
Abstract
This review summarizes neonatal meconium aspiration syndrome in light of meconium-induced inflammation and inflammatory surfactant inactivation, related to both endogenous and therapeutic exogenous surfactant. The wide effect of meconium on surfactant properties is divided into three points. Direct effect of meconium on surfactant properties refers mainly to fragmentation of dipalmitoylphosphatidylcholine and other surfactant phospholipids together with cleavage of surfactant proteins. Initiation of inflammatory response due to activation of receptors by yet unspecified compounds involves complement and Toll-like receptor activation. A possible role of lung collectins, surfactant proteins A and D, which can exert both pro- and anti-inflammatory reactions, is discussed. Initiation of inflammatory response by specified compounds in meconium reflects inflammatory functioning of cytokines, bile acids, and phospholipases contained in meconium. Unifying sketch of many interconnections in all these actions aims at providing integrated picture of inflammatory surfactant inactivation.
Similar content being viewed by others


Main
Inflammatory response during meconium aspiration syndrome (MAS) has attracted a great deal of attention in the last years.
For a long time, mechanical obstruction and subsequent chemical pneumonitis were considered as the most important reasons for neonatal mortality in MAS (1). Meanwhile, meconium-induced inhibition of surfactant had been suggested, and supplementation of exogenous surfactant provided therapeutic tool for newborns with MAS (2). Discovery of exogenous surfactant brought great hope to both scientists and neonatologists, and the discussion regarding MAS treatment turned predominantly to surfactant dose, the way of its application, and the type of ventilatory support. Although the MAS-induced inactivation of innate surfactant was found to be related to many factors including pulmonary edema fluid, plasma protein leakage, oxidative stress, or inflammation (3), possible inactivation of therapeutic exogenous surfactant by above-mentioned factors had been considered cautiously.
Then, scientific attention turned back to inflammatory pathways, as it was suggested and found that further surfactant inactivation is caused by inflammation, oxidative stress, and edema fluid (4,5,6). However, the exact molecular mechanisms are still unclear (7).
So, what kind the specifics of meconium-induced inflammation and surfactant inactivation are of? The complex composition of meconium perplexes identification of the most active factors; however, according to what we know at present, the effect of meconium can be divided into three types:
-
1. Direct effect of meconium on surfactant properties
-
2. Initiation of inflammatory response due to activation of receptors by yet unspecified compounds
-
3. Initiation of inflammatory response by specified compounds in meconium
Direct surfactant inactivation induced by meconium
Besides mechanical obstruction of airways, meconium has direct impact on surfactant surface properties (8) discussed in detail elsewhere (9). Such direct inactivation regards both the endogenous and exogenous surfactant (10,11). Being composed of amniotic fluid, desquamated epithelial cells, lanugo, vernix caseosa, mucus, blood, and gastrointestinal secretions swallowed during the intrauterine life, both hydrophobic and hydrophilic material can be found in meconium mess: cholesterol, free fatty acids (FFAs), and triglycerides in chloroform-soluble fraction and bile acids, bilirubin, dietary fiber, hemoglobin, proteins, and inorganic molecules in water- or water–methanol-soluble fraction (9,11,12,13).
When present, meconium alters structure of the major surfactant phospholipid (PL) dipalmitoylphosphatidylcholine (DPPC), causes fragmentation of its bilayer, and disrupts function of liposomes. Meconium also increases minimum surface tension of binary surfactant lipid monolayers, namely, DPPC combined with palmitoyloleoylphosphatidylcholine, phosphatidylethanolamine, or phosphatidylglycerol (13). Inability of surfactant lipids to adsorb at the air–liquid interface due to meconium affects both the endogenous and exogenous surfactant activity in a dose-dependent manner (11,13).
Both fractions of meconium impair surfactant, but the lipid-soluble fraction containing cholesterol, FFAs (mainly palmitic, stearic, and oleic acids), and triglycerides had stronger effect than the aqueous fraction (12), and lipid-soluble extract of meconium closely stimulated DPPC inhibition due to whole meconium (13).
Minimal amount of cholesterol is normally present and is also required for natural pulmonary surfactant membranes to adopt their structure and dynamics (14). Nevertheless, elevated cholesterol may form complexes with surfactant PLs, thus increasing the surfactant film fluidity. Higher fluidity results in collapse rather than multilayer formation during lateral compression in breathing cycle (15). Meconium contains a substantial amount of cholesterol, and exposure to meconium results in incorporation of cholesterol into surfactant membranes and films (16). However, surfactant surface activity did not change in the presence of cholesterol itself; mixture of cholesterol and taurocholic acid produced similar alteration to surfactant film formation to meconium, suggesting that bile acids are the mobilizing agent which facilitates incorporation of cholesterol to surfactant complexes (14). Cholesterol and bile acids co-action may explain why in vivo neither of the subfractions could reproduce the total damaging pulmonary effects of whole meconium (12).
Similar fluidizing effect was seen in the presence of oleic acid which interfered with the ability of spread surfactant films to reach low surface tensions during dynamic compression (17).
The main factor of surfactant lipid impairment in water–methanol-soluble fraction seems to be secretory phospholipases A2 (sPLA2s). sPLA2s are esterases that hydrolyze glycerophospholipids to release FFAs and lysophospholipids (LPLs). They exert direct injury to cell membranes releasing arachidonic acid (18,19,20) and direct dysfunction of surfactant due to depletion of PLs by hydrolysis of DPPC and other PLs in both endogenous and exogenous surfactant (21,22,23) and also consecutive impairment of surfactant mediated by products of hydrolysis, mainly by LPLs (21,24).
To date, about 10 enzymatically active mammalian sPLA2s are known, hydrolyzing different substrates and susceptive to different inhibitors. Of sPLA2s, the most important in meconium-induced lung injury are pancreatic sPLA2 (sPLA2-IB) being present in meconium, and then pulmonary sPLA2 (sPLA2-IIA) being induced by meconium presence, as discussed thereinafter (19,20,22). DPPC hydrolysis seems to be attributed mostly to sPLA2-IB activity since sPLA2-IIA binds extremely weakly to phosphatidylcholine vesicles (21,25). The efficiency of hydrolysis of principal PLs (DPPC, phosphatidylglycerol, and phosphatidylethanolamine) varies substantially with each enzyme subtype, and contribution of more sPLA2 subtypes in surfactant impairment cannot be ruled out (20,24). Moreover, accumulation of hydrolysis products, FFAs and LPLs, disrupts surfactant surface tension–lowering function due to altered PL packing in the surface film, with LPLs being significantly more efficient than FFAs or PLs depletion (24). LPLs also damage type I alveolar cells, increase capillary permeability, and recruit inflammatory cells into the lungs (18).
Besides enabling the insertion of cholesterol into surfactant membranes, bile acids in lungs lead also to decrease in DPPC portion and shift in the ratio between phosphatidylglycerol and sphingomyelin (26). Taurocholic acid, one of the most abundant bile acids in human, was found to affect the structure of both surfactant monolayers at the interface and surfactant aggregates in solution (27), thus contributing to loss of surfactant function. And finally, bile acids can act as nonsubstrate cofactors for sPLA2 by virtue of their incorporation into PL layer; their negative charge facilitates sPLA2 adhesion to substrate and consequent hydrolysis (21,28).
As to the direct impact of meconium to surfactant proteins, not much is known at present. Direct impact of meconium on SP-A and SP-D structure has not been described yet. There is only one reference that human SP-B and SP-C are directly destroyed in the presence of meconium, demonstrated by disappearance of SP-B and SP-C bands from western blot, and this process can be prevented by protease inhibitors (29). In contrary, bovine SP-B seems to be at least partially resistant to human meconium in vitro (VA. Ivanov, personal communication).
Cleavage of surfactant protein chain may, however, lead to alterations of its function, as it is discussed below.
The initiation of inflammatory response due to activation of receptors by unspecified compounds
In addition to direct surfactant impairment, inflammatory process is an essential part of MAS pathophysiology. In response to meconium aspiration, synthesis of various cytokines together with release of active inflammatory cells had been observed (30). Although we do not know the particular responsible component, in vitro, meconium was found to activate both the complement (31,32) and Toll-like receptors (TLR; 33,34)—two important components of the innate immune system.
The main biological effect of complement activation is induction of inflammatory reaction. Once activated, C5a product of complement triggers production of innumerable inflammatory mediators such as cytokines, chemokines, arachidonic acid metabolites, reactive oxygen metabolites, adhesion molecules, and others, resulting in interaction between endothelial cells (EnC) and leukocytes (32). In light of three possible ways of complement activation (classical, alternative, and lectin pathway), meconium was found to work in vitro via the alternative pathway (35) as well as the lectin pathway with subsequent increase of TNF-α, IL-1β, IL-6, IFN-γ, IL-8, monocyte chemoattractant protein-1, macrophage inflammatory protein-1α, macrophage inflammatory protein-1β, and eotaxin (36).
TLR are members of a family of pattern-recognition receptors which recognize molecular structures of bacteria, viruses, fungi, and protozoa and some endogenous structures and proteins released during inflammation (37,38). Some of them cooperate in complex with an accessory receptor CD14—a protein being expressed on the surface of immune cells and widely in respiratory tract on both the epithelial cells (EpC) and EnC. CD14 plays a central role in lung inflammatory processes due to recognition of a variety of bacterial and viral components, and for a long time, it had been thought to associate only with bacterial LPS (37,39). CD14 serves in CD14/TLR4/MD-2 complex present on the macrophages, EnC, and EpC (38,40,41) and was found out to be activated also by meconium (33,34). Activation of CD14/TLR4/MD-2 leads to an increase in nuclear factor κB (NF-κB), activator protein-1 (AP-1), and activation of IFNs. NF-κB translocation to the nucleus initiates simultaneously both pro-oxidative and inflammatory cascades—interlinked and complementary components of inflammatory response (38,42).
Simply, the presence of germ-free meconium in respiratory tract acts at the molecular level identically to bacterial infection in both of these ways, the complement and TLR activation. The starting compound(s) or mechanisms remain still unknown.
It remains just a matter of speculation if we try to put together some facts about the direct impact of meconium to surfactant structural properties and accompanying exposure of specific molecular regions of surfactant proteins in process of TLR and component activation.
Both SP-A and SP-D belong to collectin (collagen-lectin) family, meaning they have more binding sites: collagen region at N-terminus and carbohydrate recognition (or lectin) domain (CRD) at C-terminus (43). The protein structure and three-dimensional organization of SP-A in CRD domain is very similar to mannose-binding protein and to C1q, the initiator of classic pathway of complement system.
Despite this molecular resemblance, neither SP-A nor SP-D were shown to activate complement system. Quite the contrary, the observed effect of SP-A on complement activation was rather inhibitory due to reported bond between SP-A and C1q (43,44). Interaction between SP-A and C1q was found to influence SP-A binding to alveolar macrophages, and it was suggested that maybe the activity of SP-A is regulated via binding to C1q or vice versa (45). However, it cannot be excluded that changes of SP-A structure in the presence of meconium may lead to C1q uncoupling and thereby indirectly contribute to complement activation; in a similar way to oxidized SP-A and its nonfunctional control of complement in the aging lung (46).
Classical pathway of complement activation by meconium was not observed in vitro (36). Yet in vitro experiments with meconium and complement miss the surfactant components; and in in vivo experiments, only the terminal sC5b-9 complex had been evaluated which is common for all pathways of complement activation. The most extensive increase in sC5b-9 complex was observed in MAS animals with fatal outcome (35) and although the alternative pathway acts as an amplification loop of all initial pathways (36), we cannot rule out that in vivo there can be triggering (or at least an interplay) of all pathways. Despite the number of studies focused on meconium and complement activation, the particular triggers of discrete pathways have not yet been clarified, and there are many challenges for other investigations.
However, not only molecular structure but also orientation and exposure of binding sites are pivotal in anti-inflammatory/proinflammatory signalization of surfactant proteins, as it was demonstrated by Gardai et al. (47).
Under normal conditions, globular CRD of SP-A can bind DPPC while its collagen region provides protein–protein interactions. The interaction between SP-A and the PLs via the CRD domain might be critical to the formation of tubular myelin (16,48), a transient form of surfactant with role presumably in innate defense, as seen in knockout mice (16,48). CRD of SP-D is thought to bind to the polar head groups of phosphatidylinositol (49). It was reported that in the absence of a pathogen, SP-A binds through CRD also to signal-inhibitory regulatory protein-α and inhibits the inflammatory response via intracellular suppression of NF-κB (47,50).
However, in the presence of a foreign organism or cellular debris (which can be found in meconium), SP-A enhances inflammatory mediators production (macrophage inflammatory protein-2, TNF-α, monocyte chemoattractant protein-1) by alternative interactions of functional domains. CRD binds to pathogen or debris, and the collagen region via CD91–calreticulin (CRT) complex activates immune cells and induces production of proinflammatory mediators as well as activation of p38 mitogen-activated protein kinase in the similar way to C1q. The same picture occurs with SP-A collagenous tails or SP-A and SP-D constructs lacking head group (CRD) function of binding to signal-inhibitory regulatory protein-α (47). On the other hand, CRT also plays an important role in SP-A– and SP-D–mediated recognition and clearance of apoptotic cells. Similar to C1q and mannose-binding protein, SP-A and SP-D bound to apoptotic cells and drove apoptotic cell ingestion by phagocytes through a mechanism dependent on CRT and CD91, participating in resolution of lung inflammation (50,51). Therefore, once again, there are more possibilities of triggering different pathways if meconium gets into the lungs.
The molecular organization of surfactant proteins is pivotal in TLR activation. SP-A has ability to bind directly to TLR4/MD-2 complex via CRD, resulting in downregulation of inflammatory response to LPS, but this interaction demands specific protein structure in supratrimeric oligomerization (52), and under certain conditions, especially when lipid components of surfactant are damaged, SP-A may exhibit immunostimulatory effect mediated by TLR4 activation (53). Moreover, there is also crosstalk between TLR and the complement system, and still, there is question whether active complement fragments could directly stimulate TLR signaling (54).
As it was mentioned above, in the presence of meconium, there seems to be direct impairment of protein chain of human SP-B and SP-C (29; VA. Ivanov, unpublished observation). It is a pity we presently miss information about meconium-caused structural changes of SP-A and SP-D. Such knowledge would be probably useful, especially when it is possible that the origin of secret molecule accountable for inflammatory response via complement and TLR activation may be more “endogenous” than supposed.
As for endogenous surfactant surface activity, it seems that it is impaired by free radicals and enzymes derived from immune cells (mainly recruited neutrophils) activated by complement and cytokines (9,10), rather than cytokines and complement components themselves. However, direct measurement of surface properties of surfactant combined with cytokines or C1q would be an interesting issue.
The initiation of inflammatory response by specified compounds in meconium
Meconium is often referred to be a rich source of diverse cytokines including IL-1β, IL-6, IL-8, and TNF-α (55); however, some authors held the view that it is rather meconium-stained amniotic fluid than meconium itself, which contains above-mentioned proinflammatory substances. Meconium is considered perhaps to induce production of cytokines in maternal tissues (the uterine cervix, the placenta, the decidua, and the fetal membrane) from where they drift into amniotic fluid (56). Regardless of the original source of cytokines, meconium-stained fluid displays ability to attract polymorphonuclear leukocytes and to activate them (57,58), stimulates alveolar macrophages to generate platelet-activating factor and TNF-α (59), and increases further production of cytokines (60).
Another source of proinflammatory substances is the above-mentioned meconium-derived sPLA2 which directly damages alveolar cells by hydrolyzing membrane PLs and generates proinflammatory eicosanoids and LPLs (22). Moreover, further production of inflammatory mediators can be seen after sPLA2-mediated activation of specific lung macrophage receptors regardless of their enzymatic activity (61).
Bile acids were found to stimulate NF-κB and p38 mitogen-activated protein kinase signaling pathways in EnC, resulting in adhesion molecule expression (62), so bile acid contained in meconium may also contribute to intensified lung inflammatory response. Taken together, many substances contained in meconium may initiate additive proinflammatory signalization and exposure of damage-associated molecular patterns being recognized by TLR (7,38).
“Where the roads meet”
The inflammation and surfactant degradation due to meconium start via three above-mentioned mechanisms: direct impact on surfactant, activation of complement system and TLR receptors, and function of present cytokines. However, the subsequent processes are interrelated and promote each other.
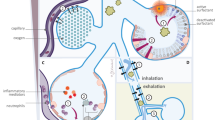
Briefly, there are two main streams in response to meconium: so-called proinflammatory (including cytokine production, expression of adhesion molecules, leukocyte sequestration and activation, degranulation of neutrophils, sPLA2s activation, etc.) and pro-oxidative one (induction of free radicals producing enzymes, changes in cell redox status, redox signaling, etc.). In both lines, TLRs, complement, NF-κB, cytokine and chemokine production, and expression of prooxidative enzymes and adhesion molecules may be roughly assigned to “mediator (or signaling) group,” while neutrophil-derived proteases, free radicals, and huge group of sPLA2s serve as “effector group.” Both reactive species and enzymes from activated cells attack surfactant components, alveolar pneumocytes, and tight junctions leading to plasma leakage and edema formation, intensified cell sequestration and increased cell signalization—all ending in necrosis and/or apoptosis together with inactivation of surfactant and its synthesis ( Figure 1 ).

Meconium-induced pathways. Besides direct surfactant inhibition, meconium activates complex of CD14 and Toll-like receptor 4/MD-2 (CD14/TLR4/MD-2), complement pathways, and triggers neutrophil and monocyte influx due to cytokine content. Intracellular pathways initiated by meconium enhance additional cytokine production, NADPH oxidase (NADPHox) activation, inducible NOS (iNOS) expression, secretory phospholipases A2 (sPLA2s) activation, and neutrophil degranulation. Subsequent oxidative stress together with enzyme activity impairs surfactant phospholipides and tight junctions and cells, resulting in edema formation, apoptosis, and decrease in surfactant protein (SP) expression and surfactant synthesis. For more details, see the section on “Where the roads meet.”
Detailed description of these processes is a hard row to hoe, as oxidative and cytokine signalization work hand-in-hand. For example, increase of free radicals activates NF-κB in several cell types (63) resulting in cytokine formation and NOS expression (64,65). Polymorphonuclear NADPH oxidase-derived reactive oxygen species (ROS) appear to regulate TLR4 gene expression in EnC which in turn leads to EnC NADPH oxidase (NADPHox) activation (38). ROS also augment TLR2-mediated adhesion molecule expression in EnC (66) and seems to be necessary for neutrophil migration efficiency (67).
Point by point we can track the main features of meconium-induced inflammation.
As a consequence of chemotactic IL-8 abundance in meconium, massive neutrophil and macrophage migration was observed (30,68). The exact time course has not yet been described; however, under conditions of LPS-induced lung injury model, neutrophil influx and activation can be seen within 2 h (69), and in experimental models of MAS, influx of polymorphonuclear leukocytes, T-lymphocytes, and monocytes is also demonstrated within few hours (22,30,31,70) which are being associated with decrease of leukocyte count in peripheral blood (6). Meanwhile, TLR and complement activation take place (32,33).
Meconium-induced TLR4 inflammation is related to different types of cells, including macrophages, EnC, and EpC and works via activation of NF-κB and AP-1, resulting in IFN and cytokine (such as TNF-α) production (34). Furthermore, TLR4 can be activated by SP-A under certain conditions (53). There is broad spectrum of evidence that complement and TLR pathways are interrelated in many cross-points, so that distinct mediators of both systems share the same signalization pathways (54). Activation of complement by meconium (36) may intensify downstream signalization, and consequent chemokines and adhesion molecules worsen the situation in the lungs by additional leukocyte sequestration (32). Along with IL-8, the complement activation product C5a is one of the most potent chemotactic factors released during inflammation with possible role of neutrophil activator as well (35).
Aspirated meconium is able to activate neutrophils rapidly (71). Once activated, neutrophils and macrophages produce huge quantities of ROS and reactive nitrogen species (71,72,73). The excess of free radicals damages surfactant PLs and proteins (74) and attacks tight junctions in EnC, facilitating extravasation (72). Moreover, rise in ROS induces gene expression of TLR2 and TLR4 in cells, thus enabling enhanced involvement of TLR signalization to inflammatory processes, while TLR4 signaling proceeds to further NADPHox activation in EnC (38).
Meanwhile, IL-1β, IL-6, and TNF-α produced in TLR4-dependent pathway and eicosanoids provoke sPLA2s expression in cells, mainly in alveolar macrophages (20,75,76,77). Aside from sPLA2-IB present in meconium, the other subtypes, such as sPLA2-IIA, sPLA2-V, or sPLA2-X are expressed in human lungs (18,20). The presence of SP-A and dioleylphosphatidylglycerol (one of surfactant PLs) downregulates sPLA2-IIA expression via inhibition of NF-κB (77) and SP-A has an ability to inhibit also sPLA2-X and sPLA2-IIA activity by direct, selective, and calcium-dependent interaction (25,75). Recently, SP-B was reported to inhibit sPLA2-IIA–mediated hydrolysis of surfactant PLs (78), suggesting that the presence (or absence) of surfactant proteins may influence susceptibility of individual preparations to meconium when compared with natural surfactant (11).
sPLA2s and their products LPLs attack cell membranes, especially those apoptotic ones, damage alveolar cells, and injure alveolar epithelium denuding basal lamina, thus enabling recruitment of inflammatory cells and plasma proteins into alveolar space (18,20).
Elastase, proteinase-3, and cathepsin released after subsequent neutrophil degranulation degrade tissue and surfactant proteins (79,80,81) thereby increasing vulnerability of surfactant to sPLA2s (25,75). Moreover, elastase may stimulate lung epithelium in further cytokine production and NF-κB–mediated cell apoptosis (72).
Once established, the oxidative–inflammatory hotchpotch directs to cell death. Apart from direct injury of pulmonary cells via lipid, protein, and nucleic acid oxidation, free radicals induce several pathways of both programmed and nonprogrammed cell death. This signalization involves activation of the Fas receptor, mitogen-activated protein kinase pathways (including extracellular signal-regulated kinase ERK1/2), caspase family activation (mainly caspase 3) and NF-κB activation (65,72,82,83,84). In human epithelial cells, these pathways are reported to be mutually interlinked and also associated with TLR path in many cross-points (85), thus proapoptotic signalization means also proinflammatory one and vice versa.
In addition to ROS-mediated apoptosis, the amount of meconium and the duration of presence of meconium in the lungs directly affect apoptotic signalization in type II pneumocytes both in vitro and in vivo. Apoptotic membranes are more subject to sPLA2 hydrolysis (18). The longer exposition to meconium or the higher meconium content, the more pronounced caspase activity in type II cells resulting in apoptosis and also cell necrosis (86) related apparently to meconium-induced activation of local renin–angiotensin system and angiotensin AT1 receptor expression (30,87,88). Moreover, increase in intracellular Ca2+ due to, for example, meconium bile salts, subsequent sPLA2 activation and other processes may contribute to apoptotic gene expression and cell membrane damage (22,89,90). And despite some contradictions in observed results, it seems that TNF-α and NO radical, especially in higher amounts, may directly decrease SP-A, SP-B, and SP-C protein synthesis in type II pneumocytes (91,92). Taken together, functional loss of endogenous surfactant cannot be recovered by its increased production.
Leakage of plasma proteins into alveolar space through damaged alveolar–capillary membrane and consequent edema formation further amplify surfactant dysfunction (10). Plasma proteins also include several enzymes able to hydrolyze surfactant, and surprisingly, not sPLA2, but rather albumin, had been found to exert the most significant PLA2-like activity in hydrolyzing surfactant phosphatidylcholine to the inactive lysophosphatidylcholine form (93). Both albumin and fibrinogen are able to decrease exogenous surfactant surface properties significantly (94,95). Formation of edema is the final step for respiratory failure.
Conclusion
Meconium-induced inflammation and surfactant inactivation is a knotty question. Many scientific papers list precisely distinct mechanisms of meconium action; however, the unifying model of concerned mechanisms and their interrelations at the molecular level was still missing.
Inflammatory processes impair endogenous as well as exogenous surfactant used in the treatment of MAS. Despite therapeutic approaches with meconium being at least partially removed by lavage (96), running inflammation still embarrasses respiratory functions and thereby poses relevant topic for future research.
Statement of Financial Support
This study was supported by project APVV-0435-11, VEGA 1/0291/12: Slovak Research and Development Agency, The Ministry of Education, Science, Research and Sport of the Slovak Republic; project “BioMed Martin” No. 26220220187 - Agency of the Ministry of Education, Science, Research and Sport of the Slovak Republic for the Structural Funds of EU, Operational Programme Research and Development, being co-financed by the European Union.
Disclosure
The authors have no conflict of interests in association with this review.
References
Bae CW, Takahashi A, Chida S, Sasaki M. Morphology and function of pulmonary surfactant inhibited by meconium. Pediatr Res 1998;44:187–91.
Findlay RD, Taeusch HW, Walther FJ. Surfactant replacement therapy for meconium aspiration syndrome. Pediatrics 1996;97:48–52.
Donn SM, Dalton J. Surfactant replacement therapy in the neonate: beyond respiratory distress syndrome. Respir Care 2009;54:1203–8.
Kobayashi T, Nitta K, Ganzuka M, Inui S, Grossmann G, Robertson B. Inactivation of exogenous surfactant by pulmonary edema fluid. Pediatr Res 1991;29:353–6.
Mokra D, Drgova A, Kopincova J, Pullmann R, Calkovska A. Anti-inflammatory treatment in dysfunction of pulmonary surfactant in meconium-induced acute lung injury. Adv Exp Med Biol 2013;756:189–96.
Kopincová J, Mokrá D, Mikolka P, Kolomazník M, Čalkovská A. N-acetylcysteine advancement of surfactant therapy in experimental meconium aspiration syndrome: possible mechanisms. Physiol Res 2014;63:Suppl 4:S629–42.
Lindenskov PH, Castellheim A, Saugstad OD, Mollnes TE. Meconium aspiration syndrome: possible pathophysiological mechanisms and future potential therapies. Neonatology 2015;107:225–30.
Moses D, Holm BA, Spitale P, Liu MY, Enhorning G. Inhibition of pulmonary surfactant function by meconium. Am J Obstet Gynecol 1991;164:477–81.
Mokra D, Calkovska A. How to overcome surfactant dysfunction in meconium aspiration syndrome? Respir Physiol Neurobiol 2013;187:58–63.
Ochs M, Schüttler M, Stichtenoth G, Herting E. Morphological alterations of exogenous surfactant inhibited by meconium can be prevented by dextran. Respir Res 2006;7:86.
Herting E, Rauprich P, Stichtenoth G, Walter G, Johansson J, Robertson B. Resistance of different surfactant preparations to inactivation by meconium. Pediatr Res 2001;50:44–9.
Tølløfsrud PA, Lindenskov PH, Drevon CA, Speer CP, Seidenspinner S, Saugstad OD. Comparison of pulmonary and inflammatory effects of lipid- and water-soluble components in meconium in newborn piglets. Biol Neonate 2003;84:330–7.
Pallem V, Kaviratna AS, Chimote G, Banerjee R. Effect of meconium on surface properties of surfactant monolayers and liposomes. Colloid Surface A 2010;370:6–14.
Lopez-Rodriguez E, Echaide M, Cruz A, Taeusch HW, Perez-Gil J. Meconium impairs pulmonary surfactant by a combined action of cholesterol and bile acids. Biophys J 2011;100:646–55.
Hiansen JQ, Keating E, Aspros A, et al. Cholesterol-mediated surfactant dysfunction is mitigated by surfactant protein A. Biochim Biophys Acta 2015;1848:813–20.
Lopez-Rodriguez E, Pérez-Gil J. Structure-function relationships in pulmonary surfactant membranes: from biophysics to therapy. Biochim Biophys Acta 2014;1838:1568–85.
Hall SB, Lu RZ, Venkitaraman AR, Hyde RW, Notter RH. Inhibition of pulmonary surfactant by oleic acid: mechanisms and characteristics. J Appl Physiol (1985) 1992;72:1708–16.
Murakami M, Kudo I. Secretory phospholipase A2. Biol Pharm Bull 2004;27:1158–64.
Kääpä P, Soukka H. Phospholipase A2 in meconium-induced lung injury. J Perinatol 2008;28:Suppl 3:S120–2.
De Luca D, Minucci A, Tripodi D, et al. Role of distinct phospholipases A2 and their modulators in meconium aspiration syndrome in human neonates. Intensive Care Med 2011;37:1158–65.
Lambeau G, Gelb MH. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem 2008;77:495–520.
Holopainen R, Aho H, Laine J, Peuravuori H, Soukka H, Kääpä P. Human meconium has high phospholipase A2 activity and induces cellular injury and apoptosis in piglet lungs. Pediatr Res 1999;46:626–32.
Schrama AJ, de Beaufort AJ, Sukul YR, Jansen SM, Poorthuis BJ, Berger HM. Phospholipase A2 is present in meconium and inhibits the activity of pulmonary surfactant: an in vitro study. Acta Paediatr 2001;90:412–6.
Hite RD, Seeds MC, Jacinto RB, Grier BL, Waite BM, Bass DA. Lysophospholipid and fatty acid inhibition of pulmonary surfactant: non-enzymatic models of phospholipase A2 surfactant hydrolysis. Biochim Biophys Acta 2005;1720:14–21.
Chabot S, Koumanov K, Lambeau G, et al. Inhibitory effects of surfactant protein A on surfactant phospholipid hydrolysis by secreted phospholipases A2. J Immunol 2003;171:995–1000.
D’Ovidio F, Mura M, Ridsdale R, et al. The effect of reflux and bile acid aspiration on the lung allograft and its surfactant and innate immunity molecules SP-A and SP-D. Am J Transplant 2006;6:1930–8.
Gross T, Zmora E, Levi-Kalisman Y, Regev O, Berman A. Lung-surfactant-meconium interaction: in vitro study in bulk and at the air-solution interface. Langmuir 2006;22:3243–50.
De Luca D, Minucci A, Zecca E, et al. Bile acids cause secretory phospholipase A2 activity enhancement, revertible by exogenous surfactant administration. Intensive Care Med 2009;35:321–6.
Ivanov VA. Meconium Toxicity to Human Surfactant Proteins. American Academy of Pediatrics, 2012. (https://aap.confex.com/aap/2012/webprogrampress/Paper16405.html.)
Vidyasagar D, Zagariya A. Studies of meconium-induced lung injury: inflammatory cytokine expression and apoptosis. J Perinatol 2008;28:Suppl 3:S102–7.
Castellheim A, Lindenskov PH, Pharo A, Fung M, Saugstad OD, Mollnes TE. Meconium is a potent activator of complement in human serum and in piglets. Pediatr Res 2004;55:310–8.
Mollnes TE, Castellheim A, Lindenskov PH, Salvesen B, Saugstad OD. The role of complement in meconium aspiration syndrome. J Perinatol 2008;28:Suppl 3:S116–9.
Salvesen B, Fung M, Saugstad OD, Mollnes TE. Role of complement and CD14 in meconium-induced cytokine formation. Pediatrics 2008;121:e496–505.
Salvesen B, Stenvik J, Rossetti C, Saugstad OD, Espevik T, Mollnes TE. Meconium-induced release of cytokines is mediated by the TRL4/MD-2 complex in a CD14-dependent manner. Mol Immunol 2010;47:1226–34.
Lindenskov PH, Castellheim A, Aamodt G, Saugstad OD, Mollnes TE. Complement activation reflects severity of meconium aspiration syndrome in newborn pigs. Pediatr Res 2004;56:810–7.
Salvesen B, Nielsen EW, Harboe M, Saugstad OD, Mollnes TE. Mechanisms of complement activation and effects of C1-inhibitor on the meconium-induced inflammatory reaction in human cord blood. Mol Immunol 2009;46:688–94.
Anas A, van der Poll T, de Vos AF. Role of CD14 in lung inflammation and infection. Crit Care 2010;14:209.
Xiang M, Fan J, Fan J. Association of Toll-like receptor signaling and reactive oxygen species: a potential therapeutic target for posttrauma acute lung injury. Mediators Inflamm 2010;2010.
Godowski PJ. A smooth operator for LPS responses. Nat Immunol 2005;6:544–6.
Andonegui G, Bonder CS, Green F, et al. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest 2003;111:1011–20.
Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol 2004;287:L143–52.
van Berlo D, Knaapen AM, van Schooten FJ, Schins RP, Albrecht C. NF-kappaB dependent and independent mechanisms of quartz-induced proinflammatory activation of lung epithelial cells. Part Fibre Toxicol 2010;7:13.
Kishore U, Greenhough TJ, Waters P, et al. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol 2006;43:1293–315.
Watford WT, Wright JR, Hester CG, Jiang H, Frank MM. Surfactant protein A regulates complement activation. J Immunol 2001;167:6593–600.
Oosting RS, Wright JR. Characterization of the surfactant protein A receptor: cell and ligand specificity. Am J Physiol 1994;267:L165–72.
Moliva JI, Rajaram MV, Sidiki S, et al. Molecular composition of the alveolar lining fluid in the aging lung. Age (Dordr) 2014;36:9633.
Gardai SJ, Xiao YQ, Dickinson M, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003;115:13–23.
Pérez-Gil J. Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim Biophys Acta 2008;1778:1676–95.
Seaton BA, Crouch EC, McCormack FX, Head JF, Hartshorn KL, Mendelsohn R. Review: structural determinants of pattern recognition by lung collectins. Innate Immun 2010;16:143–50.
Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol 2005;5:58–68.
Vandivier RW, Ogden CA, Fadok VA, et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol 2002;169:3978–86.
Yamada C, Sano H, Shimizu T, et al. Surfactant protein A directly interacts with TLR4 and MD-2 and regulates inflammatory cellular response. Importance of supratrimeric oligomerization. J Biol Chem 2006;281:21771–80.
Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol 2002;168:5989–92.
Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol 2010;31:154–63.
de Beaufort AJ, Bakker AC, van Tol MJ, Poorthuis BJ, Schrama AJ, Berger HM. Meconium is a source of pro-inflammatory substances and can induce cytokine production in cultured A549 epithelial cells. Pediatr Res 2003;54:491–5.
Yamada T, Matsubara S, Minakami H, Kohmura Y, Hiratsuka M, Sato I. Chemotactic activity for polymorphonuclear leukocytes: meconium versus meconium-stained amniotic fluid. Am J Reprod Immunol 2000;44:275–8.
Yamada T, Minakami H, Matsubara S, Yatsuda T, Kohmura Y, Sato I. Meconium-stained amniotic fluid exhibits chemotactic activity for polymorphonuclear leukocytes in vitro. J Reprod Immunol 2000;46:21–30.
Matsubara S, Yamada T, Minakami H, Takizawa T, Sato I. Meconium-stained amniotic fluid activates polymorphonuclear leukocytes ultrastructural and enzyme-cytochemical evidence. Eur J Histochem 1999;43:205–10.
Berdeli A, Akisu M, Dagci T, Akisu C, Yalaz M, Kultursay N. Meconium enhances platelet-activating factor and tumor necrosis factor production by rat alveolar macrophages. Prostaglandins Leukot Essent Fatty Acids 2004;71:227–32.
Okazaki K, Kondo M, Kato M, et al. Serum cytokine and chemokine profiles in neonates with meconium aspiration syndrome. Pediatrics 2008;121:e748–53.
Granata F, Petraroli A, Boilard E, et al. Activation of cytokine production by secreted phospholipase A2 in human lung macrophages expressing the M-type receptor. J Immunol 2005;174:464–74.
Qin P, Tang X, Elloso MM, Harnish DC. Bile acids induce adhesion molecule expression in endothelial cells through activation of reactive oxygen species, NF-kappaB, and p38. Am J Physiol Heart Circ Physiol 2006;291:H741–7.
Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 2006;72:1493–505.
Kopincová J, Púzserová A, Bernátová I. Biochemical aspects of nitric oxide synthase feedback regulation by nitric oxide. Interdiscip Toxicol 2011;4:63–8.
Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol 2012;84:581–90.
Fan J, Frey RS, Malik AB. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J Clin Invest 2003;112:1234–43.
Hattori H, Subramanian KK, Sakai J, et al. Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci USA 2010;107:3546–51.
de Beaufort AJ, Pelikan DM, Elferink JG, Berger HM. Effect of interleukin 8 in meconium on in-vitro neutrophil chemotaxis. Lancet 1998;352:102–5.
Williams JH Jr, Patel SK, Hatakeyama D, et al. Activated pulmonary vascular neutrophils as early mediators of endotoxin-induced lung inflammation. Am J Respir Cell Mol Biol 1993;8:134–44.
Mokra D, Mokry J, Drgova A, Petraskova M, Bulikova J, Calkovska A. Intratracheally administered corticosteroids improve lung function in meconium-instilled rabbits. J Physiol Pharmacol 2007;58:Suppl 5:389–98.
Soukka HR, Ahotupa M, Ruutu M, Kääpä PO. Meconium stimulates neutrophil oxidative burst. Am J Perinatol 2002;19:279–84.
Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med 2011;17:293–307.
Li YH, Yan ZQ, Brauner A, Tullus K. Meconium induces expression of inducible NO synthase and activation of NF-kappaB in rat alveolar macrophages. Pediatr Res 2001;49:820–5.
Rodríguez-Capote K, Manzanares D, Haines T, Possmayer F. Reactive oxygen species inactivation of surfactant involves structural and functional alterations to surfactant proteins SP-B and SP-C. Biophys J 2006;90:2808–21.
Arbibe L, Vial D, Rosinski-Chupin I, et al. Endotoxin induces expression of type II phospholipase A2 in macrophages during acute lung injury in guinea pigs: involvement of TNF-alpha in lipopolysaccharide-induced type II phospholipase A2 synthesis. J Immunol 1997;159:391–400.
Alaoui-El-Azher M, Wu Y, Havet N, Israël A, Lilienbaum A, Touqui L. Arachidonic acid differentially affects basal and lipopolysaccharide-induced sPLA(2)-IIA expression in alveolar macrophages through NF-kappaB and PPAR-gamma-dependent pathways. Mol Pharmacol 2002;61:786–94.
Wu YZ, Medjane S, Chabot S, et al. Surfactant protein-A and phosphatidylglycerol suppress type IIA phospholipase A2 synthesis via nuclear factor-kappaB. Am J Respir Crit Care Med 2003;168:692–9.
Hite RD, Grier BL, Waite BM, et al. Surfactant protein B inhibits secretory phospholipase A2 hydrolysis of surfactant phospholipids. Am J Physiol Lung Cell Mol Physiol 2012;302:L257–65.
Liau DF, Yin NX, Huang J, Ryan SF. Effects of human polymorphonuclear leukocyte elastase upon surfactant proteins in vitro. Biochim Biophys Acta 1996;1302:117–28.
Hirche TO, Crouch EC, Espinola M, et al. Neutrophil serine proteinases inactivate surfactant protein D by cleaving within a conserved subregion of the carbohydrate recognition domain. J Biol Chem 2004;279:27688–98.
Rubio F, Cooley J, Accurso FJ, Remold-O’Donnell E. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax 2004;59:318–23.
Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000;5:415–8.
Zhang X, Shan P, Sasidhar M, et al. Reactive oxygen species and extracellular signal-regulated kinase ½ mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am J Respir Cell Mol Biol 2003;28:305–15.
Janssen YM, Matalon S, Mossman BT. Differential induction of c-fos, c-jun, and apoptosis in lung epithelial cells exposed to ROS or RNS. Am J Physiol 1997;273:L789–96.
Galani V, Tatsaki E, Bai M, et al. The role of apoptosis in the pathophysiology of acute respiratory distress syndrome (ARDS): an up-to-date cell-specific review. Pathol Res Pract 2010;206:145–50.
Jeng MJ, Soong WJ, Lee YS, et al. Meconium exposure dependent cell death and apoptosis in human alveolar epithelial cells. Pediatr Pulmonol 2010;45:816–23.
Lukkarinen H, Laine J, Lehtonen J, et al. Angiotensin II receptor blockade inhibits pneumocyte apoptosis in experimental meconium aspiration. Pediatr Res 2004;55:326–33.
Rosenfeld CR, Zagariya AM, Liu XT, Willis BC, Fluharty S, Vidyasagar D. Meconium increases type 1 angiotensin II receptor expression and alveolar cell death. Pediatr Res 2008;63:251–6.
Zagariya A, Bhat R, Chari G, Uhal B, Navale S, Vidyasagar D. Apoptosis of airway epithelial cells in response to meconium. Life Sci 2005;76:1849–58.
Oelberg DG, Downey SA, Flynn MM. Bile salt-induced intracellular Ca++ accumulation in type II pneumocytes. Lung 1990;168:297–308.
Glumoff V, Väyrynen O, Kangas T, Hallman M. Degree of lung maturity determines the direction of the interleukin-1- induced effect on the expression of surfactant proteins. Am J Respir Cell Mol Biol 2000;22:280–8.
Lee JW, Gonzalez RF, Chapin CJ, Busch J, Fineman JR, Gutierrez JA. Nitric oxide decreases surfactant protein gene expression in primary cultures of type II pneumocytes. Am J Physiol Lung Cell Mol Physiol 2005;288:L950–7.
Damas JE, Cake MH. An albumin-associated PLA2-like activity inactivates surfactant phosphatidylcholine secreted from fetal type II pneumocytes. Am J Physiol Lung Cell Mol Physiol 2011;301:L966–74.
Park J, Bae CW, Choi YM. In vitro inhibition of biophysical surface properties and change in ultrastructures of exogenous pulmonary surfactant by albumin or fibrinogen. J Korean Med Sci 1998;13:123–30.
Seehase M, Collins JJ, Kuypers E, et al. New surfactant with SP-B and C analogs gives survival benefit after inactivation in preterm lambs. PLoS One 2012;7:e47631.
Hahn S, Choi HJ, Soll R, Dargaville PA. Lung lavage for meconium aspiration syndrome in newborn infants. Cochrane Database Syst Rev 2013;4:CD003486.
Acknowledgements
This study has been approved by the Institutional Review Board of Jessenius Faculty of Medicine in Martin, Comenius University in Bratislava, Slovak Republic.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Kopincova, J., Calkovska, A. Meconium-induced inflammation and surfactant inactivation: specifics of molecular mechanisms. Pediatr Res 79, 514–521 (2016). https://doi.org/10.1038/pr.2015.265
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.265
This article is cited by
-
Predictors of successful treatment of respiratory distress with aerosolized calfactant
Journal of Perinatology (2023)
-
Efficacy of synthetic surfactant (CHF5633) bolus and/or lavage in meconium-induced lung injury in ventilated newborn rabbits
Pediatric Research (2023)
-
The effect of gestational age, low birth weight and parity on birth asphyxia among neonates in sub-Saharan Africa: systematic review and meta-analysis: 2021
Italian Journal of Pediatrics (2022)
-
Activation of Toll-like receptors in meconium aspiration syndrome
Journal of Perinatology (2018)
