
Abstract
Our bodies are inhabited by trillions of microorganisms. The host immune system constantly interacts with the microbiota in barrier organs, including the intestines. Over decades, numerous studies have shown that our mucosal immune system is dynamically shaped by a variety of microbiota-derived signals. Elucidating the mediators of these interactions is an important step for understanding how the microbiota is linked to mucosal immune homeostasis and gut-associated diseases. Interestingly, the efficacy of cancer immunotherapies that manipulate costimulatory and coinhibitory pathways has been correlated with the gut microbiota. Moreover, adverse effects of these therapies in the gut are linked to dysregulation of the intestinal immune system. These findings suggest that costimulatory pathways in the immune system might serve as a bridge between the host immune system and the gut microbiota. Here, we review mechanisms by which commensal microorganisms signal immune cells and their potential impact on costimulation. We highlight how costimulatory pathways modulate the mucosal immune system through not only classical antigen-presenting cells but also innate lymphocytes, which are highly enriched in barrier organs. Finally, we discuss the adverse effects of immune checkpoint inhibitors in the gut and the possible relationship with the gut microbiota.
Similar content being viewed by others
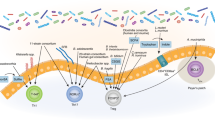


Introduction
Our body is a living place for not only host cells but also numerous microorganisms, collectively called the microbiota. Most commensal microbial colonization is symbiotic, benefiting the host. These microbes colonize barrier organs, including the intestines, where the host immune system plays an important role in balancing mucosal immunity. Recent studies have shown that the gut microbiota is closely associated with immune-related diseases such as inflammatory bowel disease (IBD), persistent antibiotic-induced colitis, atopic asthma, type 1 diabetes and autoimmunity1. Commensal microbes can regulate various immune cell types. Intestinal T-cell differentiation is significantly impacted by specific microbes. Commensal microbes can be processed by dendritic cells to induce IgA production2. The gut microbiota also regulates the cellular diversity and landscape of innate lymphoid cells (ILCs)3.
How the gut microbiota shapes immune cells has been an active field of research. Multiple pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and NOD-like receptors (NLRs), can transduce microbe-derived signals to immune cells. These innate sensing mechanisms can drive large changes in the mucosal immune system. Thus, signaling mediators and pathways connecting bacterial molecules to immune cell differentiation can play a pivotal role in host-microbiota interactions.
Unlike pathogenic infections, which induce inflammation and disease4, commensal gut microbes shape immunity in more subtle ways. Certain commensal bacteria may impact the frequency of immune cell types5 or impact immune cell function6,7, which, while not directly causing inflammation, may predispose the host to or protect the host from a variety of diseases. This review will focus specifically on commensal-immune signaling.
Gut microbiotas shape immunity by impacting cell frequencies
Perhaps the most striking effect of the gut microbiome on the immune system can be seen with the naked eye. Germ-free (GF) mice, which are raised in sterile isolators and devoid of all microbes, have visibly smaller Peyer’s patches8 and fewer T cells in their Peyer’s patches and mesenteric lymph nodes (MLNs)9. The impact of specific human bacteria on immune cell frequencies was investigated by mono-colonizing GF mice with 53 different human commensal bacteria and analyzing immune cell frequencies in the colon lamina propria, small intestine lamina propria, Peyer’s patches, mesenteric lymph nodes, and spleen5. At baseline, the frequencies and subset compositions of some immune cell types, such as RORγt+ Tregs or macrophages in the colon, are changed and highly dependent on the presence of microbes, whereas other immune cell types do not vary with colonization6,7. In other cases, the impact of colonization with specific bacteria is only observed in the context of disease settings10,11.
Scratching the surface of signaling by commensal organisms
It has been known for almost twenty years that commensals, and not just pathogens, signal through host TLRs12. Our understanding of how the host immune system distinguishes signals from commensals and pathogens and interprets signals from commensal organisms to maintain homeostasis and modulate disease is in its infancy. Here, we explore the known commensal microbe-immune signaling pathways and their impacts on health and disease. First, we will explore how secreted bacterially derived metabolites impact immune function. Next, we will discuss bacterial signaling, including that by bacterial surface molecules and other bacterial molecules that could be on surface, membrane, or in unknown locations.
Microbial metabolites
The gut microbiota produces numerous metabolites that can affect host immune function13. Although the exact immunomodulatory molecules have not been identified, gut microbiota metabolites can activate innate lymphoid cells (ILCs); the interactions between the gut microbiota and ILCs have been reviewed elsewhere14,15,16. In the mucosa, where commensal microbes reside, one of the key immune subsets is ILCs, which are distinct from conventional lymphocytes16. Similar to other lymphocytes, ILCs develop from common lymphoid progenitors (CLPs). ILCs can differentiate into five major subsets including NK cells, ILC1s, ILC2s, ILC3s, and LTi cells. ILCs play a role in modulating adaptive immunity by interacting with T cells in the intestines and other barrier organs. The lineage specification of ILCs resembles that of helper T-cell differentiation programs, and each group of ILCs shares functional similarities and transcriptional programs with T helper cell counterparts. For example, ILC1s express T-bet and IFN-γ like TH1 cells; ILC2s express GATA-3, IL-5 and IL-13 like TH2 cells; and ILC3s express RORγt and IL-17 like TH17 cells16.
Gut commensals can activate ILC3s. CX3CR1 mononuclear phagocytes sense microbial signals in a MYD88-dependent manner to induce IL-23 and IL-1β, which leads to increased release of IL-22 and GM-CSF by ILC3s17,18,19. Lactobacillus species can metabolize dietary tryptophan into AHR ligands that stimulate ILC3s20. This microbiota-ILC3/TH17 crosstalk plays an important role in intestinal barrier homeostasis, colitis, and intestinal infection17,21.
The dependence of ILC2 activity on the gut microbiota is less clear, but ILC2s can also be activated by components of the microbiota. The microbial metabolite succinate signals through Tuft cells in the gut to induce IL-25, which activates ILC2 production of IL-13 and IL-5, leading to increased goblet cell and mucous production14,15,22,23. ILC2s play an important role in immune homeostasis in the lung and the response to infection, yet how ILC2s sense microbial signals and what microbial signals impact ILC2 function remain unclear16.
Short-chain fatty acids (SCFAs), produced by many members of the gut microbiota, can signal through G-protein coupled receptors (GPRs) on epithelial cells to induce regulatory T cells. Gut microbes can metabolize bile acids in the colon to induce RORγt+ Tregs by signaling through vitamin D receptor (VDR)24, which plays an important role in protecting against colitis. Other gut microbe-derived bile acid metabolites inhibit TH17 signaling and enhance Treg signaling25. Microbial bile acids may influence bariatric surgery outcomes and obesity-associated comorbidities26.
Taken together, these examples illustrate how microbiota-derived molecules can influence immune function. Further understanding of the immunomodulatory mechanisms of the microbiota could be beneficial for developing therapies for a wide range of diseases.
Bacterial signaling molecules
Bacteroides species
The phylum Bacteroidetes is one of the two most prominent phyla that make up the human gut microbiome and is one of the early colonizers of the infant gut27,28. Therefore, it is unsurprising that Bacteroides species have multiple immunomodulatory effects. For example, polysaccharide A (PSA) isolated from the surface of the gut commensal organism Bacteroides fragilis contains a lipid A portion and a polysaccharide portion that signal through TLR1/2 heterodimers in conjunction with Dectin-1 to initiate phosphoinositide 3-kinase (PI3K) pathway signaling, leading to CREB-dependent expression of anti-inflammatory genes such as IL-10 in dendritic cell-CD4+ T-cell co-cultures9 (Fig. 1). PSA treatment protects against disease in mouse models of colitis and experimental autoimmune encephalitis (EAE)29,30,31. Lipooligosaccharides (LOS) on the outer membranes of B. fragilis and other Bacteroides species can signal through TLR4 on dendritic cells to induce interferon-β secretion, and treatment with PSA (containing the LOS) protects against murine vesicular stomatitis virus (VSV) infection32.

TLR1/2 and Dectin-1 mediate PSA-induced PI3K activation, which leads to CREB-dependent transcription of anti-inflammatory genes in dendritic cells. This signaling pathway might drive dendritic cells to differentiate T cells into IL-10-producing cells. The LOS portion of PSA can bind to TLR4 and activate the MyD88-dependent NF-kB signaling pathway and the TRIF-dependent IRF3 signaling pathway, leading to the transcription of proinflammatory cytokines (e.g., IFN-β).
PSA and LOS are not the only molecules from B. fragilis that induce immune signaling. B. fragilis also expresses alpha galactosylceramides (αGCs)33,34,35. αGCs complex with CD1d and the invariant natural killer T-cell (iNKT) cell receptor and stimulate iNKT cells to produce IL-2 and IFNγ33,35. The structure of αGCs impacts their immunomodulatory effects. For example, the synthetic iNKT cell agonist KRN7000, which robustly stimulates IL-2 production in APC-NKT cell co-cultures33, has a longer N-acyl chain than αGCs and a hydroxyl group at C4 of the sphinganine base35. These differences could at least partially explain the differences between how KRN7000 and αGCs modulate immune function. Stimulation of NKT cells with KRN7000 results in both IL-4 and IFN-γ release, but stimulation with synthetic versions of KRN7000 that have shorter N-acyl chains bias toward IL-4 release. Removal of the hydroxyl group at C4, on the other hand, results in loss of affinity for the TCR34. Additionally, the sphingosine chain branching of αGCs affects their immunomodulatory effects on gene expression and cytokine release, with branched chain αGCs stimulating more IL-2 production than straight chain αGCs36. Whether the lack of virulence factors in commensal strains, the ability of PSA and αGCs to have both immune stimulatory and inhibitory effects32,37, or some yet to be determined factors distinguish commensal strains of B. fragilis from pathogens that signal through TLRs is unclear.
Akkermansia mucinophila
Another gram-negative bacterium, Akkermansia mucinophila, is a human gut commensal that is considered beneficial because it is negatively correlated with inflammatory bowel disease and type 2 diabetes and is associated with an antitumor response to anti-PD-1 therapy in melanoma patients38,39. Recently, a phospholipid isolated from the cell membrane of A. mucinophila was shown to signal through TLR1/2 heterodimers to induce TNFα and IL-6 signaling in BMDCs. Notably, this induction was weaker than that following treatment with LPS (TLR4 agonist) or Pam3CSK4 (TLR2 agonist) and did not induce other cytokines, such as IL-23A or IL-12β, that are induced upon treatment with LPS or Pam3CSK440. This more specific signaling response could at least partially be due to the differences in structure between the A. mucinophila phospholipid and traditional TLR agonists but could also be due to other immunomodulatory effects.
Segmented filamentous bacteria
Segmented filamentous bacteria (SFB), commensal bacteria isolated from Taconic mice, specifically colonize the ileum, induce TH17 cells and stimulate type 3 innate lymphoid cells (ILC3s). SFB colonization stimulates epithelial cells in the gut to produce serum amyloid A (SAA 1 and SAA2), which promotes TH17 cells to secrete IL-17 and ILC3s to secrete IL-2218. SFB colonization protects against Citrobacter rodentium infection41 and can also promote autoimmunity42. Although the immunomodulatory molecules produced by SFB are unknown, TH17 induction is dependent on MHCII expression by dendritic cells, suggesting that SFB antigens presented by intestinal dendritic cells can drive TH17 differentiation41,43.
Modulation of costimulation by innate sensors
The two-signal model44 for T-cell activation was proposed to explain the requirement for a second signal in addition to the T-cell receptor (TCR) signal for T-cell activation45. Intensive studies of costimulation have advanced our understanding of these second signals. We now appreciate that these second signals not only regulate the initial activation of naïve T cells but also control effector, memory, and regulatory T cells. In addition, there are both positive secondary signals (costimulatory) that stimulate T-cell responses and negative secondary signals (coinhibitory) that inhibit T-cell responses46. Costimulatory and coinhibitory molecules can be expressed not only on antigen-presenting cells but also on nonhematopoietic cells, which may enable these molecules to regulate T-cell responses locally in specific tissues.
Early studies identified interactions between CD28 and its ligands B7-1 (CD80) and B7-2 (CD86) as the major secondary signal (costimulatory signal) required for initial T-cell activation. Upon TCR engagement of cognate peptide-MHC, ligation of CD28 on T cells by B7-1 or B7-2 on antigen-presenting cells (APCs) provides ‘signal 2’ that fully activates T cells. Costimulatory and coinhibitory pathways have been implicated in many disease contexts, primarily related to T-cell-mediated immune responses. These pathways not only regulate the adaptive immune system but also, as more recently reported, the functions of these molecules have expanded to the regulation of innate cells, including macrophages, dendritic cells, and innate lymphocytes. Therefore, stimuli that control the surface expression of these costimulatory and coinhibitory receptors and their ligands can shape various types of immune responses. The expression of costimulatory and coinhibitory molecules can be modulated by the tissue environment, activation states, cytokines and innate receptor signaling47.
The most well-known family of costimulatory and coinhibitory molecules is the B7 superfamily, which includes B7-1, B7-2, ICOSL (CD275, B7RP-1, B7-H2), PD-L1 (CD274, B7-H1), PD-L2 (CD273, B7-DC), B7-H3 (CD276), and B7-x (B7-H4, B7S1)48. B7 family members belong to the immunoglobulin superfamily and contain extracellular IgV and IgC domains. These molecules can be transmembrane or have a glycosylphosphatidylinositol (GPI)49 anchor. B7-1 and B7-2 bind to CD28 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4, CD152), but the binding affinity of B7-1 and CTLA-4 is much higher than that of B7-1 and CD2850,51. ICOSL binds to ICOS (CD178)52. PD-L1 and PD-L2 share PD-1 as a binding partner53, but each has a second unique binding partner. PD-L1 also binds B7-154, and PD-L2 binds repulsive guidance molecule b (RGMb)55. While the expression of B7-1, B7-2 and PD-L2 is primarily on antigen-presenting cells (APCs), PD-L1, B7-H3 and B7x can also be more broadly expressed on nonhematopoietic cells and tumors56,57. The functions of B7 family members and their receptors have been actively investigated and revealed that these pathways can transduce both positive signals and negative signals depending on the context and receptors58.
Another class of costimulatory pathways belongs to the tumor necrosis factor receptor superfamily (TNFR)59. These include the OX40 (CD134)/OX40L (CD252), CD40/CD40L, 4-1BB (CD137)/4-1BBL, CD27/CD70, Herpesvirus entry mediator (HVEM, CD270)/LIGHT (CD258)/B- and T-lymphocyte attenuator (BTLA, CD272)/CD160, CD30/CD30L, Glucocorticoid-induced TNFR-related protein (GITR, CD357)/GITRL, and Death domain receptor 3 (DR3)/Tumor necrosis factor-like cytokine 1A (TL1A) pathways60,61 (Fig. 2, Table 1). HVEM is widely expressed in all lymphocytes, including resting T and B cells, NK cells, Tregs, monocytes, and dendritic cells (DCs), as well as mesenchymal cells and epithelial cells62. LIGHT is expressed in DCs, macrophages, neutrophils, NK cells, ILCs, NKT cells and activated CD4+ and CD8+ T cells but not naïve T cells, Tregs and B cells63. BTLA, CD27 and DR3 expression on naïve T cells is low but upregulated upon activation64,65. Other members have inducible expression on APCs (e.g., OX40L, 4-1BBL) or T cells (e.g., OX40, 4-1BB)59. Most of these pathways are costimulatory for T-cell responses. However, HVEM can transduce coinhibitory signaling when binding to its ligands CD160 or BTLA, in contrast to transducing a costimulatory signal when engaging LIGHT66.

TL1A/DR3, GITRL/GITR, CD30L/CD30, HVEM/LIGHT, CD40L/CD40, 4-1BBL/4-1BB, OX40L/OX40 and CD70/CD27 interactions provide costimulatory signals to T cells, while ligation of HVEM with BTLA or CD160 provides coinhibitory signals to T cells.
In addition, other sets of pathways with costimulatory and coinhibitory functions have been identified. These include the T-cell immunoreceptor with Ig and ITIM domains (TIGIT), T-cell immunoglobulin and mucin-domain containing-3 (Tim-3, CD366) and LAG-3 (CD223) pathways. TIGIT shares the ligands CD155 and CD112 with CD226. CD226 promotes T-cell and NK cell immunity67,68. TIGIT negatively regulates NK and T-cell responses and has a higher affinity for CD155 than DNAM-1. T-cell immunoglobulin and mucin-domain containing-3 (Tim-3, CD366) is expressed on a variety of immune subsets, including T cells, myeloid cells, NK cells, mast cells and DCs, and has Galectin-9 (Gal-9), phosphatidylserine, Carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1) and High motility group Box 1 (HMGB1) as ligands. The binding of Tim-3 to phosphatidylserine is crucial for the phagocytosis of apoptotic cells and antigen presentation by DCs69. Recent studies have revealed an important role for Tim-3 in DCs in limiting inflammasome activation and showed that loss of Tim-3 in DCs can increase inflammasome activation and promote antitumor immunity70. Finally, the coinhibitory receptor LAG-3 is expressed on T cells and NK cells. The known ligands for LAG-3 are MHC class II molecules, fibrinogen-like protein 1 (FGL-1), liver sinusoidal endothelial cell lectin (LSECtin) and galectin-3 (Gal-3)71,72,73. LAG-3/Gal-3 interactions and LAG-3/LSECtin interactions inhibit IFN-γ production by CD8+ T cells72,74,75. Recent data show that LAG-3 functions as a T-cell signaling disruptor in an MHC class II-independent fashion. During the formation of the immunological synapse, LAG-3 is constitutively associated with the TCR-CD3 complex, where it interacts with the CD4 or CD8 coreceptors. The cytoplasmic tail of LAG-3 regulates the magnitude of TCR-induced signaling through the dissociation of Lck from the CD4 or CD8 coreceptors, resulting in loss of coreceptor-TCR signaling and thereby limiting T-cell activation76.
The expression of costimulatory and coinhibitory molecules can be altered by innate sensors such as TLRs, DAMPs and NODs, either as a direct downstream effect of innate receptor signaling or by an indirect effect of cellular activation. In the intestines, immune cells can be exposed to a wide range of bacterial molecules, which can stimulate these cells through these innate receptors. Of note, ligation of innate receptors on APCs often leads to activation and maturation of the cells. Below, we discuss how TLRs, DAMPs and NOD receptors regulate the expression of costimulatory and coinhibitory molecules.
Toll-like receptors
Among bacteria-derived signaling pathways, the role of TLRs in regulating the expression of B7 family members is well documented. An early study on innate recognition showed that TLR4 stimulation induces B7-1 upregulation77. In addition, LPS-induced DC maturation dramatically increases B7-1 and B7-2 expression78,79. Remarkably, MyD88, a downstream signaling adaptor of TLR signaling pathways, did not control B7-1 and B7-2 expression, while TLR4-mediated cytokine production was dependent on MyD8880,81. However, MyD88 was required for TLR9-induced expression of B7-1 and B7-2, indicating that specific TLR signaling cascades govern costimulatory molecules. Peptidoglycan and lipoteichoic acid-induced TLR2 signaling increase B7-2 expression on DCs82. TLRs also regulate ICOSL expression83. LPS/TLR4-mediated ICOSL upregulation on macrophages primarily depends on MyD88 and less on TIR-domain-containing adaptor-inducing interferon-β (TRIF). TLR3 activation by poly I:C can also increase ICOSL expression. The TLR2 agonist Pam3CSK4 increases the expression of B7-1, B7-2 and ICOSL on plasmacytoid DCs (pDCs)83.
Interestingly, TLRs not only promote costimulatory molecule expression but also induce the expression of coinhibitory molecules. For example, PD-L1 is upregulated upon LPS treatment of macrophages83, whereas PD-L2 expression remains remarkably unchanged. Stimulation of BMDCs with Schistosoma japonicum antigens elevates PD-L2, not PD-L1, expression in a TLR2-dependent manner84. Schistosoma mansoni worms, which have lipids capable of transducing signals through TLR2, selectively induce the expression of PD-L1 on macrophages but not other B7 molecules85,86. Beyond innate immune cells, TLR2 signaling by heat-killed Staphylococcus aureus or Pam3CKS4 can upregulate PD-L1 expression in head and neck squamous cell carcinoma (HNSCC) cells87. TLR4 activation drives PD-L1 expression on CD90+ colonic myofibroblasts/fibroblasts88. For B7-H3, bacterial agonists of TLR2 and TLR4 can induce its expression on human monocytes89. LPS upregulates B7x expression on renal tubular cells, podocytes, and glomerular endothelial cells90. These studies collectively suggest that modulation of coinhibitory signaling pathways by TLRs might be specific to certain contexts and ligands.
Similar to B7 superfamily members, tumor necrosis factor receptor family (TNFR) members can be upregulated on antigen-presenting cells upon TLR signaling activation. For example, CD40 expression is induced by TLRs79,80,81,83. CD70, the ligand for CD27, is upregulated on BMDCs upon LPS, Poly I:C and CpG treatment91. Zymosan, a ligand for TLR2, and LPS treatment induce CD30L expression on DCs but not on macrophages or B cells92. LPS can transiently induce GITRL expression on BMDCs and macrophages93. Additionally, LPS can induce OX40L upregulation on splenic DCs, which constitutively express OX40L on their surface, and on B cells94. BTLA expression can be induced by LPS stimulation of BMDCs95. Thus, TLR signaling can induce the expression of TNFR family members on multiple cell types.
TLR stimulation also modulates the expression of other costimulatory ligands. For example, ligands of TLR1/2, TLR3, TLR4, TLR7/8, and TLR9 can induce CD155 expression on the surface of macrophage cell lines, BMDCs, BMDMs and B cells in a MyD88- and/or TRIF-dependent manner96. CD155 upregulation by TLR agonists requires NF-kB signaling, not MAPK signaling. Tim-3 expression on peritoneal macrophages is elevated 8 h after LPS treatment and then diminishes97. Interestingly, Tim-3 signaling can inhibit TLR4-mediated NF-kB activation. The Tim-3 ligand Gal-9 can be upregulated by TLR3 and TLR4 stimulation in microglia98. These examples illustrate how TLR signaling can regulate the expression of a variety of costimulatory and coinhibitory molecules.
Notably, TLR2 signaling itself can act as a costimulator of T-cell activation99. Activated T cells express TLR2 and TLR4. Bacterial lipoprotein (BLP), a ligand of TLR2, can enhance TCR-mediated cytokine production, whereas LPS does not induce this effect. TLR1/2 activation of CD8+ T cells enhances the expression of 4-1BB, OX40L and GITR in a MyD88-dependent manner100, suggesting that TLR2 in T cells may increase T-cell responses by sensitizing T cells to other costimulatory receptors, as well as by directly enhancing T-cell activation as a costimulatory receptor. Additionally, TLR2 costimulation can increase the functionality of antitumor T cells101. Perhaps TLR2 may be an ancient form of costimulation by which bacteria can directly influence T-cell activation, but this does not occur through antigen-presenting cells.
NOD-like receptors
While TLRs can recognize gut microbes extracellularly, nucleotide-binding oligomerization domain-like receptors (NLRs) sense bacterial components inside cells. NOD1 and NOD2 are functionally well described. NOD1 detects γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP), and NOD2 recognizes muropeptides from bacterial peptidoglycan102,103,104. NLR signaling can induce the expression of costimulatory and coinhibitory molecules. For example, stimulation of NOD2 with muramyl dipeptide induces PD-L1 and ICOSL on human monocytes105,106. Interestingly, such upregulation is not observed for CD80, CD86 and PD-L2106. Intravenous injection of FK-565, a ligand of NOD1, can induce upregulation of CD86 and CD40 on CD8α + DCs in mice in vivo107. Additionally, in vitro stimulation of BMDCs with NOD ligands increases the expression of CD40, CD70 and CD86108. However, CD8α- DCs show minimal changes in expression of these costimulatory molecules, suggesting that NOD1 and NOD2 might alter costimulation capacity in a cell-type specific manner. Collectively, NOD1 and NOD2 appear to activate DCs and increase the expression levels of costimulatory molecules.
While NOD ligands augment the effect of TLR2 on costimulatory molecule expression108, several other NLR family members negatively modulate TLR signaling. NLRC3, NLRP6, NLRP12 and NLRX1 inhibit macrophage activation induced by TLR2 and TLR4 agonists109,110,111,112 and attenuate NF-kB signaling and cytokine expression. Remarkably, the effects of these inhibitory NLRs on costimulatory and coinhibitory molecules remain largely unknown. NLRP3, which can sense bacterial RNAs, can upregulate PD-L1 expression on cancer cells113,114. Given that the intratumoral microbiota has emerged as one of the key elements in regulating the tumor microenvironment, antitumor immune responses and metastasis115,116,117, NLRs in the tumor microenvironment may act as a bridge between intratumoral bacteria and immune suppression by modulating coinhibitory molecules/pathways in tumor cells.
Costimulatory pathways in innate lymphoid cells (ILCs)
Recent studies have shown that ILCs have the capability to present antigens to T cells with costimulation118. For example, a subset of ILC3s that lack natural cytotoxicity receptor (NCR) on their surface express (NCR-ILC3s) MHC class II and induce antigen-specific T-cell responses119. NCR-ILC3s can be activated by various TLR and NOD ligands. Notably, they express CD40, CD80 and CD86 upon IL-1β stimulation119. In contrast, these costimulatory molecules are expressed at low levels on NCR-ILC3s from the small intestine, where the cells interact with the gut microbiota at the steady state120,121. During gastrointestinal infection with the helminth Nippostrongilus brasiliensis, expression of PD-L1 but not PD-L2 is induced in ILC2s122. CD30L, 4-1BBL, CD70 and OX40L are highly expressed on ILC2s in the lung, and OX40L expression can be further induced by IL-33123. Interestingly, microbiota-derived NOD2 signaling by muramyl dipeptide can indirectly regulate ILC2s through IL-33 induction in macrophages in mice susceptible to Crohn’s disease124. IL-33-activated ILC2s may potentially induce ileitis by modulating costimulation, suggesting a means by which microbiota may indirectly tune ILC-mediated costimulation. In the intestines, the IBD-associated microbiota triggers CX3CR1+ mononuclear phagocytes to produce TL1A, which upregulates OX40L on all ILC3 subsets after engaging its receptor DR3125. In a T-cell colitis model, OX40L expression on ILC3s led to increased TH1 differentiation in a DR3-dependent manner. ILC2s express both ICOS and ICOSL, and their ligation is required for ILC2 functions126. The HVEM/LIGHT axis also participates in the costimulatory actions on ILCs. ILC3 expression of HVEM is critical for IFN-γ-mediated protection against enteric infection127. Collectively, these studies show how costimulatory ligands on ILCs can regulate T-cell-mediated mucosal immunity.
ILCs also express costimulatory and coinhibitory receptors that play a cell-intrinsic role in controlling ILC functions. For example, PD-1 negatively regulates a KLRG-1 + ILC2 population and IL-33-activated ILC2s128,129. TNFR2 expression on ILC2s is important for ILC2 function and survival in the lungs130. Engagement of GITR with an anti-GITR agonistic monoclonal antibody stimulates ILC2s to secrete TH2 cytokines and polarize M2 macrophages in the context of type II diabetes in RAG-/- mice131. Despite the emerging roles of ILCs as antigen-presenting cells and key cellular regulators of mucosal immunity, how innate sensing in ILCs modulates their costimulatory properties is not clear. Further work is needed to understand how commensal microbial signals influence costimulatory and coinhibitory pathways in ILCs.
Similar to T cells, TLR2 can provide costimulation to ILCs. TLR2 signaling leads to increased LTi cell proliferation and IL-22 production132. The TLR2-MyD88 pathway is required for effective NK cell responses to vaccinia virus infection133. House dust mite extract activates TLR2 signaling in ILC2s, leading to increased production of IL-5 and IL-13134. On the other hand, treatment of ILC3s with LTA, another type of ligand for TLR2, depletes ILC3s by apoptosis135. Thus, TLR2 signaling can directly regulate the function and maintenance of ILCs, with differing outcomes depending on ligand type and ILC subset.
Gut microbiota and immune checkpoint immunotherapy targeting coinhibitory pathways
One of the best examples of the association of gut microbiota and costimulation relates to immune checkpoint blockade cancer immunotherapies, one of the most successful cancer therapies. These therapies block PD-1, PD-L1, CTLA-4136 and other coinhibitory pathways. The gut microbiota has been clinically associated with these cancer immunotherapies in two ways.
First, the microbiota can modulate the efficacy of immune checkpoint blockade. Recent studies have shown the close association between gut microbial composition and responsiveness to anti-PD-1 cancer immunotherapy in cancer patients39,137,138. Initial studies demonstrated that different gut microbial species are enriched in patients who respond and those who do not respond to anti-PD-1 immunotherapy, and the same responses were reproduced in mice colonized with the patient microbiota. For example, Akkermansia mucinophila and Enterococcus hirae were found in responding patients, and mono-colonization of germ-free mice with these microbes induced a potent antitumor response to PD-1 blockade39. A defined consortium of gut microbes could induce potent responses to PD-1 blockade in tumor models139. More recently, fecal transplantation has been shown to overcome resistance to anti-PD-1 cancer immunotherapy, suggesting a causal relationship between anti-PD-1 therapy and the gut microbiota in patients140,141.
Despite ample evidence showing that the gut microbiota can regulate antitumor immune responses induced by immune checkpoint blockade, the mechanisms are not well understood. Several molecules secreted by the gut microbiota have been found to enhance the efficacy of immune checkpoint inhibitor therapy for cancer. Inosine, produced by Bifidobacter pseudolongum, was shown to promote the efficacy of anti-CTLA-4 by inducing TH1 responses through adenosine 2 A receptor signaling11. Enterococcus species secrete a hydrolase, SagA, which hydrolyzes peptidoglycan from the gut microbiota to promote an antitumor response in response to anti-PD-L1 via NOD2142. The gut microbiota can also release STING agonists to promote type 1 interferon production by monocytes and enhance antitumor responses by macrophages, DCs and NK cells143. Given that microbial signals control the expression of many costimulatory and coinhibitory molecules, gut microbes might also regulate responses to PD-1 inhibition by modulating other costimulatory and coinhibitory pathways. Our recent work showed that the gut microbiota can downregulate the PD-L2/RGMb axis and enhance antitumor immunity144. Indeed, many costimulatory and coinhibitory pathways interact with each other, and the efficacy of immune checkpoint blockade can be improved by the combined targeting of specific costimulatory and coinhibitory pathways145. Therefore, it is important to investigate how specific gut microbiota regulate costimulatory and coinhibitory pathways as a cancer immunotherapy response or resistance mechanism.
Second, immune-related adverse effects (IrAEs) that arise following immune checkpoint blockade have been correlated with microbiome composition146,147. Blocking PD-1, PD-L1 and CTLA-4 can result in these undesired inflammatory side effects148. Remarkably, a significantly high portion of patients with a high level of Streptococcus sup. developed irAEs after anti-PD-1 therapy146. Another study revealed that Bacteroides intestinalis and Intestinalibacter Bartlettii were enriched in a ≥grade 3 IrAE patient group, whereas patients who did not develop ≥grade 3 IrAE had microbiota enriched by Anaerotignum lactatifermentas and Dorea formicigenerans149. IL-1β and B. intestinalis are associated with combined immune checkpoint blockade-induced colitis in melanoma patients. The Bacteroidetes phylum is more prevalent in melanoma patients who are resistant to the development of anti-CTLA-4-associated colitis150, and metagenomic analyses in this study revealed an association of vitamin B biosynthesis pathways and polyamine transport with resistance to colitis development. Moreover, skin microbiota is linked to cutaneous IrAE151. IL-17A produced by T cells specific to the skin commensal microbe Staphylococcus aureus can mediate anti-CTLA-4-driven pathology in the skin. Impressively, a recent report showed that fecal transplantation from healthy donors to patients with immune checkpoint inhibitor-associated colitis completely resolved colitis symptoms152. Further work is needed to elucidate the mechanisms of microbiota-driven IrAEs.
Conclusion
It is widely known that signals from gut microbiota play a pivotal role in shaping mucosal immune responses in the intestines. Commensal microbes produce various molecules recognized by the mucosal immune system. The recognition of commensal-derived molecules is mediated by canonical innate sensing mechanisms such as TLRs and NODs, which are used to detect pathogen-associated molecular patterns (PAMPs). Signal transduction through these pattern recognition receptors results in changes in the transcription of immune-related genes in innate cells. One of the significant changes in innate immune cells is an alteration in the capability of antigen-presenting cells to provide costimulation and coinhibition to the adaptive immune system. In this review, we focused on the relationship between microbe-derived signals and costimulatory and coinhibitory pathways. As ILCs are one of the most pronounced populations in mucosal sites, we discussed new insights into costimulation and immune regulation by ILCs. Importantly, ILCs can act as unconventional antigen-presenting cells that regulate mucosal T-cell responses through costimulatory pathways. Finally, we highlight the clinically relevant relationship between microbiota and costimulatory and coinhibitory pathways with an emphasis on response to immune checkpoint blockade. It is remarkable that the efficacy of immune checkpoint blockade therapies depends on the gut microbiota in patients. More surprisingly, distinct microbiota determine the development of IrAEs in the intestines and other barrier organs. This recent work illustrates how costimulation can mediate host-microbe interactions to shape the mucosal immune system. A deeper understanding of the mechanisms by which T-cell costimulatory and coinhibitory pathways mediate host-microbe interactions will provide insight into the regulation of mucosal immunity at homeostasis and during disease. In conclusion, we propose that innate sensing pathways and costimulatory and coinhibitory pathways in host immune cells closely cooperate with the microbiota to shape mucosal immunity.
References
Durack, J. & Lynch, S. V. The gut microbiome: relationships with disease and opportunities for therapy. J. Exp. Med. 216, 20–40 (2019).
Macpherson, A. J. & Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665 (2004).
Gury-BenAri, M. et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell 166, 1231–1246 e1213 (2016).
Bouladoux, N., Harrison, O. J. & Belkaid, Y. The mouse model of infection with Citrobacter rodentium. Curr. Protoc. Immunol. 119, 19 15 11–19 15 25 (2017).
Geva-Zatorsky, N. et al. Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943 e911 (2017).
Sefik, E. et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 349, 993–997 (2015).
Kang, B. et al. Commensal microbiota drive the functional diversification of colon macrophages. Mucosal Immunol. 13, 216–229 (2020).
Lecuyer, E. et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 40, 608–620 (2014).
Chung, H. et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593 (2012).
Rivollier, A., He, J., Kole, A., Valatas, V. & Kelsall, B. L. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 209, 139–155 (2012).
Mager, L. F. et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489 (2020).
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004).
Rooks, M. G. & Garrett, W. S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352 (2016).
Britanova, L. & Diefenbach, A. Interplay of innate lymphoid cells and the microbiota. Immunol. Rev. 279, 36–51 (2017).
Seo, G. Y., Giles, D. A. & Kronenberg, M. The role of innate lymphoid cells in response to microbes at mucosal surfaces. Mucosal Immunol. 13, 399–412 (2020).
Artis, D. & Spits, H. The biology of innate lymphoid cells. Nature 517, 293–301 (2015).
Longman, R. S. et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 211, 1571–1583 (2014).
Sano, T. et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 163, 381–393 (2015).
Mortha, A. et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343, 1249288 (2014).
Zelante, T. et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385 (2013).
Talbot, J. et al. Feeding-dependent VIP neuron-ILC3 circuit regulates the intestinal barrier. Nature 579, 575–580 (2020).
Howitt, M. R. et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333 (2016).
Nadjsombati, M. S. et al. Detection of succinate by intestinal tuft cells triggers a Type 2 innate immune circuit. Immunity 49, 33–41 e37 (2018).
Song, X. et al. Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 577, 410–415 (2020).
Hang, S. et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 576, 143–148 (2019).
Sipe, L. M., Chaib, M., Pingili, A. K., Pierre, J. F. & Makowski, L. Microbiome, bile acids, and obesity: How microbially modified metabolites shape anti-tumor immunity. Immunol. Rev. 295, 220–239 (2020).
Wexler, A. G. & Goodman, A. L. An insider’s perspective: bacteroides as a window into the microbiome. Nat. Microbiol. 2, 17026 (2017).
Stewart, C. J. et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588 (2018).
Erturk-Hasdemir, D. et al. Symbionts exploit complex signaling to educate the immune system. Proc. Natl Acad. Sci. USA 116, 26157–26166 (2019).
Ochoa-Reparaz, J. et al. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 3, 487–495 (2010).
Mazmanian, S. K., Round, J. L. & Kasper, D. L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625 (2008).
Stefan, K. L., Kim, M. V., Iwasaki, A. & Kasper, D. L. Commensal microbiota modulation of natural resistance to virus infection. Cell 183, 1312–1324 e1310 (2020).
An, D. et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156, 123–133 (2014).
Savage, P. B., Teyton, L. & Bendelac, A. Glycolipids for natural killer T cells. Chem. Soc. Rev. 35, 771–779 (2006).
Wieland Brown, L. C. et al. Production of alpha-galactosylceramide by a prominent member of the human gut microbiota. PLoS Biol. 11, e1001610 (2013).
Oh, S. F. et al. Host immunomodulatory lipids created by symbionts from dietary amino acids. Nature 600, 302–307 (2021).
Blander, J. M. & Sander, L. E. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat. Rev. Immunol. 12, 215–225 (2012).
Zhou, K. Strategies to promote abundance of Akkermansia muciniphila, an emerging probiotics in the gut, evidence from dietary intervention studies. J. Funct. Foods 33, 194–201 (2017).
Routy, B. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Bae, M. et al. Akkermansia muciniphila phospholipid induces homeostatic immune responses. Nature 608, 168–173 (2022).
Ivanov, I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Flannigan, K. L. & Denning, T. L. Segmented filamentous bacteria-induced immune responses: a balancing act between host protection and autoimmunity. Immunology 154, 537–546 (2018).
Goto, Y. et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 40, 594–607 (2014).
Mitchison, N. A. The carrier effect in the secondary response to hapten-protein conjugates. II. Cellular cooperation. Eur. J. Immunol. 1, 18–27 (1971).
Mueller, D. L., Jenkins, M. K. & Schwartz, R. H. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol. 7, 445–480 (1989).
Baumeister, S. H., Freeman, G. J., Dranoff, G. & Sharpe, A. H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 34, 539–573 (2016).
Chen, L. & Flies, D. B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242 (2013).
Sharpe, A. H. & Freeman, G. J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2, 116–126 (2002).
Collins, M., Ling, V. & Carreno, B. M. The B7 family of immune-regulatory ligands. Genome Biol. 6, 223 (2005).
van der Merwe, P. A., Bodian, D. L., Daenke, S., Linsley, P. & Davis, S. J. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 185, 393–403 (1997).
Collins, A. V. et al. The interaction properties of costimulatory molecules revisited. Immunity 17, 201–210 (2002).
Yoshinaga, S. K. et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature 402, 827–832 (1999).
Latchman, Y. et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 (2001).
Chaudhri, A. et al. PD-L1 binds to B7-1 only In Cis on the same cell surface. Cancer Immunol. Res. 6, 921–929 (2018).
Xiao, Y. et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J. Exp. Med. 211, 943–959 (2014).
Zang, X. et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod Pathol 23, 1104–1112 (2010).
Zhong, X., Tumang, J. R., Gao, W., Bai, C. & Rothstein, T. L. PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur. J. Immunol. 37, 2405–2410 (2007).
Sharpe, A. H. Mechanisms of costimulation. Immunol. Rev. 229, 5–11 (2009).
Croft, M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity. Nat. Rev. Immunol. 3, 609–620 (2003).
Valatas, V., Kolios, G. & Bamias, G. TL1A (TNFSF15) and DR3 (TNFRSF25): a co-stimulatory system of cytokines with diverse functions in gut mucosal immunity. Front Immunol 10, 583 (2019).
Elgueta, R. et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 229, 152–172 (2009).
Seo, G. Y. et al. Epithelial HVEM maintains intraepithelial T cell survival and contributes to host protection. Sci. Immunol. 7, eabm6931 (2022).
Ware, C. F., Croft, M. & Neil, G. A. Realigning the LIGHT signaling network to control dysregulated inflammation. J Exp Med 219, e20220236 (2022),
Beier, K. C., Kallinich, T. & Hamelmann, E. Master switches of T-cell activation and differentiation. Eur. Respir. J. 29, 804–812 (2007).
Pappu, B. P. et al. TL1A-DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J. Exp. Med. 205, 1049–1062 (2008).
Cai, G. & Freeman, G. J. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol. Rev. 229, 244–258 (2009).
Anderson, A. C., Joller, N. & Kuchroo, V. K. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44, 989–1004 (2016).
Chiang, E. Y. & Mellman, I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J. Immunother. Cancer 10, e004711 (2022).
Nakayama, M. et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113, 3821–3830 (2009).
Dixon, K. O. et al. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 595, 101–106 (2021).
Goldberg, M. V. & Drake, C. G. LAG-3 in cancer immunotherapy. Curr. Top. Microbiol. Immunol. 344, 269–278 (2011).
Wang, J. et al. Fibrinogen-like protein 1 Is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347 e312 (2019).
Andrews, L. P. et al. Molecular pathways and mechanisms of LAG3 in cancer therapy. Clin. Cancer Res. 28, 5030–5039 (2022).
Kouo, T. et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res 3, 412–423 (2015).
Xu, F. et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 74, 3418–3428 (2014).
Guy, C. et al. LAG3 associates with TCR-CD3 complexes and suppresses signaling by driving co-receptor-Lck dissociation. Nat. Immunol. 23, 757–767 (2022).
Medzhitov, R., Preston-Hurlburt, P. & Janeway, C. A. Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 (1997).
Winzler, C. et al. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J. Exp Med. 185, 317–328 (1997).
Sallusto, F., Cella, M., Danieli, C. & Lanzavecchia, A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J. Exp. Med. 182, 389–400 (1995).
Kawai, T., Adachi, O., Ogawa, T., Takeda, K. & Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11, 115–122 (1999).
Kaisho, T., Takeuchi, O., Kawai, T., Hoshino, K. & Akira, S. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J. Immunol. 166, 5688–5694 (2001).
Michelsen, K. S. et al. The role of toll-like receptors (TLRs) in bacteria-induced maturation of murine dendritic cells (DCS). Peptidoglycan and lipoteichoic acid are inducers of DC maturation and require TLR2. J. Biol. Chem. 276, 25680–25686 (2001).
Zhou, Z. et al. Antagonism between MyD88- and TRIF-dependent signals in B7RP-1 up-regulation. Eur. J. Immunol. 35, 1918–1927 (2005).
Gao, Y. et al. TLR2 directing PD-L2 expression inhibit T cells response in Schistosoma japonicum infection. PLoS ONE 8, e82480 (2013).
Smith, P. et al. Schistosoma mansoni worms induce anergy of T cells via selective up-regulation of programmed death ligand 1 on macrophages. J. Immunol. 173, 1240–1248 (2004).
van der Kleij, D. et al. A novel host-parasite lipid cross-talk. Schistosomal lyso-phosphatidylserine activates toll-like receptor 2 and affects immune polarization. J. Biol. Chem. 277, 48122–48129 (2002).
Mann, J. E. et al. Microbe-mediated activation of toll-like receptor 2 drives PDL1 expression in HNSCC. Cancers (Basel) 13, 4782 (2021).
Beswick, E. J. et al. TLR4 activation enhances the PD-L1-mediated tolerogenic capacity of colonic CD90+ stromal cells. J. Immunol. 193, 2218–2229 (2014).
Yoon, B. R. et al. Preferential induction of the T cell auxiliary signaling molecule B7-H3 on synovial monocytes in rheumatoid arthritis. J. Biol. Chem. 291, 4048–4057 (2016).
Pawar, R. D. et al. B7x/B7-H4 modulates the adaptive immune response and ameliorates renal injury in antibody-mediated nephritis. Clin. Exp. Immunol. 179, 329–343 (2015).
Bullock, T. N. & Yagita, H. Induction of CD70 on dendritic cells through CD40 or TLR stimulation contributes to the development of CD8+ T cell responses in the absence of CD4+ T cells. J. Immunol. 174, 710–717 (2005).
Han, S. et al. Modulation of TNFSF expression in lymphoid tissue inducer cells by dendritic cells activated with Toll-like receptor ligands. BMB Rep. 44, 129–134 (2011).
Tone, M. et al. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc. Natl Acad. Sci. USA 100, 15059–15064 (2003).
Satake, Y. et al. Characterization of rat OX40 ligand by monoclonal antibody. Biochem. Biophys. Res. Commun. 270, 1041–1048 (2000).
Han, P., Goularte, O. D., Rufner, K., Wilkinson, B. & Kaye, J. An inhibitory Ig superfamily protein expressed by lymphocytes and APCs is also an early marker of thymocyte positive selection. J. Immunol. 172, 5931–5939 (2004).
Kamran, N. et al. Toll-like receptor ligands induce expression of the costimulatory molecule CD155 on antigen-presenting cells. PLoS ONE 8, e54406 (2013).
Yang, X. et al. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J. Immunol. 190, 2068–2079 (2013).
Steelman, A. J. & Li, J. Astrocyte galectin-9 potentiates microglial TNF secretion. J. Neuroinflammation 11, 144 (2014).
Komai-Koma, M., Jones, L., Ogg, G. S., Xu, D. & Liew, F. Y. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc. Natl Acad. Sci. USA 101, 3029–3034 (2004).
Joseph, A. M., Srivastava, R., Zabaleta, J. & Davila, E. Cross-talk between 4-1BB and TLR1-TLR2 signaling in CD8+ T cells regulates TLR2’s costimulatory effects. Cancer Immunol. Res. 4, 708–716 (2016).
Salerno, F., Freen-van Heeren, J. J., Guislain, A., Nicolet, B. P. & Wolkers, M. C. Costimulation through TLR2 drives polyfunctional CD8(+) T cell Rresponses. J. Immunol. 202, 714–723 (2019).
Girardin, S. E. et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300, 1584–1587 (2003).
Chamaillard, M. et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707 (2003).
Caruso, R., Warner, N., Inohara, N. & Nunez, G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41, 898–908 (2014).
Hedl, M., Lahiri, A., Ning, K., Cho, J. H. & Abraham, C. Pattern recognition receptor signaling in human dendritic cells is enhanced by ICOS ligand and modulated by the Crohn’s disease ICOSLG risk allele. Immunity 40, 734–746 (2014).
Hewitt, R. E. et al. Immuno-inhibitory PD-L1 can be induced by a peptidoglycan/NOD2 mediated pathway in primary monocytic cells and is deficient in Crohn’s patients with homozygous NOD2 mutations. Clin. Immunol. 143, 162–169 (2012).
Asano, J. et al. Nucleotide oligomerization binding domain-like receptor signaling enhances dendritic cell-mediated cross-priming in vivo. J. Immunol. 184, 736–745 (2010).
Wagner, C. S. & Cresswell, P. TLR and nucleotide-binding oligomerization domain-like receptor signals differentially regulate exogenous antigen presentation. J. Immunol. 188, 686–693 (2012).
Schneider, M. et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat. Immunol. 13, 823–831 (2012).
Zaki, M. H. et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660 (2011).
Anand, P. K. et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488, 389–393 (2012).
Xia, X. et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity 34, 843–853 (2011).
Kanneganti, T. D. et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006).
Lu, F. et al. NLRP3 inflammasome upregulates PD-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett. 497, 178–189 (2021).
Fu, A. et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell 185, 1356–1372 e1326 (2022).
Galeano Nino, J. L. et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature 611, 810–817 (2022).
Nejman, D. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 (2020).
Sonnenberg, G. F. & Hepworth, M. R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 19, 599–613 (2019).
von Burg, N. et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc. Natl Acad. Sci. USA 111, 12835–12840 (2014).
Lehmann, F. M. et al. Microbiota-induced tissue signals regulate ILC3-mediated antigen presentation. Nat. Commun. 11, 1794 (2020).
Hepworth, M. R. et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science 348, 1031–1035 (2015).
Schwartz, C. et al. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J. Exp. Med. 214, 2507–2521 (2017).
Halim, T. Y. F. et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on Group 2 innate lymphoid cells. Immunity 48, 1195–1207 e1196 (2018).
De Salvo, C. et al. NOD2 drives early IL-33-dependent expansion of group 2 innate lymphoid cells during Crohn’s disease-like ileitis. J. Clin. Invest. 131, e140624 (2021).
Castellanos, J. G. et al. Microbiota-induced TNF-like ligand 1A drives Group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during Colitis. Immunity 49, 1077–1089 e1075 (2018).
Maazi, H. et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity 42, 538–551 (2015).
Seo, G. Y. et al. LIGHT-HVEM signaling in innate lymphoid cell subsets protects against enteric bacterial infection. Cell Host Microbe 24, 249–260 e244 (2018).
Taylor, S. et al. PD-1 regulates KLRG1(+) group 2 innate lymphoid cells. J. Exp. Med. 214, 1663–1678 (2017).
Helou, D. G. et al. PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatment ameliorates airway hyperreactivity. Nat. Commun. 11, 3998 (2020).
Hurrell, B. P. et al. TNFR2 signaling enhances ILC2 survival, function, and induction of airway hyperreactivity. Cell Rep. 29, 4509–4524 e4505 (2019).
Galle-Treger, L. et al. Costimulation of type-2 innate lymphoid cells by GITR promotes effector function and ameliorates type 2 diabetes. Nat. Commun. 10, 713 (2019).
Crellin, N. K. et al. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 33, 752–764 (2010).
Martinez, J., Huang, X. & Yang, Y. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLoS Pathog. 6, e1000811 (2010).
Ishii, T. et al. Activation through toll-like receptor 2 on group 2 innate lymphoid cells can induce asthmatic characteristics. Clin. Exp. Allergy 49, 1624–1632 (2019).
Xu, H., Wang, X., Lackner, A. A. & Veazey, R. S. Type 3 innate lymphoid cell depletion is mediated by TLRs in lymphoid tissues of simian immunodeficiency virus-infected macaques. FASEB J. 29, 5072–5080 (2015).
Zang, X. & Allison, J. P. The B7 family and cancer therapy: costimulation and coinhibition. Clin. Cancer Res. 13, 5271–5279 (2007).
Gopalakrishnan, V. et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018).
Matson, V. et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018).
Tanoue, T. et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605 (2019).
Davar, D. et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371, 595–602 (2021).
Baruch, E. N. et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371, 602–609 (2021).
Griffin, M. E. et al. Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 373, 1040–1046 (2021).
Lam, K. C. et al. Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 184, 5338–5356 e5321 (2021).
Park, J. S. et al. Targeting PD-L2-RGMb overcomes microbiome-related immunotherapy resistance. Nature 617, 377–385 (2023).
Curran, M. A., Montalvo, W., Yagita, H. & Allison, J. P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl Acad. Sci. USA 107, 4275–4280 (2010).
McCulloch, J. A. et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat. Med. 28, 545–556 (2022).
Wang, Y., Jenq, R. R., Wargo, J. A. & Watowich, S. S. Microbiome influencers of checkpoint blockade-associated toxicity. J. Exp. Med. 220, e20220948 (2023).
Postow, M. A., Sidlow, R. & Hellmann, M. D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 378, 158–168 (2018).
Andrews, M. C. et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 27, 1432–1441 (2021).
Dubin, K. et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 7, 10391 (2016).
Hu, Z. I. et al. Immune checkpoint inhibitors unleash pathogenic immune responses against the microbiota. Proc. Natl Acad. Sci. USA 119, e2200348119 (2022).
Wang, Y. et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat. Med. 24, 1804–1808 (2018).
Acknowledgements
The authors would like to thank the members of the Sharpe lab for insightful discussion. JSP was supported by 5F32CA247072-02 (National Institute of Health/National Cancer Institute). FSG was supported by 5T32HD55148-10 (National Institutes of Health/National Institute of Childhood Health and Human Disease). AHS was supported by P01 AI56299. JSP, FSG, DLK, and AHS received funding from Quark Ventures A31696 unrelated to this review. All figures were created with BioRender.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A.H.S. currently has funding from Quark, AbbVie, Moderna and Erasca unrelated to this Review. A.H.S. serves on advisory boards for SQZ Biotechnologies, Selecta, Elpiscience, Monopteros, Bicara, Fibrogen, IOME, Alixia, Corner Therapeutics, Bioventre, Glaxo Smith Kline, Amgen, and Janssen. She is also on scientific advisory boards for the Massachusetts General Cancer Center, Program in Cellular and Molecular Medicine at Boston Children’s Hospital, the Human Oncology and Pathogenesis Program at Memorial Sloan Kettering Cancer Center, the Gladstone Institute, and the Johns Hopkins Bloomberg-Kimmel Institute for Cancer Immunotherapy. She is an academic editor for the Journal of Experimental Medicine. A.H.S. has patents/pending royalties on the PD-1 pathway from Roche and Novartis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, J.S., Gazzaniga, F.S., Kasper, D.L. et al. Microbiota-dependent regulation of costimulatory and coinhibitory pathways via innate immune sensors and implications for immunotherapy. Exp Mol Med 55, 1913–1921 (2023). https://doi.org/10.1038/s12276-023-01075-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01075-0
