone marrow, the vascular niche is thought to be split into an arteriolar and a sinusoidal-megakaryocyte component. The arteriolar components were first identified as the preferential location of quiescent HSCs in the endosteal region of the bone marrow88. In addition to endothelial cells and mesenchymal stem cells, the arteriolar niche includes sympathetic neurons and nonmyelinating Schwann cells, each with their own niche contribution. Sympathetic neurons alter CXCL12, angiopoietin 1, KIT ligand, and VCAM-1 expression in mesenchymal stem cells through β3-adrenergic receptor signaling and thus enhance mobilization89,90. Schwann cells contribute to the activation of TGFβ, a regulator of HSC quiescence91,92. In addition to the endothelial and mesenchymal stem cell components, the venous sinusoidal niche also contains megakaryocytes that reduce HSC proliferation through CXCL4 and TGFβ but also promote recovery after radioablation through FGF193,94,95,96. Macrophages also effect the bone marrow niche through their regulation of CXCL12 expression on mesenchymal stem cells97,98. The bone marrow niche also responds to a variety of signals, including circadian rhythms, prostaglandins, pathogen-associated molecular patterns, and hormones89,99,100,101,102. Ironically, in a model of primary myelofibrosis in the bone marrow niche, the overgrowth of mesenchymal stem cells reduces the amount of the marrow space available for hematopoietic cells103.
Additionally, the hematopoietic niche undergoes remodeling in response to myelopoietic stimuli during aging or in patients with obesity104,105,106,107,108. Phenotypes associated with aging can be rescued by altering sympathetic signaling within the bone marrow, indicating that sympathetic nervous system activity may play a part in age-related changes in hematopoiesis106,107. In obesity, the role of adipocytes in modulating the niche has been an important topic of study109. However, varying effects of adipocytes on HSPC maintenance and differentiation have been reported. Initial studies linked BM with high adiposity to lower hematopoietic output110,111. Other data have shown that BM adipose tissue is capable of producing important hematopoietic cytokines, such as KITL and CXCL12, while being able to support hematopoiesis in vitro112,113,114. Additionally, adipocytes are recognized as contributing myeloid-biasing cytokines such as TNFα and IL-6115,116. When taken together, the components of the bone marrow niche supply many factors, often in conflict with each other, that drive and alter continued hematopoietic function.
Extramedullary hematopoietic niche
EMH is an important topic in clinical medicine and offers numerous opportunities to further our understanding of hematopoiesis itself. One area of interest is understanding the extramedullary niche as a unique tissue that does not necessarily mimic the bone marrow but nevertheless recapitulates the principal factors involved in hematopoietic development. However, the diverse niche components that are involved in hematopoiesis in nonbone marrow sites are poorly characterized. While several aspects of the splenic hematopoietic niche in adult animals have been studied, comparatively few studies have evaluated the liver, lymph node, or skin niche. In addition, EMH of the spleen is localized to sinusoids of the red pulp, where both mesenchymal stem cells and endothelial cells produce the KIT ligand and only mesenchymal stem cells produce CXCL12117. In a liver model of EMH, CXCL12 appeared to be upregulated in sinusoidal endothelial cells118. Moreover, KIT ligand and CXCL12 double-positive cells was present within the lesion of an adult patient with nodular, cutaneous EMH119.
Despite these similarities, tissue-specific differences have also been identified. Splenic mesenchymal stem cells are leptin receptor-negative and express Tlx1120. Additionally, some supporting cell types in the spleen appear to be different from those in the bone marrow. For instance, a decrease in NK cells in the spleen was associated with increased myeloid progenitors, suggesting that NK cells negatively regulate splenic hematopoiesis. However, some studies support the notion that T cells in the spleen act as hematopoietic niche cells121,122. Finally, macrophages play a role in supporting erythropoiesis and hematopoiesis in the spleen123,124,125,126. Taken together, what is known about EMH suggests that certain core factors are required for hematopoiesis in any organ and that there are organ-specific cell types or factors that can modulate these core processes.
Cytokines with EMH potential
Below, this article provides a detailed look at individual cytokines with the potential to induce hematopoiesis and attempts to shed light on the mechanisms underlying the interaction between cancer and hematopoiesis.
KIT ligand
KIT ligand is a fundamental hematopoietic growth factor and ligand of the receptor c-KIT127,128. KIT ligand has soluble and transmembrane forms129. Mice that lack function in both forms have severe or fatal anemia due to hematopoietic failure and pigment and germ cell deficiencies130. Mice lacking only the transmembrane form were still anemic, lacked pigmentation in the coat and were sterile129. Furthermore, the low body weight, anemia, and bone marrow cellularity in mice lacking transmembrane KIT ligands could not be mitigated by the overexpression of soluble KIT ligands but could be ameliorated with membrane-restricted KIT ligands131. Conversely, overexpressing membrane-restricted KIT ligands did not rescue reduced bone marrow myeloid progenitors in mice lacking transmembrane KIT ligands, but the overexpression of soluble KIT did131, and the number of total peripheral blood leukocytes were rescued by both131. Finally, compared to wild-type mice, mice expressing KIT ligand lacking the main cleavage site, and therefore lacking the majority of soluble KIT ligands, did not exhibit differences in the number of hematopoietic progenitors in the bone marrow or in mature cells in the blood132.
The binding of KIT ligand to c-KIT dimerizes the receptor and activates its intrinsic tyrosine kinase activity133,134,135. The signaling characteristics vary between the different KIT ligand forms. Soluble KIT ligand signals rapidly and transiently followed by receptor degradation, while the membrane-associated form has sustained signaling135. Compared to soluble KIT ligands, membrane-associated KIT ligands induce longer-lasting downstream ERK1/2 and MAP kinase activity136. The inability of c-KIT to be internalized when bound to a membrane-associated KIT ligand may cause its sustained signaling, as immobilized anti-c-KIT antibodies also exhibit sustained signaling137. These data indicate that both forms of the KIT ligand are unique and functionally important and that the transmembrane form may play more important roles. Despite the importance of the c-KIT/KIT ligand for maintaining hematopoiesis, c-KIT signaling is also implicated in the quiescence of HSCs138. Additionally, c-KIT has been found to enhance the signaling of the EPO receptor and the IL-7 receptor, two important hematopoietic cytokines, as well as PDGFRα139,140,141. The KIT ligand and its receptor c-KIT clearly play crucial roles in hematopoiesis and have biological benefits befitting its centrality. The expression of c-Kit is likely to be a critical player in EMH, and studies investigating its regulation in the context of EMH are needed.
CXCL12
CXCL12 is a pleotropic chemokine that plays various roles in development, hematopoiesis, inflammation, and injury repair. In hematopoiesis, CXCL12 is considered the major cytokine produced by the stem cell niche to retain HSPCs in the niche142,143. CXCL12 belongs to the CXC family of chemokines. Structurally, CXC family members have two conserved N-terminal cysteine residues that are separated by one variable residue, and these chemokines signal primarily through GPCRs144,145 The receptor predominantly recognized for binding CXCL12 is CXCR4, while binding to ACKR3 also occurs146,147,148,149,150. CXCR4 knockout animals die perinatally due to a combination of hematopoietic, neurogenic, vascular, and cardiogenic defects151. In contrast, ACKR3 knockout mice still die perinatally but exhibit normal hematopoiesis152,153. Signaling through CXCR4 is complicated and involves the activation of various Gα proteins, leading to the activation of the MAPK, PLC, and PI3K pathways154,155. Additionally, G protein-independent signaling pathways were identified. Although JAK-STAT signaling in CXCL12 cells has been reported156,157,158, other evidence challenges the importance of this pathway159. Activation of signaling downstream of beta-arrestin has also been reported160,161,162. CXCL12 and its receptor CXCR4 are uniquely functionally related in the context of hematopoiesis. However, the contribution of CXCL12 to the induction and maintenance of EMH is currently unknown, and given its role in maintaining hematopoiesis in the bone marrow, CXCL12 may even be a counterregulatory cytokine to EMH.
Granulocyte colony-stimulating factor
Granulocyte colony-stimulating factor (G-CSF) is a cytokine that has potent hematopoiesis activity163. G-CSF signals through the homodimeric receptor G-CSFR164. Downstream of receptor activation is signaling by the JAK-STAT pathway, particularly through STAT3 and STAT5, which both promote proliferation, while STAT3 promotes differentiation165. G-CSF is thought to be produced primarily by stromal cells, such as fibroblasts and bone marrow niche cells, and by activated myeloid cells166. G-CSF levels are low during homeostasis but can increase during an immune response and subsequently decrease to baseline167,168. Mice lacking either G-CSF or its receptor have severe neutropenia but still produce small amounts of neutrophils37,169. G-CSF is also able to mobilize HSCs into circulation and establish EMH18,170.
IL-1α and IL-1β
The IL-1 cytokine superfamily has 11 members corresponding to 10 different receptors. Within this cytokine family, there are three subfamilies: IL-1, IL-18, and IL-36. The unifying features of cytokines in this family include their lack of a signal peptide for secretion, cytoplasmic distribution as precursor molecules and the presence of a β-trefoil pyramidal barrel structure composed of six two-stranded β hairpins171,172. However, IL-1 receptor antagonist differs from the other family members regarding these shared features, as it lacks all of them. IL-1 signaling is transmitted through a trimeric complex comprised of the IL-1 receptor, the IL-1 family cytokines, and a coreceptor. When cytokines bind to primary receptors, they recruit coreceptors, and signaling can occur on the cytoplasmic side. For receptors with Toll-IL-1 receptor domains, MyD88 and downstream NF-κB signaling are activated. Additionally, the coreceptor can be present in a soluble form, either cleaved from the cell surface or produced by the liver. Both IL-1α and IL-1β signal through the IL-1R1 receptor and the coreceptor IL-1R3, both of which have Toll-IL-1 receptor domains. IL-1β binds to IL-1R2 with IL-1R3 as its coreceptor. IL-1β is a prototypical IL-1 family member. It is a very potent inflammatory molecule that plays a role in numerous diseases, including atherosclerosis and cancer173,174. IL-1β transcripts are induced by TLR ligands and by IL-1 itself. Following translation, IL-1β is in an inactive form until it is cleaved intracellularly by proteases such as the inflammasome or extracellularly by proteases such as proteinase 3, elastase, MMP-9, granzyme A, and mast cell chymase171,175. When recognized by HSCs, IL-1β stimulates proliferation and myelopoiesis39. IL-1α is considered a classic damage-associated molecular pattern that can initiate immune responses, and its biology reflects this process176,177. IL-1α is an unusual member of the IL-1 family because it is constitutively present in epithelial and mesenchymal cell types and does not require proteolysis for activation171. When proteolytically processed from its pro-form by proteases such as granzyme B, IL-1α becomes up to 10 times more potent178. IL-1α also has a nuclear localization signal, and when apoptosis occurs, IL-1α traffics to the nucleus, binds to chromatin, and becomes immunologically silent. In contrast, when necrosis occurs, IL-1α migrates to the cytoplasm, where it becomes immunologically active after the cell dies179. Given its unique biological activity, it is not surprising that IL-1α has been reported to contribute to autoimmune disease, microbial infections, and cancer177. IL-1 signaling has recently been shown to be associated with inflammatory changes in the bone marrow niche in that impair hematopoietic function during aging108. We recently showed that IL-1α induces TNFα expression in splenic HSPCs, which subsequently activates splenic niche activity24. In the present study, peripheral neutrophils were decreased in tumor-bearing mice treated with an IL-1R-blocking antibody. Overall, the unique biology of IL-1β and IL-1α results in potent initiators of nonsterile and sterile inflammation, respectively, suggesting that these family members might be potent initiators of EMH.
Leukemia inhibitory factor
Leukemia inhibitory factor (LIF) is an IL-6 family member that signals through gp130 and the LIF receptor180,181. Signaling through the LIF receptor is also shared with other IL-6 family members, including OSM, CTNF, CT-1, and CLC, although all of these cytokines have additional receptors or coreceptors for signaling180,182. Signaling downstream of LIFR:gp130 is thought to be most prominent through JAK1, although signals can be transmitted through JAK2 and TYK2 as well183,184,185,186. JAK1 activation leads to the activation of STAT3, MAP kinase pathways, and PI3K in amounts that appear to be cell type-specific187,188,189. LIF is best known for its ability to maintain mouse embryonic stem cells in vitro180. STAT3 and PI3 kinase both lead to enhanced self-renewal and inhibition of differentiation in mouse embryonic stem cells, while MAP kinase activity activates differentiation190,191,192,193. LIF has several interesting functions outside of embryonic stem cells. Mutations in LIF have been reported in infertile women, and these reports concur with the failure of blastocyst implantation in LIF knockout dams and with the observation of high LIF expression in the endometrial glands194,195,196. In neuronal and stromal cell types, LIF also seems to increase growth while altering differentiation. Overexpression of LIF in an injected cell line led to bone marrow fibrosis and elevated osteoblast numbers197. Similarly, osteoblasts express the LIF receptor, and LIF enhances osteoblast differentiation while inhibiting adipocyte differentiation198. LIF overexpression by adenovirus injection enhances neural stem cell self-renewal, induces astrocyte differentiation in culture, and stimulates the proliferation of oligodendrocyte precursor cells when overexpressed by adenovirus199,200,201. In vitro-stimulated myoblasts exhibit increased proliferation but did not differentiate into myotubes, while LIF enhances muscle injury recovery in vivo202,203. In cancer, LIF expression has been reported in many solid tumors, such as colorectal, nasopharyngeal, skin, and breast cancer, and has been reported to support a variety of tumor functions180,204. Although the exploration of the role of LIF is ongoing, several lines of evidence suggest that LIF is a potent cytokine that impacts the proliferation and differentiation of stromal cells throughout the body. We recently reported that LIF is critical for the proliferation of cells localized in the EMH niche24, highlighting how inflammatory pathologies may interact with stromal components to regulate EMH during disease.
We believe that additional stromal active cytokines beyond LIF may also play important roles in EMH. Oncostatin M is an IL-6 family cytokine related to LIF that can signal through the LIF receptor and its own oncostatin M receptor205. Oncostatin M has been shown to promote myelopoiesis and to be active on stromal cells206,207. However, oncostatin M differs from LIF in important ways. Most importantly, oncostatin M can signal through STAT1 in addition to STAT3205. In regard to clinical disease, oncostatin M has been widely associated with joint disease and has been found in the synovial fluid of patients with rheumatoid arthritis208,209,210. Additionally, oncostatin M was recently identified as a biomarker of failure to respond to anti-TNFα therapy in patients with inflammatory bowel disease211. Taken together, these data indicate that oncostatin M is an important but understudied stromal active cytokine that is associated with human pathology and expanded hematopoiesis and has overlapping yet distinct effects compared with those of LIF.
Tumor necrosis factor alpha
Tumor necrosis factor alpha (TNFα) is a quintessential inflammatory cytokine with wide-ranging and pleiotropic functions and effects212. TNFα is a trimeric member of the TNF family of cytokines. TNFα is produced as a membrane bound protein, primarily by immune cells such as monocytes and macrophages, that can be released as a soluble factor by the action of a specific protease, TACE or TNFα-converting enzyme213. TNFα can signal through two receptors, TNFR1 and TNFR2. The downstream signaling pathway of TNFα is complicated and involves several signaling mediators. The outcomes of TNFα signaling are highly context-dependent but can include NF-κB activation, MAPK activation, and cell death. In the context of cancer, TNFα plays roles in nearly every stage, including tumorigenesis, tumor growth, angiogenesis, and metastasis but also has anti-oncogenic effects and can be used as an anticancer treatment212. At the intersection of cancer and hematopoiesis, TNFα has been implicated in inducing myelopoiesis in the context of cancer214. With this link to myelopoiesis, local TNFα has also been shown to potentially be linked to EMH in the context of cancer.
CXCR2 ligands
CXCR2 is a GPCR chemokine receptor with a wide repertoire of ligands that are highly conserved among vertebrates215. CXCR2 shares 77% amino acid identity with CXCR1 in humans and is positioned nearby on the same chromosome216. Ligands that bind to CXCR2 share a glutamic acid-leucine-arginine (ELR) motif and include CXCL1-3 and CXCL5-8216. CXCR2 is classically expressed on neutrophils, including G-MDSCs, and oligodendrocytes, although a wide selection of cells has been reported215. Upon activation of the receptor and after initiation of GPCR signaling, intracellular calcium stores are released into the cytoplasm, and gradient chemotaxis, degranulation, and MAPK activation can occur217,218. In the context of cancer, CXCL1-3 has been found to be directly expressed on human melanoma cells, and CXCL8 has been found to be produced by human prostate cancer cells219,220. Immune cells within the TME, such as macrophages, have also been shown to recruit G-MDSCs through CXCL2 expression60. Although not fully understood, CXCR2 ligands can mobilize HSPCs from the bone marrow despite the apparent lack of CXCR2 expression by these cells221. This ability of CXCR2 ligands to mobilize stem cells may be an important contributor to the induction of EMH.
Concluding remarks and future perspectives
While hematopoiesis is essential for maintaining immune system function, it is also increasingly recognized as an important contributor to organismal pathology. Here, we have provided an overview of the interaction between solid tumors and hematopoiesis through extramedullary production of an immunosuppressive granulocyte subset and the systemic and niche factors enabling that production. Although this is still an emerging area of interest, the study of EMH holds promise for providing a better understanding of the alterations in hematopoiesis that occur in cancer and for better therapeutic approaches to cancer.
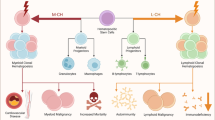
In this review, we present studies contributing to a broad understanding of the initiation and maintenance of EMH. Initially, solid tumors or noncancerous TME cells begin to produce CXCR2, either endogenously or through inflammation. These CXCR2 ligands recruit neutrophils to tumors while mobilizing HSPCs from the bone marrow. Cytokines are released simultaneously by the tumor and the TME induces the stromal cells in extramedullary sites to initiate hematopoiesis. Due to the unique biology of the extramedullary niche, circulating cytokines, or both, hematopoiesis in the extramedullary site becomes skewed toward the production of immunosuppressive granulocytes, G-MDSCs. These G-MDSCs can return to the TME via CXCR2 and support tumor growth. This broad overview is consistent with the available data and provides multiple avenues for therapeutic development and further exploration.
In addition to cancer, G-MDSCs have been found to be involved in many chronic inflammatory pathologies, suggesting that the processes underlying EMH may operate more broadly than previously appreciated222. G-MDSCs have been identified in human patients with viral and bacterial sepsis, where they are thought to efficiently resolve the infection223,224. Unlike their role in cancer, some studies have theorized that G-MDSCs promote disease resolution in the proper context. In rheumatoid arthritis, G-MDSCs have been identified in the synovial fluid of humans and mouse models, where their immunosuppressive function may be beneficial225. A study of dextran sulfate sodium (DSS)-induced colitis showed that G-MDSCs improved recovery when transplanted into mice just before the DSS model was established226. G-MDSC administration was also able to ameliorate experimental autoimmune encephalitis (EAE) in mice227. Another study revealed that multiple sclerosis patients with active disease had functional G-MDSCs227. The diversity of pathologies and both the beneficial and detrimental effects of G-MDSCs in various diseases suggest that controlling the induction and elimination of EMH is a potent target for therapeutic research in a broad range of clinically relevant inflammatory diseases.
The data suggest that additional extramedullary sites outside the liver and spleen deserve study. In the case of solid tumors, several unexpected places, such as the paraspinal region, peritoneum, bronchia, adrenal gland, endometrium, pancreas, and ureter, were also found to have EMH activity11,13. The breadth of the sites capable of conducting hematopoiesis is surprising and hints at a broader relevance of this phenomenon than is commonly appreciated. Additionally, as has been discussed in the literature, EMH is likely to be underreported because it does not produce distinguishing features on imaging and requires biopsy for confirmation. Moreover, clinicians are not often aware of the possibility of hematopoiesis occurring outside of the bone marrow and therefore may not be looking for it when treating patients228. Lymph node hematopoiesis is of interest because evidence suggests that these cells are the predominant site of EMH in cancer patients11. Conceptually, the lymph nodes and spleen share a function as organs of immune surveillance for different fluid compartments in the body, the lymph and blood, respectively. Additionally, on a more detailed level, the microanatomical organization of the two organs also rhymes. However, one key difference in the context of solid tumors is that the lymph node exhibits an increased load of cytokines from the tumor mass. Assuming that the lymph node stroma is as reactive as the spleen is, one would expect that lymph node hematopoiesis would be more dramatic than in the spleen itself. However, draining lymph nodes are also common primary sites of metastasis. Therefore, tumor growth in the lymph node may obscure any ongoing hematopoiesis.
The generation of treatments that manipulate the immune system to favor anticancer immunity has been an exciting development in modern cancer therapy. Central to the understanding of anticancer immune activation is that mutations in the cancer genome can produce novel antigens recognized by the adaptive immune system and reduce the expression of immune surveillance genes, leading to NK cell activation229,230. Procancer immune suppression is mediated by intrinsic mechanisms preventing chronic immune activation and cancer-induced immune cell phenotypic skewing away from activation229. These competing signals set a balance that is now being manipulated by therapeutics such as immune checkpoint inhibitors, recombinant cytokine therapy, and chimeric antigen receptor T-cell and NK-cell therapies229,231,232,233. However, care must be taken as the overactivation of the immune system via these therapies can lead to severe side effects234,235. Therapeutics developed with an understanding of cancer–immune interactions have been revolutionary, but progress still remains in terms of improving efficacy and minimizing side effects. Central to improving the efficacy of immune-targeting therapeutics is local activation of the immune system, a state modulated by systemically derived myeloid cells produced by EMH.
In conclusion, emerging studies on the initiation and maintenance of EMH have highlighted how cellular products from this process alter the course of clinically important diseases such as cancer. We look forward to future studies reporting the components of the extracellular niche and to clinical studies broadening the relevance of this pathological process to other disease states.
References
Jemal, A., Thomas, A., Murray, T. & Thun, M. Cancer statistics, 2002. CA Cancer J. Clin. 52, 23–47 (2002).
Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 24, 711–720 (2018).
Oduro, K. A. Jr. et al. Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood J. Am. Soc. Hematol. 120, 2203–2213 (2012).
Wu, C. et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J. Clin. Invest. 128, 3425–3438 (2018).
Corbeau, I., Jacot, W. & Guiu, S. Neutrophil to lymphocyte ratio as prognostic and predictive factor in breast cancer patients: a systematic review. Cancers 12, 958 (2020).
Iwai, N. et al. Neutrophil to lymphocyte ratio predicts prognosis in unresectable pancreatic cancer. Sci. Rep. 10, 1–7 (2020).
Templeton, A. J. et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J. Natl Cancer Inst. 106, dju124 (2014).
Zhang, Y. et al. Prognostic value of neutrophil-to-lymphocyte ratio and platelet-to-lymphocyte ratio in gastric cancer. Medicine 97, e0144 (2018).
De Cicco, P., Ercolano, G. & Ianaro, A. The new era of cancer immunotherapy: targeting myeloid-derived suppressor cells to overcome immune evasion. Front. Immunol. 11, 1680 (2020).
O’Malley, D. P. Benign extramedullary myeloid proliferations. Mod. Pathol. 20, 405–415 (2007).
Bao, Y. et al. Extramedullary hematopoiesis secondary to malignant solid tumors: a case report and literature review. Cancer Manag. Res. 10, 1461 (2018).
Khalil, S., Ariel Gru, A. & Saavedra, A. P. Cutaneous extramedullary haematopoiesis: Implications in human disease and treatment. Exp. Dermatol. 28, 1201–1209 (2019).
Fan, N., Lavu, S., Hanson, C. A. & Tefferi, A. Extramedullary hematopoiesis in the absence of myeloproliferative neoplasm: Mayo Clinic case series of 309 patients. Blood Cancer J. 8, 1–4 (2018).
Nagai, Y. et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24, 801–812 (2006).
Regan-Komito, D. et al. GM-CSF drives dysregulated hematopoietic stem cell activity and pathogenic extramedullary myelopoiesis in experimental spondyloarthritis. Nat. Commun. 11, 1–15 (2020).
Peters, M. et al. Extramedullary expansion of hematopoietic progenitor cells in interleukin (IL)-6–sIL-6R double transgenic mice. J. Exp. Med. 185, 755–766 (1997).
Khan, K. N. M. et al. Recombinant human interleukin-3 induces extramedullary hematopoiesis at subcutaneous injection sites in cynomolgus monkeys. Toxicol. Pathol. 24, 391–397 (1996).
Dagdas, S. et al. Unusual extramedullary hematopoiesis in a patient receiving granulocyte colony-stimulating factor. Acta Haematol. 116, 198–202 (2006).
Cenariu, D. et al. Extramedullary hematopoiesis of the liver and spleen. J. Clin. Med. 10, 5831 (2021).
Conor O’keane, J., Wolf, B. C. & Neiman, R. S. The pathogenesis of splenic extramedullary hematopoiesis in metastatic carcinoma. Cancer 63, 1539–1543 (1989).
Cortez-Retamozo, V. et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl Acad. Sci. USA 109, 2491–2496 (2012).
Wu, W.-C. et al. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc. Natl Acad. Sci. USA 111, 4221–4226 (2014).
Lu, I.-N. et al. Tumor-associated hematopoietic stem and progenitor cells positively linked to glioblastoma progression. Nat. Commun. 12, 1–16 (2021).
Barisas, D. A. et al. Tumor-derived interleukin-1α and leukemia inhibitory factor promote extramedullary hematopoiesis. PLoS Biol. 21, e3001746 (2023).
Koch, C. A., Li, C.-Y., Mesa, R. A. & Tefferi, A. Nonhepatosplenic extramedullary hematopoiesis: associated diseases, pathology, clinical course, and treatment. In Mayo Clinic Proceedings. Elsevier; 2003. pp. 1223–1233.
Blais, M. È., Louis, I. & Perreault, C. T‐cell development: an extrathymic perspective. Immunol. Rev. 209, 103–114 (2006).
Mitroulis, I., Kalafati, L., Hajishengallis, G. & Chavakis, T. Myelopoiesis in the context of innate immunity. J. Innate Immun. 10, 365–372 (2018).
Chiba, Y. et al. Regulation of myelopoiesis by proinflammatory cytokines in infectious diseases. Cell. Mol. Life Sci. 75, 1363–1376 (2018).
Rosenbauer, F. & Tenen, D. G. Transcription factors in myeloid development: balancing differentiation with transformation. Nat. Rev. Immunol. 7, 105–117 (2007).
Klemsz, M. J., McKercher, S. R., Celada, A., Van Beveren, C. & Maki, R. A. The macrophage and B cell-specific transcription factor PU. 1 is related to the ets oncogene. Cell 61, 113–124 (1990).
Zhang, D.-E. et al. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein α-deficient mice. Proc. Natl Acad. Sci. USA 94, 569–574 (1997).
Tanaka, T. et al. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell 80, 353–361 (1995).
Yamanaka, R. et al. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein ɛ-deficient mice. Proc. Natl Acad. Sci. USA 94, 13187–13192 (1997).
Hock, H. et al. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 18, 109–120 (2003).
Holtschke, T. et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 87, 307–317 (1996).
Hirai, H. et al. C/EBPβ is required for’emergency’granulopoiesis. Nat. Immunol. 7, 732–739 (2006).
Lieschke, G. J. et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 84, 1737–1746 (1994).
Basu, S. et al. “Emergency” granulopoiesis in G-CSF–deficient mice in response to Candida albicans infection. Blood J. Am. Soc. Hematol. 95, 3725–3733 (2000).
Pietras, E. M. et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 18, 607–618 (2016).
Zhan, Y. & Cheers, C. Haemopoiesis in mice genetically lacking granulocyte–macrophage colony stimulating factor during chronic infection with Mycobacterium avium. Immunol. Cell Biol. 78, 118–123 (2000).
Seita, J. et al. Interleukin-27 directly induces differentiation in hematopoietic stem cells. Blood J. Am. Soc. Hematol. 111, 1903–1912 (2008).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020).
Mantovani, A., Marchesi, F., Jaillon, S., Garlanda, C. & Allavena, P. Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell. Mol. Immunol. 18, 566–578 (2021).
Veglia, F., Sanseviero, E. & Gabrilovich, D. I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 21, 485–498 (2021).
Alshetaiwi, H. et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 5, eaay6017 (2020).
Condamine, T. et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 1, aaf8943–aaf8943 (2016).
Condamine, T., Mastio, J. & Gabrilovich, D. I. Transcriptional regulation of myeloid‐derived suppressor cells. J. Leukoc. Biol. 98, 913–922 (2015).
Gabrilovich, D. I. & Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174 (2009).
Condamine, T. & Gabrilovich, D. I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 32, 19–25 (2011).
Movahedi, K. et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell–suppressive activity. Blood, J. Am. Soc. Hematol. 111, 4233–4244 (2008).
Waight, J. D., Hu, Q., Miller, A., Liu, S. & Abrams, S. I. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS ONE 6, e27690 (2011).
Li, W. et al. G-CSF is a key modulator of MDSC and could be a potential therapeutic target in colitis-associated colorectal cancers. Protein Cell 7, 130–140 (2016).
Meyer, M. A. et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat. Commun. 9, 1–19 (2018).
Casbon, A.-J. et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl Acad. Sci. USA 112, E566–E575 (2015).
Stadtmann, A. & Zarbock, A. CXCR2: from bench to bedside. Front. Immunol. 3, 263 (2012).
Yuen, K. C. et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 26, 693–698 (2020).
Schalper, K. A. et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 26, 688–692 (2020).
Tazawa, H. et al. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am. J. Pathol. 163, 2221–2232 (2003).
Spicer, J. D. et al. Neutrophils promote liver metastasis via mac-1–mediated interactions with circulating tumor cellsneutrophil/tumor cell adhesion promotes metastasis. Cancer Res. 72, 3919–3927 (2012).
Shang, K. et al. Crucial involvement of tumor-associated neutrophils in the regulation of chronic colitis-associated carcinogenesis in mice. PLoS ONE 7, e51848 (2012).
Jamieson, T. et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 122, 3127–3144 (2012).
Katoh, H. et al. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 24, 631–644 (2013).
Wang, Y., Ding, Y., Guo, N. & Wang, S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front. Immunol. 10, 172 (2019).
Li, P. et al. Lung mesenchymal cells elicit lipid storage in neutrophils that fuel breast cancer lung metastasis. Nat. Immunol. 21, 1444–1455 (2020).
Lin, P. et al. Expansion of myeloid immune suppressor cells in Tumor-bearing host directly promotes tumor angiogenesis, tumor growth, and metastasis. Clin. Cancer Res. 13, PL02-02–PL02-02 (2007).
Kowanetz, M. et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+ Ly6C+ granulocytes. Proc. Natl Acad. Sci. USA 107, 21248–21255 (2010).
Shojaei, F. et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450, 825–831 (2007).
Kusmartsev, S. et al. Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J. Immunol. 181, 346–353 (2008).
Toh, B. et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 9, e1001162 (2011).
Cools-Lartigue, J. et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 123, 3446–3458 (2013).
Mendelson, A. & Frenette, P. S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 20, 833–846 (2014).
Calvi, L. M. & Link, D. C. The hematopoietic stem cell niche in homeostasis and disease. Blood J. Am. Soc. Hematol. 126, 2443–2451 (2015).
Sugiyama, T., Kohara, H., Noda, M. & Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988 (2006).
Ding, L., Saunders, T. L., Enikolopov, G. & Morrison, S. J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012).
Méndez-Ferrer, S. et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834 (2010).
Pinho, S. et al. PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 210, 1351–1367 (2013).
Greenbaum, A. et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230 (2013).
Zhang, J. et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425, 836–841 (2003).
Calvi, L. et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 (2003).
Ding, L. & Morrison, S. J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235 (2013).
Ding, D.-C., Shyu, W.-C. & Lin, S.-Z. Mesenchymal stem cells. Cell Transplant. 20, 5–14 (2011).
Kennedy, M. et al. A common precursor for primitive erythropoiesis and definitive haematopoiesis. Nature 386, 488–493 (1997).
Kobayashi, H. et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat. Cell Biol. 12, 1046–1056 (2010).
Shalaby, F. et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66 (1995).
Doan, P. L. et al. Epidermal growth factor regulates hematopoietic regeneration after radiation injury. Nat. Med. 19, 295–304 (2013).
Himburg, H. A. et al. Pleiotrophin regulates the retention and self-renewal of hematopoietic stem cells in the bone marrow vascular niche. Cell Rep. 2, 964–975 (2012).
Hooper, A. T. et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 4, 263–274 (2009).
Kunisaki, Y. et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013).
Méndez-Ferrer, S., Lucas, D., Battista, M. & Frenette, P. S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442–447 (2008).
Katayama, Y. et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124, 407–421 (2006).
Yamazaki, S. et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147, 1146–1158 (2011).
Yamazaki, S. et al. TGF-β as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood J. Am. Soc. Hematol. 113, 1250–1256 (2009).
Heazlewood, S. Y. et al. Megakaryocytes co-localise with hemopoietic stem cells and release cytokines that up-regulate stem cell proliferation. Stem Cell Res. 11, 782–792 (2013).
Bruns, I. et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 20, 1315–1320 (2014).
Zhao, M. et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 20, 1321–1326 (2014).
Olson, T. S. et al. Megakaryocytes promote murine osteoblastic HSC niche expansion and stem cell engraftment after radioablative conditioning. Blood J. Am. Soc. Hematol. 121, 5238–5249 (2013).
Chow, A. et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 208, 261–271 (2011).
Christopher, M. J., Rao, M., Liu, F., Woloszynek, J. R. & Link, D. C. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J. Exp. Med. 208, 251–260 (2011).
Andonegui, G. et al. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J. Clin. Investig. 119, 1921–1930 (2009).
Day, R. B., Bhattacharya, D., Nagasawa, T. & Link, D. C. Granulocyte colony-stimulating factor reprograms bone marrow stromal cells to actively suppress B lymphopoiesis in mice. Blood J. Am. Soc. Hematol. 125, 3114–3117 (2015).
Ikushima, Y. M. et al. Prostaglandin E2 regulates murine hematopoietic stem/progenitor cells directly via EP4 receptor and indirectly through mesenchymal progenitor cells. Blood J. Am. Soc. Hematol. 121, 1995–2007 (2013).
Itkin, T., Kaufmann, K. B., Gur-Cohen, S., Ludin, A. & Lapidot, T. Fibroblast growth factor signaling promotes physiological bone remodeling and stem cell self-renewal. Curr. Opin. Hematol. 20, 237–244 (2013).
Decker, M. et al. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 19, 677–688 (2017).
Dykstra, B., Olthof, S., Schreuder, J., Ritsema, M. & De Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 208, 2691–2703 (2011).
Sudo, K., Ema, H., Morita, Y. & Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 192, 1273–1280 (2000).
Maryanovich, M. et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 24, 782–791 (2018).
Ho, Y.-H. et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell 25, 407–418. e406 (2019).
Mitchell, C. A. et al. Stromal niche inflammation mediated by IL-1 signalling is a targetable driver of haematopoietic ageing. Nat. Cell Biol. 25, 30–41 (2023).
Bowers, E. & Singer, K. Obesity-induced inflammation: the impact of the hematopoietic stem cell niche. JCI insight 6, e145295 (2021).
Naveiras, O. et al. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460, 259–263 (2009).
Zhu, R.-J., Wu, M.-Q., Li, Z.-J., Zhang, Y. & Liu, K.-Y. Hematopoietic recovery following chemotherapy is improved by BADGE-induced inhibition of adipogenesis. Int. J. Hematol. 97, 58–72 (2013).
Mattiucci, D. et al. Bone marrow adipocytes support hematopoietic stem cell survival. J. Cell. Physiol. 233, 1500–1511 (2018).
Spindler, T. J., Tseng, A. W., Zhou, X. & Adams, G. B. Adipocytic cells augment the support of primitive hematopoietic cells in vitro but have no effect in the bone marrow niche under homeostatic conditions. Stem Cells Dev. 23, 434–441 (2014).
Corre, J. et al. Human bone marrow adipocytes support complete myeloid and lymphoid differentiation from human CD34+ cells. Br. J. Haematol. 127, 344–347 (2004).
Liu, L.-F., Shen, W.-J., Ueno, M., Patel, S. & Kraemer, F. B. Characterization of age-related gene expression profiling in bone marrow and epididymal adipocytes. BMC Genomics 12, 1–18 (2011).
Miggitsch, C. et al. Human bone marrow adipocytes display distinct immune regulatory properties. EBioMedicine 46, 387–398 (2019).
Inra, C. N. et al. A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature 527, 466–471 (2015).
Mendt, M. & Cardier, J. E. Role of SDF-1 (CXCL12) in regulating hematopoietic stem and progenitor cells traffic into the liver during extramedullary hematopoiesis induced by G-CSF, AMD3100 and PHZ. Cytokine 76, 214–221 (2015).
Kogame, T. et al. Presence of SCF/CXCL 12 double‐positive large blast‐like cells at the site of cutaneous extramedullary haematopoiesis. J. Eur. Acad. Dermatol. Venereol. 32, e465–e466 (2018).
Oda, A. et al. Niche-induced extramedullary hematopoiesis in the spleen is regulated by the transcription factor Tlx1. Sci. Rep. 8, 1–16 (2018).
Broxmeyer, H. E. et al. Th1 cells regulate hematopoietic progenitor cell homeostasis by production of oncostatin M. Immunity 16, 815–825 (2002).
Lee, J. H., Wang, C. & Kim, C. H. FoxP3+ regulatory T cells restrain splenic extramedullary myelopoiesis via suppression of hemopoietic cytokine-producing T cells. J. Immunol. 183, 6377–6386 (2009).
Chow, A. et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 19, 429–436 (2013).
Liu, M. et al. Macrophages support splenic erythropoiesis in 4T1 tumor-bearing mice. PLoS ONE 10, e0121921 (2015).
Dutta, P. et al. Macrophages retain hematopoietic stem cells in the spleen via VCAM-1. J. Exp. Med. 212, 497–512 (2015).
Ulyanova, T., Phelps, S. R. & Papayannopoulou, T. The macrophage contribution to stress erythropoiesis: when less is enough. Blood J. Am. Soc. Hematol. 128, 1756–1765 (2016).
Huang, E. et al. The hematopoietic growth factor KL is encoded by the SI locus and is the ligand of the c-kit receptor, the gene product of the W locus. Cell 63, 225–233 (1990).
Fong, Y. & Blumgart, L. H. Surgery Of The Liver And Biliary Tract (WB Saunders, 2000).
Brannan, C. et al. Developmental abnormalities in Steel17H mice result from a splicing defect in the steel factor cytoplasmic tail. Genes Dev. 6, 1832–1842 (1992).
Russell, E. S. Hereditary anemias of the mouse: a review for geneticists. Adv. Genet. 20, 357–459 (1979).
Kapur, R. et al. Signaling through the interaction of membrane-restricted stem cell factor and c-kit receptor tyrosine kinase: genetic evidence for a differential role in erythropoiesis. Blood J. Am. Soc. Hematol. 91, 879–889 (1998).
Tajima, Y. et al. Consequences of exclusive expression in vivo of Kit-ligand lacking the major proteolytic cleavage site. Proc. Natl Acad. Sci. USA 95, 11903–11908 (1998).
Qiu, F. et al. Primary structure of c‐kit: relationship with the CSF‐1/PDGF receptor kinase family–oncogenic activation of v‐kit involves deletion of extracellular domain and C terminus. EMBO J. 7, 1003–1011 (1988).
Yarden, Y. et al. Human proto‐oncogene c‐kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 6, 3341–3351 (1987).
Lev, S., Blechman, J. M., Givol, D. & Yarden, Y. Steel factor and c-kit protooncogene: genetic lessons in signal transduction. Crit. Rev. Oncog. 5, 141–168 (1994).
Kapur, R., Chandra, S., Cooper, R., McCarthy, J. & Williams, D. A. Role of p38 and ERK MAP kinase in proliferation of erythroid progenitors in response to stimulation by soluble and membrane isoforms of stem cell factor. Blood J. Am. Soc. Hematol. 100, 1287–1293 (2002).
Kurosawa, K. et al. Immobilized anti-KIT monoclonal antibody induces ligand-independent dimerization and activation of Steel factor receptor: biologic similarity with membrane-bound form of Steel factor rather than its soluble form. Blood 87, 2235–2243 (1996).
Thorén, L. A. et al. Kit regulates maintenance of quiescent hematopoietic stem cells. J. Immunol. 180, 2045–2053 (2008).
Wu, H., Klingmüller, U., Acurio, A., Hsiao, J. G. & Lodish, H. F. Functional interaction of erythropoietin and stem cell factor receptors is essential for erythroid colony formation. Proc. Natl Acad. Sci. USA 94, 1806–1810 (1997).
Jahn, T. et al. Direct interaction between Kit and the interleukin-7 receptor. Blood J. Am. Soc. Hematol. 110, 1840–1847 (2007).
Zhu, M. et al. KIT oncoprotein interactions in gastrointestinal stromal tumors: therapeutic relevance. Oncogene 26, 6386–6395 (2007).
Kim, C. H. & Broxmeyer, H. E. In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: stromal cell–derived factor-1, steel factor, and the bone marrow environment. Blood J. Am. Soc. Hematol. 91, 100–110 (1998).
Brightman, F. A., Leahy, D. E., Searle, G. E. & Thomas, S. Application of a generic physiologically based pharmacokinetic model to the estimation of xenobiotic levels in human plasma. Drug Metab. Dispos. 34, 94–101 (2006).
Bachelerie, F. et al. International Union of Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 66, 1 (2014).
Zlotnik, A. & Yoshie, O. Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127 (2000).
Oberlin, E. et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 382, 833–835 (1996).
Bleul, C. C. et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382, 829–833 (1996).
Balabanian, K. et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 280, 35760–35766 (2005).
Burns, J. M. et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 203, 2201–2213 (2006).
Cruz-Orengo, L. et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J. Exp. Med. 208, 327–339 (2011).
Ma, Q. et al. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4-and SDF-1-deficient mice. Proc. Natl Acad. Sci. USA 95, 9448–9453 (1998).
Gerrits, H. et al. Early postnatal lethality and cardiovascular defects in CXCR7‐deficient mice. Genesis 46, 235–245 (2008).
Sierro, F. et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl Acad. Sci. USA 104, 14759–14764 (2007).
Rubin, J. B. Chemokine signaling in cancer: one hump or two? In Seminars in Cancer Biology. Elsevier; 2009. pp. 116–122.
Busillo, J. M. & Benovic, J. L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta (BBA)-Biomembr. 1768, 952–963 (2007).
Soldevila, G. et al. Impaired chemokine‐induced migration during T‐cell development in the absence of Jak 3. Immunology 112, 191–200 (2004).
Zhang, X.-F., Wang, J.-F., Matczak, E., Proper, J. & Groopman, J. E. Janus kinase 2 is involved in stromal cell–derived factor-1α–induced tyrosine phosphorylation of focal adhesion proteins and migration of hematopoietic progenitor cells. Blood J. Am. Soc. Hematol. 97, 3342–3348 (2001).
Vila‐Coro, A. J. et al. The chemokine SDF‐lα triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 13, 1699–1710 (1999).
Moriguchi, M. et al. CXCL12 signaling is independent of Jak2 and Jak3. J. Biol. Chem. 280, 17408–17414 (2005).
Thelen, M. Dancing to the tune of chemokines. Nat. Immunol. 2, 129–134 (2001).
Sun, Y., Cheng, Z., Ma, L. & Pei, G. β-Arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 277, 49212–49219 (2002).
Shukla, A. K., Xiao, K. & Lefkowitz, R. J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 36, 457–469 (2011).
Bendall, L. J. & Bradstock, K. F. G-CSF: From granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev. 25, 355–367 (2014).
Fukunaga, R., Ishizaka-Ikeda, E. & Nagata, S. Purification and characterization of the receptor for murine granulocyte colony-stimulating factor. J. Biol. Chem. 265, 14008–14015 (1990).
Dong, F. et al. Stimulation of Stat5 by granulocyte colony-stimulating factor (G-CSF) is modulated by two distinct cytoplasmic regions of the G-CSF receptor. J. Immunol. 161, 6503–6509 (1998).
Cornish, A. L., Campbell, I. K., McKenzie, B. S., Chatfield, S. & Wicks, I. P. G-CSF and GM-CSF as therapeutic targets in rheumatoid arthritis. Nat. Rev. Rheumatol. 5, 554–559 (2009).
Kawakami, M. et al. Levels of serum granulocyte colony-stimulating factor in patients with infections. Blood 76, 1962–1964 (1990).
Cebon, J., Layton, J. E., Maher, D. & Morstyn, G. Endogenous haemopoietic growth factors in neutropenia and infection. Br. J. Haematol. 86, 265–274 (1994).
Liu, F., Wu, H. Y., Wesselschmidt, R., Kornaga, T. & Link, D. C. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor–deficient mice. Immunity 5, 491–501 (1996).
Tamura, M. et al. Induction of neutrophilic granulocytosis in mice by administration of purified human native granulocyte colony-stimulating factor (G-CSF). Biochem. Biophys. Res. Commun. 142, 454–460 (1987).
Dinarello, C. A. Overview of the IL‐1 family in innate inflammation and acquired immunity. Immunol. Rev. 281, 8–27 (2018).
Towne, J. E. et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 286, 42594–42602 (2011).
Ridker, P. M. et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Pyrillou, K., Burzynski, L. C. & Clarke, M. C. Alternative pathways of IL-1 activation, and its role in health and disease. Front. Immunol. 11, 613170 (2020).
Kim, B. et al. The interleukin-1α precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front. Immunol. 4, 391 (2013).
Malik, A. & Kanneganti, T. D. Function and regulation of IL‐1α in inflammatory diseases and cancer. Immunol. Rev. 281, 124–137 (2018).
Afonina, I. S. et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol. Cell 44, 265–278 (2011).
Werman, A. et al. The precursor form of IL-1α is an intracrine proinflammatory activator of transcription. Proc. Natl Acad. Sci. USA 101, 2434–2439 (2004).
Nicola, N. A. & Babon, J. J. Leukemia inhibitory factor (LIF). Cytokine Growth Factor Rev. 26, 533–544 (2015).
Metcalf, D. The unsolved enigmas of leukemia inhibitory factor. Stem Cells 21, 5–14 (2003).
Houben, E., Hellings, N., Broux, B. & Oncostatin, M. an underestimated player in the central nervous system. Front. Immunol. 10, 1165 (2019).
Stahl, N. et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 β receptor components. Science 263, 92–95 (1994).
Rodig, S. J. et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 93, 373–383 (1998).
Takahashi, Y. et al. Leukemia inhibitory factor regulates trophoblast giant cell differentiation via Janus kinase 1-signal transducer and activator of transcription 3-suppressor of cytokine signaling 3 pathway. Mol. Endocrinol. 22, 1673–1681 (2008).
Chung, B. M. et al. Jak2 and Tyk2 are necessary for lineage-specific differentiation, but not for the maintenance of self-renewal of mouse embryonic stem cells. Biochem. Biophys. Res. Commun. 351, 682–688 (2006).
Heinrich, P. C. et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 (2003).
Fahmi, A. et al. p42/p44-MAPK and PI3K are sufficient for IL-6 family cytokines/gp130 to signal to hypertrophy and survival in cardiomyocytes in the absence of JAK/STAT activation. Cell. Signal. 25, 898–909 (2013).
Oh, H. et al. Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J. Biol. Chem. 273, 9703–9710 (1998).
Niwa, H., Burdon, T., Chambers, I. & Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12, 2048–2060 (1998).
Paling, N. R., Wheadon, H., Bone, H. K. & Welham, M. J. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J. Biol. Chem. 279, 48063–48070 (2004).
Meloche, S., Vella, F. D., Voisin, L., Ang, S.-L. & Saba-El-Leil, M. Erk2 signaling and early embryo stem cell self-renewal. Cell Cycle 3, 239–241 (2004).
Burdon, T., Stracey, C., Chambers, I., Nichols, J. & Smith, A. Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev. Biol. 210, 30–43 (1999).
Stewart, C. L. et al. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 359, 76–79 (1992).
Giess, R., Tanasescu, I., Steck, T. & Sendtner, M. Leukaemia inhibitory factor gene mutations in infertile women. Mol. Hum. Reprod. 5, 581–586 (1999).
Charnock-Jones, D., Sharkey, A., Fenwick, P. & Smith, S. Leukaemia inhibitory factor mRNA concentration peaks in human endometrium at the time of implantation and the blastocyst contains mRNA for the receptor at this time. Reproduction 101, 421–426 (1994).
Metcalf, D. & Gearing, D. Fatal syndrome in mice engrafted with cells producing high levels of the leukemia inhibitory factor. Proc. Natl Acad. Sci. USA 86, 5948–5952 (1989).
Walker, E. C. et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J. Clin. Investig. 120, 582–592 (2010).
Deverman, B. E. & Patterson, P. H. Exogenous leukemia inhibitory factor stimulates oligodendrocyte progenitor cell proliferation and enhances hippocampal remyelination. J. Neurosci. 32, 2100–2109 (2012).
Yoshida, T., Satoh, M., Nakagaito, Y., Kuno, H. & Takeuchi, M. Cytokines affecting survival and differentiation of an astrocyte progenitor cell line. Dev. Brain Res. 76, 147–150 (1993).
Bauer, S. & Patterson, P. H. Leukemia inhibitory factor promotes neural stem cell self-renewal in the adult brain. J. Neurosci. 26, 12089–12099 (2006).
Barnard, W., Bower, J., Brown, M., Murphy, M. & Austin, L. Leukemia inhibitory factor (LIF) infusion stimulates skeletal muscle regeneration after injury: injured muscle expresses lif mRNA. J. Neurol. Sci. 123, 108–113 (1994).
Austin, L. & Burgess, A. Stimulation of myoblast proliferation in culture by leukaemia inhibitory factor and other cytokines. J. Neurol. Sci. 101, 193–197 (1991).
Wu, L. et al. HIF-2α mediates hypoxia-induced LIF expression in human colorectal cancer cells. Oncotarget 6, 4406 (2015).
Richards, C. D. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm. 2013, 512103 (2013).
Metcalfe, S. LIF in the regulation of T-cell fate and as a potential therapeutic. Genes Immun. 12, 157–168 (2011).
Albiero, M. et al. Diabetes-associated myelopoiesis drives stem cell mobilopathy through an OSM-p66Shc signaling pathway. Diabetes 68, 1303–1314 (2019).
Langdon, C., Leith, J., Richards, C. D. & Smith, F. Oncostatin M stimulates monocyte chemoattractant protein‐1‐and interleukin‐1‐induced matrix metalloproteinase‐1 production by human synovial fibroblasts in vitro. Arthritis Rheum. 40, 2139–2146 (1997).
Hui, W., Bell, M. & Carroll, G. Detection of oncostatin M in synovial fluid from patients with rheumatoid arthritis. Ann. Rheum. Dis. 56, 184–187 (1997).
Cawston, T. et al. The role of oncostatin M in animal and human connective tissue collagen turnover and its localization within the rheumatoid joint. Arthritis Rheum. 41, 1760–1771 (1998).
West, N. R. et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor–neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 23, 579–589 (2017).
Wang, X. & Lin, Y. Tumor necrosis factor and cancer, buddies or foes? 1. Acta Pharmacol. Sin. 29, 1275–1288 (2008).
Cheng, X., Shen, Y. & Li, R. Targeting TNF: a therapeutic strategy for Alzheimer’s disease. Drug Discov. today 19, 1822–1827 (2014).
Al Sayed, M. F. et al. T-cell–secreted TNFα induces emergency myelopoiesis and myeloid-derived suppressor cell differentiation in cancer. Cancer Res. 79, 346–359 (2019).
Veenstra, M. & Ransohoff, R. M. Chemokine receptor CXCR2: physiology regulator and neuroinflammation controller? J. Neuroimmunol. 246, 1–9 (2012).
Ahuja, S. K., Özçelik, T., Milatovitch, A., Francke, U. & Murphy, P. M. Molecular evolution of the human interleukin–8 receptor gene cluster. Nat. Genet. 2, 31–36 (1992).
Knall, C. et al. Interleukin-8 regulation of the Ras/Raf/mitogen-activated protein kinase pathway in human neutrophils. J. Biol. Chem. 271, 2832–2838 (1996).
Murdoch, C. & Finn, A. Chemokine receptors and their role in inflammation and infectious diseases. Blood J. Am. Soc. Hematol. 95, 3032–3043 (2000).
Luan, J. et al. Mechanism and biological significance of constitutive expression of MGSA/GRO chemokines in malignant melanoma tumor progression. J. Leukoc. Biol. 62, 588–597 (1997).
Moore, B. B. et al. Distinct CXC chemokines mediate tumorigenicity of prostate cancer cells. Am. J. Pathol. 154, 1503–1512 (1999).
Karpova, D. et al. Targeting VLA4 integrin and CXCR2 mobilizes serially repopulating hematopoietic stem cells. J. Clin. Investig. 129, 2745–2759 (2019).
Sendo, S., Saegusa, J. & Morinobu, A. Myeloid-derived suppressor cells in non-neoplastic inflamed organs. Inflamm. Regen. 38, 1–11 (2018).
Coudereau, R. et al. Emergence of immunosuppressive LOX‐1+ PMN‐MDSC in septic shock and severe COVID‐19 patients with acute respiratory distress syndrome. J. Leukoc. Biol. 111, 489–496 (2022).
De Zuani, M. et al. Human myeloid‐derived suppressor cell expansion during sepsis is revealed by unsupervised clustering of flow cytometric data. Eur. J. Immunol. 51, 1785–1791 (2021).
Kurkó, J. et al. Identification of myeloid-derived suppressor cells in the synovial fluid of patients with rheumatoid arthritis: a pilot study. BMC Musculoskelet. Disord. 15, 1–7 (2014).
Zhang, R. et al. Dextran sulphate sodium increases splenic Gr1+ CD11b+ cells which accelerate recovery from colitis following intravenous transplantation. Clin. Exp. Immunol. 164, 417–427 (2011).
Ioannou, M. et al. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J. Immunol. 188, 1136–1146 (2012).
Orphanidou-Vlachou, E., Tziakouri-Shiakalli, C. & Georgiades, C. S. Extramedullary hemopoiesis. In Seminars in Ultrasound, CT and MRI. Elsevier; 2014. pp. 255–262.
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 541, 321–330 (2017).
Bald, T., Krummel, M. F., Smyth, M. J. & Barry, K. C. The NK cell–cancer cycle: advances and new challenges in NK cell–based immunotherapies. Nat. Immunol. 21, 835–847 (2020).
Gong, Y., Klein Wolterink, R. G., Wang, J., Bos, G. M. & Germeraad, W. T. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 14, 1–35 (2021).
Yu, S. et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J. Hematol. Oncol. 10, 1–13 (2017).
García-Martínez, E. et al. Trial Watch: Immunostimulation with recombinant cytokines for cancer therapy. Oncoimmunology 7, e1433982 (2018).
Brudno, J. N. & Kochenderfer, J. N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood J. Am. Soc. Hematol. 127, 3321–3330 (2016).
Morad, G., Helmink, B. A., Sharma, P. & Wargo, J. A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 184, 5309–5337 (2021).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barisas, D.A.G., Choi, K. Extramedullary hematopoiesis in cancer. Exp Mol Med 56, 549–558 (2024). https://doi.org/10.1038/s12276-024-01192-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-024-01192-4
