
Abstract
Classical studies have shown that neuronal immediate-early genes (IEGs) play important roles in synaptic processes critical for key brain functions. IEGs are transiently activated and rapidly upregulated in discrete neurons in response to a wide variety of cellular stimuli, and they are uniquely involved in various aspects of synapse development. In this review, we summarize recent studies of a subset of neuronal IEGs in regulating synapse formation, transmission, and plasticity. We also discuss how the dysregulation of neuronal IEGs is associated with the onset of various brain disorders and pinpoint key outstanding questions that should be addressed in this field.
Similar content being viewed by others

Introduction
Numerous studies have shown that neural activity plays an important role in regulating synaptic strength, neuronal membrane properties, and neural circuit refinement1,2,3. In particular, sensory experiences continually influence brain development at synaptic, circuit, and organismal levels, as Hubel4 and Wiesel5 elegantly demonstrated in their work on visual cortex organization during the critical period5. The effects of sensory experience are manifested by neurotransmitter release at presynaptic terminals, their reception at postsynaptic membranes, and depolarization of postsynaptic neurons by increased concentration of cytoplasmic calcium. This increase in cytoplasmic calcium activates a program of gene expression in the nucleus6,7,8,9. Including classic immediately-early genes (IEGs) such as Fos and Jun, a number of activity-regulated transcription factors have been identified and extensively investigated3,10,11. These studies have led to an important hypothesis that transcriptional regulation is a key mechanism by which neuronal activity can trigger experience-dependent synaptic changes and maturation of neural circuits.
More than dozens of neuronal IEGs were identified by Paul Worley, Elly Nedivi, and colleagues12, owing to the use of the subtractive hybridization method, in combination with various neural stimulation protocols, most notably seizure paradigms. Among them, cAMP-responsive element-binding protein (CREB) has been the most studied in regulating activity-dependent synapse development and plasticity13. Its physiological significance in neuronal development and cognitive behaviors has been consistently shown across a variety of model organisms including mouse, Drosophila, and Aplysia13. In addition to CREB, other IEGs have been extensively investigated12. Intriguingly, distinct IEGs are influenced by neuronal stimulation or activity blockade in a pathway-specific and stimulus-specific manner. For example, Egr1 mRNA levels are N-methyl-d-aspartate-type glutamate receptor-dependent, whereas c-fos levels are not14,15. Moreover, different IEGs exhibit distinct temporal window with different activation kinetics. Synaptic activity also drives long-term changes in neuronal structure and function, contributing to learning and memory, via mechanisms involving transcriptional activation through second messenger systems16. A mechanistic concept for differential regulation of neuronal IEGs was utilized for mapping specific neural functions onto neuronal activity in different brain regions17.
In the current review, we discuss three exemplary IEGs (activity-regulated cytoskeleton-associated protein (Arc/Arg3.1), neuronal Per/Arnt/Sim (PAS) domain protein 4 (Npas4), and Homer protein homolog 1a (Homer1a)) that play important roles in distinct facets of synapse and neural circuit development, as excellent reviews for the other neuronal IEGs are available. We describe the involvement of the three IEGs in synapse formation, transmission, plasticity, and cognitive behaviors. We also discuss the possible implications of these proteins in some brain disorders.
Activity-regulated cytoskeleton-associated protein
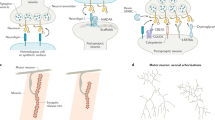
Arc (also known as Arg3.1) is one of the most tightly regulated molecules18,19,20. Neuronal activity regulates its transcription, translation, trafficking, localization, and stability21,22. Unlike other IEG products, Arc is not a transcription factor, but acts as an effector involved in various neuronal signaling pathways. Thus, Arc mRNA is rapidly transcribed in response to neuronal activity, and precisely targeted to activated synapses in neuronal dendrites23,24,25.
Induction of Arc levels in an activity-dependent manner significantly elevates the proportion of thin spines and reduces surface α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor density, which prevents network hyperexcitability, and contributes to maintenance of neuronal circuit homeostasis26. Arc induction is also required for late long-term potentiation (LTP) and memory consolidation27,28,29, which is regulated by local actin polymerization28. Arc knockout (KO) mice exhibited a failure to form long-term memories, while their short-term memories were intact29. These behavioral defects are associated with the alteration in LTP in vivo. In addition, Arc mediates metabotropic glutamate receptor (mGluR)-dependent long-term depression through facilitation of AMPA receptor endocytosis30,31,32,33 (Fig. 1). One of the proposed mechanisms by which Arc plays a regulatory role in AMPA receptor trafficking is via its interaction with components of the endocytic machinery, dynamin-2 and endophilin-330. Calcium/calmodulin-dependent protein kinase IIβ plays a role in the targeting of synaptic activity-induced Arc to strong stimulation experienced-inactive synapses where Arc mediates AMPA receptor clearance34. Arc is also required for homeostatic plasticity35,36,37. In particular, visual experience-induced Arc controls internalization of AMPA receptors, and as such, regulates homeostatic plasticity of excitatory synaptic transmission in layer 2/3 neurons of the visual cortex36. Arc KO mice displayed abnormal ocular dominance plasticity38, suggesting that Arc is required for the experience-dependent processes that establish synaptic connections in visual cortex.

Neuronal activation induces rapid transcription of immediate-early genes (IEGs) such as Npas4, Arc, and Homer1a. Npas4 regulates the transcription of its downstream target genes including BDNF to regulate inhibitory synapse development. Arc is targeted to activated excitatory synapses and facilitates AMPA receptor endocytosis through interaction with dynamin and endophilin, resulting in long-term depression. Homer1a is also specifically targeted to excitatory synapses, where it competes with other Homer proteins to interact with Homer-binding proteins such as mGluRs, Shanks, and IP3R and negatively acts in regulating excitatory synapse structure, function, and plasticity
In addition to regulating synaptic plasticity, Arc is involved in synaptic pruning. Specifically, Arc mediates elimination of redundant climbing axons to Purkinje cell in the developing cerebellum39. Neuronal activity-dependent myocyte enhancer factor 2 (MEF2) generates Arc transcripts, and subsequently activates local dendritic mGluR5 proteins to promote the translation of MEF2-induced Arc mRNAs, which are prerequisite for Arc’s mediation of structural and functional synapse elimination in hippocampal neurons40.
Arc is expressed rapidly and operates in the nucleus. Nuclear Arc level is modulated by synaptic activity41, and controls the homeostatic response by increasing promyelocytic leukemia protein expression and decreasing AMPA receptor GluA1 subunit transcription41,42. In addition, nuclear Arc interacts with histone acetyltransferase TIP60 and modifies chromatin structures43,44, suggesting that nuclear Arc mediates an epigenetic pathway to orchestrate neuronal activity-dependent transcription programs.
Mounting evidence supports the diverse roles of Arc at synapses and within the nucleus, but a precise mechanism for Arc localization is largely elusive. A recent study showed that extracellular signal-regulated kinase-mediated phosphorylation of Arc facilitates its cytosolic localization45. In addition to phosphorylation, Arc is rapidly sumoylated during LTP consolidation in the dentate gyrus of the hippocampus, which allows Arc to concentrate at the synapse to modulate actin cytoskeletal dynamics46. Moreover, Arc expression is subject to ubiquitination47,48, and glycogen synthase kinase-3-mediated phosphorylation49. In particular, RING domain-containing ubiquitin ligase Triad3A/RNF216 ubiquitylates Arc, which is then rapidly degraded via proteasome48, suggesting that loss of Triad3A might preclude Arc-dependent forms of synaptic plasticity. A recent study showed that missense mutations in Triad3 identified in patients with Gordon Homes Syndrome (GHS) resulted in defective Arc ubiquitination, thereby leading to impaired spatial learning and memory50. Therefore, Arc dysregulation may contribute to cognitive impairment and dementia observed in patients with GHS.
Neuronal PAS domain protein 4
Npas4 is a brain-specific transcription factor, the expression of which is regulated by neuronal activity, similar to Arc51. Npas4 plays a role in inhibitory synapse development by regulating the activity-dependent gene programs in cultured neurons52. More specifically, an enriched environment-induced Npas4 leads to an increase in the number of inhibitory synapses on the soma, and concurrently, a decrease in the number of inhibitory synapses on the apical dendrites of CA1 hippocampal pyramidal neurons53. Mechanistically, the inhibition mode within these subcellular compartments appears to be differentially tuned by distinct sets of Npas4 downstream targets. For instance, brain-derived neurotrophic factor (BDNF) specifically mediates Npas4-mediated somatic inhibition (Fig. 1), while other downstream targets (mostly uncharacterized) are thought to mediate dendritic inhibition. In addition, Npas4 mediates activity-dependent neurite outgrowth through cyclin-dependent kinase 5-dependent phosphorylation of synapsin I proteins in hippocampal cultured neurons54. In olfactory bulb interneurons, sensory experience immediately increases Npas4 protein levels, which is pivotal for dendritic spine formation55.
In addition to regulating inhibitory synapse structure and transmission, Npas4 also controls the homeostatic inhibitory–excitatory balance by regulating adult visual cortical plasticity56. For excitatory–inhibitory balance within neural circuits, Npas4 activates distinct programs of late-response genes in a cell-type-specific manner57. The late-responsive Npas4 downstream genes differentially regulate the patterns of synaptic inputs onto inhibitory and excitatory neurons, promoting inhibitory actions toward excitatory neurons while inducing excitatory actions toward inhibitory neurons. Moreover, Npas4 shapes the structure and function of prefrontal inhibitory circuits during adolescence in a sex-specific manner58, suggesting that Npas4 might be involved in the pathophysiological pathways underlying neuropsychiatric disorders that emerge during adolescence.
Npas4 is known as one of the activity-regulated inhibitor of death (AID) genes59. AID genes are the core components of a genomic survival program that is induced by nuclear calcium signaling in response to neuronal activity. Intriguingly, synaptotagmin-10 (Syt10) was first identified as the neuroprotective downstream target of Npas460, suggesting a role for the Npas4-Syt10 pathway in neuronal survival against excessive synaptic activity leading to excitotoxicity.
Npas4 has been linked to cognitive functions. Specifically, in the lateral nucleus of the amygdala, Npas4 level is elevated in a learning-dependent manner, and is responsible for the fear memory formation without affecting innate fear and expression of fear memory61. In CA3 hippocampal neurons, Npas4 level is specifically increased after contextual learning, regulating transcriptional programs required for contextual memory formation62. In addition, Npas4 mRNA levels are decreased in the hippocampus of aged memory-impaired, but not memory-unimpaired, mice, suggesting that Npas4 may contribute to preservation of hippocampus-related cognition63. Npas4 deficiency was characterized by impaired spatial recognition memory, decreased anxiety-like behavior, and increased depression-like behavior64. These KO phenotypes suggest that Npas4 may be a prominent candidate factor for anxiety, depression, and related cognitive disorders. The role of Npas4 in regulating social behaviors has not been clearly determined; Coutellier et al.64 showed that Npas4 KO mice were less social than wild-type littermates, whereas Jaehne et al.65 reported that the Npas4 KO mice exhibited normal social behaviors. Intriguingly, Npas4 KO mice exhibited long-lasting stress-related cognitive defects during adolescence66. The cognitive impairments observed in Npas4 KO mice were accompanied by reduced migration of neuroblast cells in the subventricular zone to cortical regions66. Further investigations should determine whether cell biological and behavioral phenotypes reported in Npas4 KO mice (e.g., cognitive deficits and stress-induced altered neurogenesis) are causally linked, and associated with brain disorders implicated in Npas4 dysfunctions.
Homer protein homolog 1a
The Homer protein family consists of three members (Homer1–3)67. Each Homer transcript generates several alternative splicing variants. One of two major splice variants, Homer1a (a shorter variant of Homer1), is induced by neuronal activity and is therefore considered an IEG68. Homer proteins typically possess a conserved N-terminal Ena/VASP homology I (EVH1) domain, and a coiled-coil domain at the C terminus. The EVH1 domain of Homer1 binds to a PPXXF motif present in various synaptic molecules, such as SH3 and multiple ankyrin repeat domains proteins (Shanks), group I mGluRs, and inositol-1,4,5-triphosphate (IP3) receptors69,70. The coiled-coil domain of Homer1 mediates its dimerization, which enables Homers to physically and functionally link mGluRs with IP3 receptors71. However, Homer1a lacks the coiled-coil domain and thus acts in a dominant-negative manner to disengage endogenous Homers with other effectors of mGluRs68 (Fig. 1). Homer1a is expressed both in excitatory and inhibitory neurons across various brain regions, such as amygdala, hippocampus, primary somatosensory cortex, and dorsal striatum72. Subcellularly, Homer1a is primarily located at the postsynaptic density where Homer1a competes with other constitutively expressed Homer proteins for interactions with Homer-binding proteins. As such, Homer1a negatively regulates excitatory synapse structure and function73. Expression of Homer1a following neuronal activation leads to the dissociation of the mGluRs/IP3 receptor complex and a reduced mGluR-mediated calcium response68.
While Homer1a KO mice showed normal spine number, morphology, and memory acquisition, and intact short-term memory, they exhibited impaired fear memory consolidation and reconsolidation74, suggesting a specific role of Homer1a in various stages of long-term, but not short-term, fear memory formation. Homer1a is epigenetically upregulated in the hippocampus and amygdala during the consolidation of cued fear conditioning mediated by BDNF75. In addition, Homer1a prevents the development of pain-related synaptic plasticity in the amygdala through disruption of mGluR1 signaling within the basolateral amygdala in an animal model of arthritis pain76.
Homer1a is crucial for the mGluR-mediated homeostatic scaling process77,78. In particular, Homer1a as a molecular integrator of arousal and sleep drives homeostatic downscaling, which is active during sleep for synaptic remodeling79. Similarly, Homer1a regulates calcium homeostasis80, activity-induced presynaptic and postsynaptic structural plasticity81, and dendritic targeting of mGluR582.
Several animal models of epilepsy exhibit a marked increase in Homer1a mRNA levels in hippocampal dentate gyrus granule neurons83,84, and epileptiform stimulus-induced synapse loss involves elevated Homer1a expression85. These results suggest that Homer1a may participate in a negative feedback loop to reduce network excitability. In addition to epilepsy, Homer1a could potentially be involved in depression-like behavior86,87. Intriguingly, knockdown of Homer1a by RNA interference in medial prefrontal cortex (mPFC) enhanced depressive-like behavior in mice, and overexpression of Homer1a in mPFC showed anti-depressant effects88. Given that various anti-depressant treatments increase Homer1a expression in mPFC88, Homer1a may be a key molecular player in anti-depressant therapy, although the mechanism(s) underlying its anti-depressant action require further elucidation.
Alteration of Homer1a levels was observed in the hippocampus and cingulate gyrus of human patients with various neuropsychiatric disorders, including schizophrenia, bipolar disorder, and major depression89. Intriguingly, in Fmr1 KO mouse model of fragile X syndrome, mGluR5 is more associated with shorter Homer1a rather than the longer Homer isoforms90,91. Deletion of Homer1a restores the association of mGluR5 with the longer Homer isoforms, leading to correction of a subset of phenotypes observed in the Fmr1 KO mice90. It remains to be solved how Fmr1 deficiency induces disrupted mGluR5–Homer interactions, but the altered interactions of mGluR5 with Homer may be an important mechanism underlying cognitive brain disorders involving mGluR5 dysfunction. Furthermore, Homer1a was reported to counteract amyloid-β-induced downregulation of potassium channel activity, hinting its potential as a therapeutic target for Alzheimer’s disease92. Supporting this notion, transcranial magnetic stimulation triggers effects on calcium-activated potassium channel facilitating large conductance and leading to enhanced hippocampal LTP and reduced cortical excitability, in Homer1a-dependent manner93. Collectively, these studies pinpoint Homer1a as a prominent biomarker and potential therapeutic target for certain brain disorders.
Conclusions and perspectives
It is increasingly apparent that IEGs are critical for the structure, function, and plasticity of both excitatory and inhibitory synapses across various cell types and brain regions. In this review, we highlighted only a subset of IEGs that function in distinct manners to regulate specific aspects of synapse development. Although IEGs have been continually utilized as markers for labeling active population of neurons, their physiological significance remains to be fully clarified. In the future, other neuronal IEGs beyond those discussed in this review should be intensively studied towards a fuller understanding of how IEGs organize synapse development and by extension neural circuit development. Unquestionably, these future directions will aid in clearly defining the role of environment in development of various neurodevelopmental and neuropsychiatric disorders.
References
Chaudhury, S., Sharma, V., Kumar, V., Nag, T. C. & Wadhwa, S. Activity-dependent synaptic plasticity modulates the critical phase of brain development. Brain. Dev. 38, 355–363 (2016).
Walmsley, B., Berntson, A., Leao, R. N. & Fyffe, R. E. Activity-dependent regulation of synaptic strength and neuronal excitability in central auditory pathways. J. Physiol. 572, 313–321 (2006).
West, A. E. & Greenberg, M. E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3, 1–21 (2011).
Hubel, D. H. Exploration of the primary visual cortex, 1955–78. Nature 299, 515–524 (1982).
Wiesel, T. N. Postnatal development of the visual cortex and the influence of environment. Nature 299, 583–591 (1982).
Greenberg, M., Ziff, E. & Greene, L. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science 234, 80–83 (1986).
Lanahan, A. & Worley, P. Immediate-early genes and synaptic function. Neurobiol. Learn. Mem. 70, 37–43 (1998).
Nedivi, E., Wu, G. Y. & Cline, H. T. Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science 281, 1863–1866 (1998).
Sheng, M. & Greenberg, M. E. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron 4, 477–485 (1990).
Chandra, R. & Lobo, M. K. Beyond neuronal activity markers: delect immediate early genes in striatal neuron subtypes functionally mediate psychostimulant addiction. Front. Behav. Neurosci. 11, 112 (2017).
Chen, L. F., Zhou, A. S. & West, A. E. Transcribing the connectome: roles for transcription factors and chromatin regulators in activity-dependent synapse development. J. Neurophysiol. 118, 755–770 (2017).
Loebrich, S. & Nedivi, E. The function of activity-regulated genes in the nervous system. Physiol. Rev. 89, 1079–1103 (2009).
Kandel, E. R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 5, 14 (2012).
Cole, A. J., Saffen, D. W., Baraban, J. M. & Worley, P. F. Rapid increase of an immediate early gene messenger RNA in hippocampal neurons by synaptic NMDA receptor activation. Nature 340, 474–476 (1989).
Wisden, W. et al. Differential expression of immediate early genes in the hippocampus and spinal cord. Neuron 4, 603–614 (1990).
Abraham, W. C., Dragunow, M. & Tate, W. P. The role of immediate early genes in the stabilization of long-term potentiation. Mol. Neurobiol. 5, 297–314 (1991).
Kubik, S., Miyashita, T. & Guzowski, J. F. Using immediate-early genes to map hippocampal subregional functions. Learn. Mem. 14, 758–770 (2007).
Guzowski, J. F., McNaughton, B. L., Barnes, C. A. & Worley, P. F. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat. Neurosci. 2, 1120–1124 (1999).
Link, W. et al. Somatodendritic expression of an immediate early gene is regulated by synaptic activity. Proc. Natl. Acad. Sci. USA 92, 5734–5738 (1995).
Lyford, G. L. et al. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 14, 433–445 (1995).
Korb, E. & Finkbeiner, S. Arc in synaptic plasticity: from gene to behavior. Trends Neurosci. 34, 591–598 (2011).
Shepherd, J. D. & Bear, M. F. New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 14, 279–284 (2011).
Farris, S., Lewandowski, G., Cox, C. D. & Steward, O. Selective localization of Arc mRNA in dendrites involves activity- and translation-dependent mRNA degradation. J. Neurosci. 34, 4481–4493 (2014).
Na, Y. et al. Real-time imaging reveals properties of glutamate-induced Arc/Arg 3.1 translation in neuronal dendrites. Neuron 91, 561–573 (2016).
Steward, O., Wallace, C. S., Lyford, G. L. & Worley, P. F. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron 21, 741–751 (1998).
Peebles, C. L. et al. Arc regulates spine morphology and maintains network stability in vivo. Proc. Natl. Acad. Sci. USA 107, 18173–18178 (2010).
Guzowski, J. F. et al. Inhibition of activity-dependent Arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J. Neurosci. 20, 3993–4001 (2000).
Messaoudi, E. et al. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J. Neurosci. 27, 10445–10455 (2007).
Plath, N. et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444 (2006).
Chowdhury, S. et al. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459 (2006).
Jakkamsetti, V. et al. Experience-induced Arc/Arg3.1 primes CA1 pyramidal neurons for metabotropic glutamate receptor-dependent long-term synaptic depression. Neuron 80, 72–79 (2013).
Park, S. et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83 (2008).
Waung, M. W., Pfeiffer, B. E., Nosyreva, E. D., Ronesi, J. A. & Huber, K. M. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97 (2008).
Okuno, H. et al. Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIβ. Cell 149, 886–898 (2012).
Béïque, J. C., Na, Y., Kuhl, D., Worley, P. F. & Huganir, R. L. Arc-dependent synapse-specific homeostatic plasticity. Proc. Natl. Acad. Sci. USA 108, 816–821 (2011).
Gao, M. et al. A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. J. Neurosci. 30, 7168–7178 (2010).
Shepherd, J. D. et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484 (2006).
McCurry, C. L. et al. Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat. Neurosci. 13, 450–457 (2010).
Mikuni, T. et al. Arc/Arg3.1 is a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum. Neuron 78, 1024–1035 (2013).
Wilkerson Julia, R. et al. A role for dendritic mGluR5-mediated local translation of Arc/Arg3.1 in MEF2-dependent synapse elimination. Cell Rep. 7, 1589–1600 (2014).
Korb, E., Wilkinson, C. L., Delgado, R. N., Lovero, K. L. & Finkbeiner, S. Arc in the nucleus regulates PML-dependent GluA1 transcription and homeostatic plasticity. Nat. Neurosci. 16, 874–883 (2013).
Honjoh, S. et al. Higher Arc nucleus-to-cytoplasm ratio during sleep in the superficial layers of the mouse cortex. Front. Neural Circuits 11, 60 (2017).
Oey, N. E. et al. A neuronal activity-dependent dual function chromatin-modifying complex regulates Arc expression(1,2,3). eNeuro 2, 0020–14.2015 (2015). pii: ENEURO.
Wee, C. L. et al. Nuclear Arc interacts with the histone acetyltransferase Tip60 to modify H4K12 acetylation(1,2,3). eNeuro 1, 0019–14.2014 (2014).
Nikolaienko, O., Eriksen, M. S., Patil, S., Bito, H. & Bramham, C. R. Stimulus-evoked ERK-dependent phosphorylation of activity-regulated cytoskeleton-associated protein (Arc) regulates its neuronal subcellular localization. Neuroscience 360, 68–80 (2017).
Nair, R. R. et al. Dynamic Arc SUMOylation and selective interaction with F-actin-binding protein Drebrin A in LTP consolidation in vivo. Front .Synaptic Neurosci. 9, 8 (2017).
Greer, P. L. et al. The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating Arc. Cell 140, 704–716 (2010).
Mabb Angela, M. et al. Triad3A regulates synaptic strength by ubiquitination of Arc. Neuron 82, 1299–1316 (2014).
Gozdz, A. et al. GSK3α and GSK3β phosphorylate Arc and regulate its degradation. Front. Mol. Neurosci. 10, 192 (2017).
Husain, N. et al. TRIAD3/RNF216 mutations associated with Gordon Holmes syndrome lead to synaptic and cognitive impairments via Arc misregulation. Aging Cell. 16, 281–292 (2017).
Sun, X. & Lin, Y. Npas4: linking neuronal activity to memory. Trends Neurosci. 39, 264–275 (2016).
Lin, Y. et al. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204 (2008).
Bloodgood, B. L., Sharma, N., Browne, H. A., Trepman, A. Z. & Greenberg, M. E. The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503, 121–125 (2013).
Yun, J. et al. Neuronal Per Arnt Sim (PAS) domain protein 4 (NPAS4) regulates neurite outgrowth and phosphorylation of synapsin I. J. Biol. Chem. 288, 2655–2664 (2013).
Yoshihara, S. et al. Npas4 regulates Mdm2 and thus Dcx in experience-dependent dendritic spine development of newborn olfactory bulb Interneurons. Cell Rep. 8, 843–857 (2014).
Maya-Vetencourt, J. F. et al. Experience-dependent expression of NPAS4 regulates plasticity in adult visual cortex. J. Physiol. 590, 4777–4787 (2012).
Spiegel, I. et al. Npas4 regulates excitatory–inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 157, 1216–1229 (2014).
Shepard, R., Heslin, K. & Coutellier, L. The transcription factor Npas4 contributes to adolescent development of prefrontal inhibitory circuits, and to cognitive and emotional functions: Implications for neuropsychiatric disorders. Neurobiol. Dis. 99, 36–46 (2017).
Zhang, S. J. et al. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 5, e1000604 (2009).
Woitecki, A. M. H. et al. Identification of synaptotagmin 10 as effector of NPAS4-mediated protection from excitotoxic neurodegeneration. J. Neurosci. 36, 2561–2570 (2016).
Ploski, J. E., Monsey, M. S., Nguyen, T., DiLeone, R. J. & Schafe, G. E. The neuronal PAS domain protein 4 (Npas4) is required for new and reactivated fear memories. PLoS ONE 6, e23760 (2011).
Ramamoorthi, K. et al. Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science 334, 1669–1675 (2011).
Qiu, J. et al. Decreased Npas4 and Arc mRNA levels in the hippocampus of aged memory-impaired wild-type but not memory preserved 11beta-HSD1 deficient mice. J. Neuroendocrinol. 28, 1–10 (2016).
Coutellier, L., Beraki, S., Ardestani, P. M., Saw, N. L. & Shamloo, M. Npas4: a neuronal transcription factor with a key role in social and cognitive functions relevant to developmental disorders. PLoS ONE 7, e46604 (2012).
Jaehne, E. J., Klarić, T. S., Koblar, S. A., Baune, B. T. & Lewis, M. D. Effects of Npas4 deficiency on anxiety, depression-like, cognition and sociability behaviour. Behav. Brain. Res. 281, 276–282 (2015).
Coutellier, L., Gilbert, V. & Shepard, R. Npas4 deficiency increases vulnerability to juvenile stress in mice. Behav. Brain. Res. 295, 17–25 (2015).
Shiraishi-Yamaguchi, Y. & Furuichi, T. The Homer family proteins. Genome Biol. 8, 206 (2007).
Xiao, B. et al. Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron 21, 707–716 (1998).
Brakeman, P. R. et al. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 386, 284–288 (1997).
Tu, J. C. et al. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21, 717–726 (1998).
Xiao, B., Cheng, Tu. J. & Worley, P. F. Homer: a link between neural activity and glutamate receptor function. Curr Opin Neurobiol 10, 370–374 (2000).
Imamura, N., Nonaka, A., Yamamoto, H., Matsuki, N. & Nomura, H. Experience-dependent Homer1a expression in excitatory and inhibitory neurons. Neuroreport 22, 353–357 (2011).
Sala, C. et al. Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J. Neurosci. 23, 6327–6337 (2003).
Inoue, N. et al. Requirement of the immediate early gene vesl-1S/homer-1a for fear memory formation. Mol. Brain 2, 7 (2009).
Mahan, A. L. et al. Epigenetic modulation of Homer1a transcription regulation in amygdala and hippocampus with pavlovian fear conditioning. J. Neurosci. 32, 4651–4659 (2012).
Tappe-Theodor, A., Fu, Y., Kuner, R. & Neugebauer, V. Homer1a signaling in the amygdala counteracts pain-related synaptic plasticity, mGluR1 function and pain behaviors. Mol. Pain 7, 38 (2011).
Hu, J. H. et al. Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron 68, 1128–1142 (2010).
Siddoway, B., Hou, H. & Xia, H. Molecular mechanisms of homeostatic synaptic downscaling. Neuropharmacology 78, 38–44 (2014).
Diering, G. H. et al. Homer1a drives homeostatic scaling-down of excitatory synapses during sleep. Science 355, 511–515 (2017).
Worley, P. F. et al. Homer proteins in Ca2+ signaling by excitable and non-excitable cells. Cell Calcium 42, 363–371 (2007).
Inoue, Y., Udo, H., Inokuchi, K. & Sugiyama, H. Homer1a regulates the activity-induced remodeling of synaptic structures in cultured hippocampal neurons. Neuroscience 150, 841–852 (2007).
Ango, F. et al. Dendritic and axonal targeting of type 5 metabotropic glutamate receptor is regulated by Homer1 proteins and neuronal excitation. J. Neurosci. 20, 8710–8716 (2000).
Morioka, R., Kato, A., Fueta, Y. & Sugiyama, H. Expression of vesl-1S/homer-1a, a gene associated with long-term potentiation, in the brain of the epileptic EI mouse. Neurosci. Lett. 313, 99–101 (2001).
Potschka, H. et al. Kindling-induced overexpression of Homer 1A and its functional implications for epileptogenesis. Eur. J. Neurosci. 16, 2157–2165 (2002).
Li, Y., Popko, J., Krogh, K. A. & Thayer, S. A. Epileptiform stimulus increases Homer 1a expression to modulate synapse number and activity in hippocampal cultures. J. Neurophysiol. 109, 1494–1504 (2013).
Rietschel, M. et al. Genome-wide association-, replication-, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol. Psychiatry 68, 578–585 (2010).
Sun, P. et al. Increase in cortical pyramidal cell exciability accompanies depression-like behavior in mice: a transcranial magnetic stimulation study. J. Neurosci. 31, 16464–16472 (2011).
Serchov, T. et al. Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of Homer1a. Neuron 87, 549–562 (2015).
Leber, S. L. et al. Homer1a protein expression in schizophrenia, bipolar disorder, and major depression. J. Neural Transm. 124, 1261–1273 (2017).
Ronesi, J. A. et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci. 15, 431–440 (2012).
Giuffrida, R. et al. A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J. Neurosci. 25, 8908–8916 (2005).
Yamamoto, K. et al. Suppression of a neocortical potassium channel activity by intracellular amyloid-β and its rescue with Homer1a. J. Neurosci. 31, 11100–11109 (2012).
Wang, F. et al. Improvement of spatial learning by facilitating large-conductance calcium-activated potassium channel with transcranial magnetic stimulation in Alzheimer’s disease model mice. Neuropharmacology 97, 210–219 (2015).
Acknowledgements
This work was supported by grants from the Ministry for Health and Welfare Affairs, Republic of Korea (HI15C3026 to J.W.U.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, S., Kim, H. & Um, J.W. Synapse development organized by neuronal activity-regulated immediate-early genes. Exp Mol Med 50, 1–7 (2018). https://doi.org/10.1038/s12276-018-0025-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-018-0025-1
This article is cited by
-
Repetitive and compulsive behavior after Early-Life-Pain associated with reduced long-chain sphingolipid species
Cell & Bioscience (2023)
-
Analysis of sleep deprivation-associated Homer1 gene and protein acting on synaptic plasticity by bioinformatics and animal experiments
Anesthesiology and Perioperative Science (2023)
-
SRF depletion in early life contributes to social interaction deficits in the adulthood
Cellular and Molecular Life Sciences (2022)
-
Arc silence aggravates traumatic neuronal injury via mGluR1-mediated ER stress and necroptosis
Cell Death & Disease (2020)
-
Immediate-Early Genes Detection in the CNS of Terrestrial Snail
Cellular and Molecular Neurobiology (2020)
