
Abstract
Intestinal microbes promote the injurious effects of radiation on those tissues. However, the molecular factors mediating this effect are largely unknown. In this work, we explored the effects of orally administered antibiotics and MyD88, a key adapter molecule in toll-like receptor signaling, on molecular and cellular responses of mouse colon to radiation. Results show that oral antibiotics lowered radiation-induced colonic damage by protecting epithelial cells against radiation-induced apoptosis, leading to increased survival of crypts. MyD88 deficiency partially phenocopied the effects of oral antibiotics on apoptosis and crypt survival, suggesting that colonic microbes exert their injurious effects in part via that molecule. Analysis of DNA double-strand breaks, the primary genotoxic lesions induced by radiation, showed that their early induction in mouse colon was unaffected by MyD88. However, MyD88 deficiency resulted in the later disappearance of DNA double-strand breaks. Loss of DNA double-strand breaks was accompanied by the evidence of increased activation of both the non-homologous end-joining and homologous recombination pathways of DNA repair in MyD88-deficient mice. These results show that colonic microbes and MyD88 regulate DNA double-strand break repair in irradiated mouse colon, effects which exert significant control over radiation-induced apoptosis and crypt survival.
Similar content being viewed by others


Introduction
During the radiotherapy of abdominal or pelvic malignancies, ionizing radiation can damage the mucosal surface of the gastrointestinal tract, leading to symptoms that may necessitate a reduction in radiation dose or the cessation of therapy. Furthermore, accidents at nuclear-energy facilities or following nuclear warfare lead to serious life-threatening intestinal injury. To date, biological strategies to limit intestinal radiation injury have been hindered by an insufficient understanding of the pathophysiology of this condition.
Studies in mice have provided mechanistic insights into some aspects of intestinal radiation injury. At the cellular level, ionizing radiation induces DNA double-strand breaks, which trigger a p53-mediated response.1 If double-strand breaks are sufficiently abundant, the p53 response can result in apoptosis. However, if double-strand breaks are few, the p53 response may halt cell cycle progression and following DNA double-strand break repair, allow the cell to survive.2 In the intestinal epithelium, the rapidly dividing crypt transit cells are the most sensitive to radiation-induced apoptosis, which can be detected at doses of <1 Gy, within minutes of radiation. At radiation doses of 8–12 Gy, the relatively more radioresistant stem cells of the crypts undergo apoptosis.3 The death of these cells is assessed by using the crypt microcolony assay that measures the survival and repopulation of damaged crypts several days after radiation.4 The early induction of intestinal epithelial cell apoptosis by radiation, followed by later mucosal damage due to progressive loss of crypts leads to the mucositis that follows several days after the beginning of abdominal or pelvic radiotherapy. The sensitivity of intestinal crypt epithelial stem cells to undergo apoptosis following radiation therefore determines the severity of injury.
Soluble factors in the microenvironment of the intestinal crypt, notably prostaglandins, have been shown to regulate radiation-induced injury.5, 6, 7 Work using germ-free mice has also shown an important role for the intestinal microflora in stimulating radiation-induced crypt loss and lethality.8, 9 This suggests that intestinal microbes or their products influence the sensitivity of intestinal epithelial stem cells to undergo radiation-induced apoptosis. However, the mechanism by which microbes might signal to intestinal epithelial cells to promote their apoptosis is not known. Moreover, little is understood about whether microbial signaling affects the sensitivity of intestinal epithelial cells to radiation-induced apoptosis via an interaction with the DNA double-strand break response, or via other effects.
Toll-like receptors (TLRs) have been extensively characterized as cell-surface molecules that sense substances derived from microbes and thus are candidates for components of a microbial signaling pathway regulating the intestinal radiation response. Most TLRs, with the exception of TLR3, recruit MyD88, a critical adaptor molecule required for signalling from TLRs (reviewed by Fitzgerald and Chen10). With a mechanism not completely understood, different TLR ligands induce distinct cellular responses, including TLR-2/-4-mediated cellular apoptosis.11, 12
In order to gain insights into how microbial factors control radiation-induced colonic mucosal injury, we used antibiotic-treated mice and MyD88 knockout mice, and analyzed the cardinal features of radiation injury: cell death and DNA damage. The results show that colonic microbes and MyD88 regulate radiation-induced DNA damage responses that control the sensitivity of colonic epithelial cells to undergo apoptosis.
Results
Antibiotics attenuate the GI radiation syndrome by promoting the survival of colonic epithelial crypts
Previous work in germ-free mice indicated that intestinal microbes promoted the induction of radiation-induced injury to the small intestine.8, 9 We first treated mice with broad spectrum orally administered antibiotics, according to established protocols (see Materials and methods) to determine if pharmacological modulation of the colonic microbiota could alter colonic radiation responses.13, 14 Before radiation, there are no significant differences in water or food intake between the antibiotic-treated mice and control mice. In the 4.5 days following exposure to low doses (4 or 6 Gy) of radiation, similar decreases in water and food intake were observed in both antibiotic-treated mice and controls (Figures 1a and b). However, at higher doses of radiation (8, 10, 14 or 20 Gy), no further reductions in the amount of either water or food intake were observed in antibiotic-treated mice, but in control mice without antibiotics, both water and food intake were lower. The radiation-induced reduction in food and water intake in control mice was accompanied by weight loss, which was also more pronounced in the control than in the antibiotic-treated mice (Figure 1c). These results suggested that oral antibiotics might mitigate radiation-induced gastrointestinal (GI) syndrome.

Antibiotics attenuate the GI radiation syndrome by promoting the survival of colonic epithelial crypts. (a) Daily consumption of water and food (b) in antibiotic-treated and control mice in the 4.5 days following the indicated doses of whole-body radiation. (c) Body weight at baseline and 4.5 days following 14 Gy whole-body radiation in antibiotic-treated and control mice. (d) H&E-stained sections of colon at baseline and 4.5 days post 14 Gy whole-body radiation in antibiotic-treated and control mice. (e) Results of the colon crypt microcolony assay at 4.5 days following whole-body radiation in antibiotic-treated and control mice. (f) Representative PCNA immunofluorescence of colon sections, at baseline and 4.5 days following 14 Gy whole-body radiation in antibiotic-treated and control mice. Blue is DAPI nuclear staining. Red is PCNA staining. Quantification of PCNA staining at 4.5 days post radiation in antibiotic-treated and control mice. (g) Assessment of PCNA staining by crypt cell position at 4.5 days post radiation. In all panels, means±standard error are shown, **P<0.01 antibiotic-treated versus control mice.
To assess the degree of colonic injury caused by radiation, we examined histological sections of colon obtained from non-irradiated mice or from mice 4.5 days following irradiation. Appearances were identical in antibiotic-treated and control mice without radiation (Figure 1d). Antibiotic-treated mice 4.5 days after irradiation also had an almost normal appearing colonic architecture even at high doses of radiation up to 14 or 20 Gy. However, control mice exhibited a severe loss of normal crypt architecture and cellularity accompanied by a striking inflammatory cell infiltrate into the lamina propria. Consistent with established paradigms of radiation-induced colon crypt killing, the crypt microcolony assay revealed a progressive dose-dependent crypt loss in control mice that received doses of ⩾10 Gy (Figure 1e). In contrast, very little crypt loss occurred in antibiotic-treated mice, even with 20 Gy radiation, a dose that is sufficient to almost completely kill all colonic crypts of control mice.
The loss of cells within colonic crypts in control mice could have resulted from diminished epithelial proliferation and/or increased cell death. Therefore, we assessed colonic epithelial cell proliferation by quantifying S-phase cells with proliferating cellular nuclear antigen (PCNA) immunohistochemistry. In control mice, we observed a radiation dose-dependent decline in the average number of PCNA-positive cells 4.5 days following irradiation (Figure 1f). In contrast, antibiotic-treated mice were protected against radiation-induced loss of PCNA-positive cells. We localized the position of PCNA-positive cells in non-irradiated control mice and found that most were at positions 1–6 from the crypt base, the stem and transit cell zones (Figure 1g). Fourteen Gray of radiation almost completely abolished PCNA positivity in control mice, but antibiotic treatment restored the pattern of PCNA positivity to that observed in non-irradiated control mice. Together, these results indicate that the luminal microbiota imparts radiosensitivity to colonic epithelial stem cells, thereby promoting the development of the radiation GI syndrome
We further aimed to distinguish if the protection from radiation-induced colonic injury conferred by oral antibiotics is due to a reduction in radiation-induced apoptosis. As shown in Figure 2a, at 4.5 h post exposure to 14 Gy, the typical morphological characteristics of apoptosis, such as membrane blebbing, cytoplasmic and nuclear shrinkage, DNA fragmentation and chromatin condensation, were observed in non-antibiotic-treated control mice. In contrast, in antibiotic-treated mice, no or sometimes one or two apoptotic cells could be found in a colonic crypt section. We further assessed colonic epithelial cellular apoptosis by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. In both antibiotic-treated and control mice without radiation, the baseline of TUNEL staining is low (Figure 2b). At 4.5 h post exposure to 14 Gy, we observed a striking increase in the number of TUNEL-positive cells in control mice. In antibiotic-treated mice there also was an increase in the number of TUNEL-positive cells, but to a lesser degree than in control mice. Positional analysis of TUNEL-positive cells showed that they were mostly located at positions 1 to 6 from the crypt base. Antibiotic treatment significantly lowered the rate of TUNEL-positive cells at all those positions, almost to the rates observed in non-irradiated mice. These data show that antibiotics protect colonic crypt cells from radiation-induced apoptosis. When taken together with the results of the crypt microcolony assay, it appears that oral antibiotics block the apoptotic killing of crypt stem cells by radiation, which results in significantly lower crypt loss and colonic mucosal injury.

Antibiotics protect the colonic epithelial cells located in the stem and progenitor cell zone against radiation-induced apoptosis. (a) Representative images of H&E-stained colon sections in antibiotic-treated and control mice at baseline and 4.5 h after 14 Gy radiation. (b) Representative images of DAPI and TUNEL stained sections of colon in antibiotic-treated and control mice at baseline and 4.5 h after 14 Gy radiation. Blue is DAPI nuclear staining and TUNEL is green. Quantification of TUNEL-positive cells at baseline and 4.5 h post 14 Gy radiation. (c) Assessment of TUNEL positivity by crypt cell position. In all panels, means±standard error are shown, **P<0.01 antibiotic-treated versus control mice.
MyD88 has a pathogenic role in the colonic GI syndrome and crypt survival
As oral antibiotic-treated mice are resistant to radiation-induced damage in the colon, this suggested that luminal microbes modulate colonic epithelial radiosensitivity. Previous studies suggested that gut microbes or their products acting via TLRs could induce cellular apoptosis in vitro or modulate intestinal radiosensitivity in vivo.8, 9 Therefore, we employed MyD88 knockout mice which lack most TLR-dependent signaling, as well as signaling from interleukin-1 and -18 to determine if those receptors are responsible for gut microbial-enhanced apoptotic response to radiation.
We used a radiation dose of 14 Gy, one that is efficient at inducing a high degree of DNA damage and reliably results in GI syndrome in the colon.15 As assessed by loss of body weight, both wild-type and MyD88 knockout mice were affected by the GI syndrome in the 4.5 days following 14 Gy irradiation (Figure 3a). However, the degree of weight loss in wild-type mice was significantly greater than the weight loss in MyD88 knockout mice. The average daily food intake in wild-type mice also fell by more than that of MyD88 knockout mice (Figure 3b), with corresponding changes in water intake (Figure 3c). These clinical parameters indicated that wild-type mice were affected more severely by the damaging effects of radiation. We also examined the colons of mice at baseline and 4.5 days following radiation. Hematoxylin and eosin (H&E) staining showed identical normal-appearing morphology of the colonic epithelium in MyD88 knockout and wild-type mice (Figure 3d). At 4.5 days following 14 Gy irradiation, wild-type mice suffered colonic mucosal injury. Crypts were lost, and there was dramatic inflammation in the lamina propria. MyD88 knockout mice also suffered some colonic injury with loss of crypts and milder mucosal inflammation. Results of the crypt microcolony assay showed almost complete loss of viable colon crypts in the wild-type mice (Figure 3e). In marked contrast, MyD88 knockout mice only suffered ∼50% crypt loss. PCNA staining was also done to check the viability of the residual epithelial cells. Similar numbers of PCNA-positive cells per crypt section were observed in wild-type and MyD88 knockout mice in the baseline condition (Figure 3f). However, numbers were dramatically lowered by 4.5 days following radiation in the wild-type mice. In contrast, there was significantly higher survival of PCNA-positive cells in the MyD88 knockout mice, a finding that is consistent with the preserved viability of epithelial cells in those mice.

MyD88 deficiency decreases the radiation-induced GI syndrome through increasing the survival of colon crypts. (a) Body weight at baseline and 4.5 days following 14 Gy whole-body radiation in MyD88-deficient and wild-type mice. (b) Daily consumption of food and water (c) in MyD88-deficient and wild-type mice in the 4.5 days following 14 Gy whole-body radiation. (d) H&E-stained sections of colon at baseline and 4.5 days post 14 Gy whole-body radiation in MyD88-deficient and wild-type mice. (e) Results of the colon crypt microcolony assay at baseline and 4.5 days following 14 Gy whole-body radiation in MyD88-deficient and wild-type mice. (f) Representative PCNA immunofluorescence of colon sections, at baseline and 4.5 days following 14 Gy whole-body radiation in MyD88-deficient and wild type mice. Quantification of PCNA staining at baseline and 4.5 days post 14 Gy radiation. Blue is DAPI nuclear staining. Red is PCNA staining. In all panels, means±standard error are shown, *P<0.05, **P<0.01 MyD88-deficient versus wild-type mice.
We next assessed the effects of MyD88 deficiency on radiation-induced apoptosis. As shown in Figure 4a, wild-type mice developed apoptotic cells at 4.5 h post exposure to 14 Gy, whereas MyD88 knockout mice displayed markedly fewer apoptotic cells. TUNEL-positive cells were induced by radiation in both wild-type and MyD88 knockout mice, but the abundance of TUNEL positivity was two- to three-fold higher in wild-type than in MyD88 knockout mice (Figure 4b). Taken together, these findings indicate that MyD88 deficiency partly phenocopies the effects of antibiotics as regards colonic crypt epithelial responses to radiation.

MyD88 deficiency decreases radiation-induced apoptosis in mouse colon following whole-body radiation. (a) Representative images of H&E-stained colon sections in MyD88-deficient and wild-type mice 4.5 h after 14 Gy whole-body radiation. (b) Representative images of consecutive DAPI, TUNEL and γ-H2ax-stained sections of colon in MyD88-deficient and wild-type mice 4.5 h after 14 Gy radiation. Blue is DAPI nuclear staining, TUNEL is green and γ-H2ax is red. In the merged image, yellow indicates colocalization of TUNEL-γ-H2ax double-positive nuclei. (c) Quantification of TUNEL-positive cells at baseline and 4.5 h post 14 Gy radiation. In all panels, means±standard error are shown, **P<0.01 MyD88-deficient versus wild-type mice.
As DNA double-strand break is the primary genotoxic lesion induced by ionizing radiation, we next assessed any correlation between them and apoptosis. We used immunofluorescent staining of γ-H2ax, which is a molecular marker that accumulates at DNA double-strand breaks, resulting in immunofluorescent foci that are a reliable indicator of the abundance of strand breaks.16, 17 As shown in Figure 4b, double staining with γ-H2ax and TUNEL at 4.5 hours post radiation in wild-type mice showed that most TUNEL-positive cells also showed γ-H2ax immunoreactivity. Over 95% of the double-labeled cells showed colocalization between TUNEL and γ-H2ax. In contrast, only few γ-H2ax-positive cells could be seen in the MyD88 knockout mice. These findings indicate an important consequence of MyD88-dependent regulation of DNA damage responses, suggesting that DNA damage repair machinery may underlie the molecular mechanism of radiation-induced epithelial apoptosis.
Disappearance of radiation-induced DNA double-strand breaks in MyD88 knockout mouse colon.
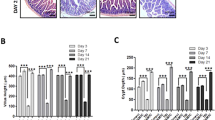
We next explored further how DNA double-strand breaks correlated with the differential responses of colon epithelial cells to radiation conferred by MyD88, by assessing the kinetics of double-strand break formation and resolution in colon crypt epithelial cells of mice following radiation. As early as 15 min following radiation, there was a dramatic induction of γ-H2ax foci in epithelial cells of the colon crypts (Figure 5a). The number of γ-H2ax foci that were induced by radiation was initially similar between MyD88-deficient and wild-type mice and increased up to about 1.5 h following radiation. However, by 4.5 h post radiation, the number of γ-H2ax foci further increased in wild-type mice, but fell dramatically in the MyD88 knockout mice. At that time following radiation, γ-H2ax foci were ∼10 times lower in MyD88 knockout than wild-type mice. This shows that the initial responses of colon epithelial cells to radiation was not affected by MyD88, but the significant disappearance of γ-H2ax foci at the later time indicates a regulatory role for MyD88 in DNA damage responses.

MyD88 deficiency results in differentiated γ-H2ax and p-s1981-Atm kinetics in mouse colon following whole-body radiation. (a) Representative γ-H2ax foci immunofluorescence of colon sections, at the indicated times following 14 Gy whole-body radiation in MyD88-deficient and wild-type mice. Quantification of γ-H2ax foci and their time course following 14 Gy radiation. (b) Representative p-s1981-Atm foci immunofluorescence of colon sections, at baseline and 4.5 h following 14 Gy radiation in MyD88-deficient and wild-type mice. (c) Quantification of p-s1981-Atm foci at baseline and 4.5 h post 14 Gy radiation. Blue is DAPI nuclear staining. Red is γ-H2ax or p-s1981-Atm staining. In both panels, means±standard error are shown, **P<0.01 MyD88-deficient versus wild-type mice.
The mutated in ataxia telangiectasia kinase, ATM is an early transducer of intracellular signaling from DNA double-strand breaks,18 and can be functionally assessed by immunofluorescent detection of serine 1981 phosphorylation (P-s1981-Atm).19 We observed similar basal levels of P-s1981-Atm foci in wild-type and MyD88 knockout mice (Figure 5b). However, at 4.5 h following radiation, P-s1981-Atm foci were approximately two-times higher in wild-type than in MyD88 knockout mice. Thus, by 4.5 h following radiation, not only is the abundance of DNA double-strand breaks lower in MyD88 knockout than in wild-type mice but this difference is also accompanied by a reduced DNA damage signaling.
Increased DNA repair protein activation in MyD88 knockout mice
The observation of lower γ-H2ax and P-s1981-Atm foci by 4.5 h post radiation in MyD88 knockout mouse colon compared to that in wild-type suggested that MyD88 might inhibit DNA double-strand break repair. To assess this possibility, we compared markers of DNA repair pathway activation in wild-type and MyD88 knockout mice. We used the appearance of Ku70–80 foci as a marker of the non-homologous end-joining pathway,20 and Rad51 foci as a marker of the homologous recombination pathway of DNA double-strand break repair.21 At baseline, few Ku70–80 foci were present in either mice (Figure 6a). However, 4.5 h post radiation, there were clear Ku70–80-positive foci mostly localized at the bottom of colon crypts in MyD88 knockout mice. In contrast, few Ku70–80-positive cells were noted in wild-type mice. Similar to the results obtained with Ku70–80, we found comparable baseline numbers, but significantly lower 4.5 h post-radiation numbers of Rad51 foci in wild-type compared to that in MyD88 knockout mice (Figure 6b). Therefore, these results suggest that MyD88-mediated signaling controls DNA double-strand break repair pathways, both non-homologous end-joining and homologous recombination.

MyD88 deficiency results in enhanced DNA repair in mouse colon following whole-body radiation. (a) Representative Ku 70–80 immunofluorescence of colon sections, at baseline and 4.5 h following 14 Gy whole-body radiation, in MyD88-deficient and wild-type mice. Quantification of Ku 70–80 foci at baseline and 4.5 h post 14 Gy radiation. (b) Representative Rad51 immunofluorescence of colon sections, at baseline and 4.5 h following 14 Gy whole-body radiation, MyD88-deficient and wild-type mice. Quantification of Rad51 foci at baseline and 4.5 h post 14 Gy radiation. Blue is DAPI nuclear staining. Red is Ku 70–80 or Rad51 staining. In both panels, means±standard error are shown, **P<0.01 MyD88-deficient versus wild-type mice.
Discussion
Intestinal microbial signaling is increasingly recognized to have important roles in the pathogenesis of many gastrointestinal diseases including colon cancer, colitis and radiation enteritis.22, 23, 24, 25 Our results show that exposure of mice to a regime of oral antibiotics that significantly alters the intestinal microbial flora13, 14 affects the sensitivity of the colon epithelial cells to radiation-induced apoptosis, crypt loss and GI syndrome. The effects of antibiotics were partially phenocopied by deficiency of MyD88, indicating an injurious role of MyD88, which is accompanied by a dampening or inhibitory effect on DNA double-strand break repair in colon epithelial cells. Greater DNA double-strand break repair in MyD88-deficient mice led to lower levels of apoptosis, less-severe GI syndrome and significantly superior crypt survival. Together, these findings suggest that normal colonic microbes and MyD88 inhibit the repair of radiation-induced DNA damage resulting in the prolonged induction of DNA damage signaling that results in apoptosis, crypt loss and GI injury.
Our results have shed some light on the molecular mechanism by which colonic microbes and MyD88 regulate the colonic radiation response of the colon. We found that γ-H2ax-positive DNA double-strand breaks, the earliest molecular lesions induced by ionizing radiation, were induced to a similar extent both in the presence and absence of MyD88. However, there followed a loss of double-strand breaks and a concomitant increase in the activation of DNA double-strand break repair pathways in MyD88 knockout mice, which was lower than that observed in control mice. It has been suggested that γ-H2ax foci are functionally linked to apoptosis after cytotoxic treatments.26, 27 Other work showed that radiosensitive tumor cells retained γ-H2ax longer than did radioresistant cells following exposure to radiation.28 These findings suggest that MyD88 is involved in the maintenance of DNA double-strand breaks, possibly by inhibiting their repair. Interestingly, both the non-homologous end-joining and homologous recombination repair pathways were over-activated in the absence of MyD88, suggesting that MyD88 does not exert a specific inhibitory effect on either pathway. Furthermore, we showed that the preservation of radiation-induced DNA double-strand breaks under the influence of MyD88 is followed by apoptosis of those cells. From these data, we suggest that epithelial stem cell fate following radiation is regulated by MyD88, as the effects on double-strand breaks and apoptosis were followed by less-severe GI syndrome and crypt loss in knockout mice.
We did not measure the effects of the antibiotic treatment on the abundance or species of microbes in the mice. However, a previous publication showed that the cocktail of antibiotics we used produced very significant reductions in bacteria across a broad spectrum.14 Thus, we do not know if the observed phenomena are explained by an overall change in microbial numbers or by more specific effects of some species. It is also possible that the beneficial effects of antibiotics in our experiments were in part due to the prevention of systemic sepsis that could occur due to bacterial translocation following radiation-induced colonic injury.
From our data, we cannot ascertain whether the effects of microbes or MyD88 on colonic radiation responses are epithelial cell-autonomous or dependent on its role in other cells. However, the latter mechanism is perhaps more likely as evidence has already accumulated that the responses of cancer cells to radiation injury are influenced by MyD88-dependent signaling in other cells in their microenvironment.29 Other work has shown that radiation damages the microvasculature of the intestinal mucosa that resulted in perfusion defects that contribute to epithelial cell apoptosis.30 Our results show that MyD88 promotes colonic radiation injury, but prior work has shown that the systemic administration of two TLR ligands has an opposite radioprotective effect. Bacterial lipopolysaccharide, a ligand for TLR-4, protects crypts from radiation,31, 32 whereas flagellin, a ligand for TLR-5 is radioprotective to the intestinal epithelium in a mechanism involving MyD88-dependent signaling in bone marrow-derived cells.33, 34 Similar results have recently been shown for a TLR-9 ligand and for a probiotic acting via TLR-2 and cyclo-oxygenase.35, 36 It thus appears that selective systemic activation of TLR-4, -5 or -9 can decrease the radiation-induced apoptosis of intestinal epithelial cells, acting via MyD88 and NF-κB, but that in the absence of pre-radiation activation, MyD88 has a different role, promoting radiation-induced apoptosis. These findings point to the importance of the crypt microenvironment in the regulation of radiation responses, and suggest that MyD88, probably acting in multiple cell types in response to different TLR ligands or cytokines such as interleukin-1 or -18, regulates intestinal epithelial cell fate. Further work will be needed to explore these possibilities.
Our observations on the functions of MyD88 in colonic radiation injury provide interesting parallels to other models of colon disease that have been previously reported. For example, in colitis induced by feeding dextran sulfate sodium14, 23 or by infection with C. rodentium,37, 38 significantly lower acute inflammation was observed in the absence of MyD88. However, less-severe colitis in MyD88 knockout mice was accompanied by worse bleeding with dextran sulfate sodium, or bacteremia with C. rodentium. This finding is similar to our observation of the relative absence of mucosal inflammation 4.5 days following radiation of MyD88 knockout mice. Other work showed that the development of colitis-associated tumors in IL-10 knockout mice was attenuated in the absence of MyD88,39 as was tumorigenesis in ApcMin/+ mice.25 The pro-tumorigenic effect of MyD88 may reflect its role in regulating the inflammatory makeup of the crypt microenvironment. Taken together with our results, a critical role for MyD88 can be found in controlling the responses of colonic crypts to many types of injuries.
The significance of our findings relates to the possibility of therapeutic manipulation of the colonic microbial-mucosal immune interface to mitigate the unwanted effects of radiation on colonic epithelium during radiotherapy of extra-intestinal tumors, or following exposure to single high-radiation doses in accidents or warfare. Our results suggest that the reduction of colonic microbial signaling or its modulation might protect the colon from the damaging effects of radiation. Promising results with probiotics in the prevention of radiation proctitis40, 41 suggest that this paradigm may be valid.
Materials and methods
Animals and radiation treatment
The animals used in these studies were MyD88-deficient mice42 or wild-type C57Bl/6J mice (the Jackson Laboratory, USA). Mice were kept under standard conditions (ambient temperature 22±2 °C, air humidity 40–70% and a 12/12-h light/dark cycle) and continuous access to a standard diet and water. Animal protocols were approved by the NUI, Galway Committee for Animal Studies. To lower the abundance of colonic microbial colonization, established protocols using 4 weeks treatment with combination of antibiotics including neomycin, metronidazole, ampicillin and vancomycin in drinking water were applied to normal mice.14 Mice received 14 Gy dose of whole-body radiation, using a 137 Cesium γ-source radiator at a dose rate of 23.5 Gy min−1 (Mainance Engineering, Waterlooville, UK). Non-anesthestized mice were immobilized in a specific steel chamber, and radiation dose was monitored by a dose rate meter. The mice were killed at various time points after radiation. Segments of colons were excised 4 cm from the distal rectum, fixed in 4% paraformaldehyde for 16–24 h, penetrated with 70% ethanol and paraffin-embedded.
Crypt survival assay
The crypt microcolony assay4 was employed to evaluate the radiosensitivity of colonic crypts 4.5 days after whole-body radiation. Five-micrometer sections were deparaffinised, rehydrated and stained with haematoxylin & eosin (H&E). The total number of intact crypts per section was quantified under light microscopy (Olympus, Southend on Sea, UK).
Immunofluorescence
Five-micrometer colon tissue sections were permeabilised with 0.1% Triton X-100/0.1% sodium citrate solution for 10 min, blocked in 1% normal goat serum in PBS for 30 min, washed in PBS, and then respectively immunostained using either antibodies to phospho-Histone H2ax-Ser139 (1:200, JBW301, Upstate Millipore corporation, Darmstadt, Germany), proliferating cell nuclear antigen (1:100, ab2426, Abcam), Ku70–80 (1:100, ab3108, Abcam), phospho-s1981-Atm (1:100, ab2888, Abcam) or Rad51 (1:100, ab63801, Abcam). Immunostaining was visualized using mouse- or rabbit- secondary antibodies conjugated to Alexa-Fluor-488 (Invitrogen) and sections were mounted using Glycerol gelatin mounting media (Sigma) plus 4′, 6-diamidino-2-phenylindole (DAPI) (Sigma). A negative control without primary antibody was performed in all cases. Samples were analyzed under a fluorescence microscope (Olympus). The number of positive nuclei per crypt section was quantified by counting the number of 50–100 well-oriented intact crypts with a visible lumen in adjacent nuclei.
Apoptosis analysis
TUNEL assay was performed using in situ Cell Death Detection kit, AP (Roche, Dublin, Ireland), as recommended. The number of TUNEL-positive nuclei per crypt section was quantified by counting 50–100 well-oriented intact crypts with a visible lumen in adjacent nuclei.
Statistical analysis
Statistical significance was evaluated by Student’s t-test or analysis of variance as appropriate, with a P-value of <0.05 considered statistically significant.
References
Wilson JW, Pritchard DM, Hickman JA, Potten CS . Radiation-induced p53 and p21WAF-1/CIP1 expression in the murine intestinal epithelium: apoptosis and cell cycle arrest. Am J Pathol 1998; 153: 899–909.
Nelson WG, Kastan MB . DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol 1994; 14: 1815–1823.
Potten CS . A comprehensive study of the radiobiological response of the murine (BDF1) small intestine. Int J Radiat Biol 1990; 58: 925–973.
Withers HR, Elkind MM . Microcolony survival assay for cells of mouse intestinal mucosa exposed to radiation. Int J Radiat Biol Relat Stud Phys Chem Med 1970; 17: 261–267.
Cohn SM, Schloemann S, Tessner T, Seibert K, Stenson WF . Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J Clin Invest 1997; 99: 1367–1379.
Houchen CW, Stenson WF, Cohn SM . Disruption of cyclooxygenase-1 gene results in an impaired response to radiation injury. Am J Physiol Gastrointest Liver Physiol 2000; 279: G858–G865.
Tessner TG, Muhale F, Riehl TE, Anant S, Stenson WF . Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism involving AKT activation and bax translocation. J Clin Invest 2004; 114: 1676–1685.
Onoue M, Uchida K, Yokokura T, Takahashi T, Mutai M . Effect of intestinal microflora on the survival time of mice exposed to lethal whole-body gamma irradiation. Radiat Res 1981; 88: 533–541.
Crawford PA, Gordon JI . Microbial regulation of intestinal radiosensitivity. Proc Natl Acad Sci USA. 2005; 102: 13254–13259.
Fitzgerald KA, Chen ZJ . Sorting out Toll signals. Cell 2006; 125: 834–836.
Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A . The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J 2000; 19: 3325–3336.
Roses RE, Xu M, Koski GK, Czerniecki BJ . Radiation therapy and Toll-like receptor signaling: implications for the treatment of cancer. Oncogene 2008; 27: 200–207.
Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T . Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science 2002; 298: 1424–1427.
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R . Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004; 118: 229–241.
Cai WB, Roberts SA, Bowley E, Hendry JH, Potten CS . Differential survival of murine small and large intestinal crypts following ionizing radiation. Int J Radiat Biol 1997; 71: 145–155.
Rogakou EP, Boon C, Redon C, Bonner WM . Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146: 905–916.
Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM . Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res 2002; 158: 486–492.
Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 2006; 173: 195–206.
Kurose A, Tanaka T, Huang X, Halicka HD, Traganos F, Dai W et al. Assessment of ATM phosphorylation on Ser-1981 induced by DNA topoisomerase I and II inhibitors in relation to Ser-139-histone H2AX phosphorylation, cell cycle phase, and apoptosis. Cytometry A 2005; 68: 1–9.
Smith GC, Divecha N, Lakin ND, Jackson SP . DNA-dependent protein kinase and related proteins. Biochem Soc Symp 1999; 64: 91–104.
Haaf T, Golub EI, Reddy G, Radding CM, Ward DC . Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci USA 1995; 92: 2298–2302.
Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med 2010; 207: 1625–1636.
Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007; 133: 1869–1881.
Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 2005; 288: G1055–G1065.
Rakoff-Nahoum S, Medzhitov R . Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 2007; 317: 124–127.
Banath JP, Olive PL . Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res 2003; 63: 4347–4350.
Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell 2006; 23: 121–132.
Taneja N, Davis M, Choy JS, Beckett MA, Singh R, Kron SJ et al. Histone H2AX phosphorylation as a predictor of radiosensitivity and target for radiotherapy. J Biol Chem 2004; 279: 2273–2280.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13: 1050–1059.
Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 2001; 293: 293–297.
Riehl T, Cohn S, Tessner T, Schloemann S, Stenson WF . Lipopolysaccharide is radioprotective in the mouse intestine through a prostaglandin-mediated mechanism. Gastroenterology 2000; 118: 1106–1116.
Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci USA. 2004; 101: 2452–2457.
Vijay-Kumar M, Aitken JD, Sanders CJ, Frias A, Sloane VM, Xu J et al. Flagellin treatment protects against chemicals, bacteria, viruses, and radiation. J Immunol 2008; 180: 8280–8285.
Burdelya LG, Krivokrysenko VI, Tallant TC, Strom E, Gleiberman AS, Gupta D et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science 2008; 320: 226–230.
Saha S, Bhanja P, Liu L, Alfieri AA, Yu D, Kandimalla ER et al. TLR9 agonist protects mice from radiation-induced gastrointestinal syndrome. PLoS One 2012; 7: e29357.
Ciorba MA, Riehl TE, Rao MS, Moon C, Ee X, Nava GM et al. Lactobacillus probiotic protects intestinal epithelium from radiation injury in a TLR-2/cyclo-oxygenase-2-dependent manner. Gut 2012; 61: 829–838.
Gibson DL, Ma C, Bergstrom KS, Huang JT, Man C, Vallance BA . MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol 2008; 10: 618–631.
Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D . TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J Immunol 2007; 179: 566–577.
Uronis JM, Muhlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C . Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One 2009; 4: e6026.
Fuccio L, Guido A, Eusebi LH, Laterza L, Grilli D, Cennamo V et al. Effects of probiotics for the prevention and treatment of radiation-induced diarrhea. J Clin Gastroenterol 2009; 43: 506–513.
Maria-Aggeliki KS, Nikolaos KL, Kyrias GM, Vassilis KE . The potential clinical impact of probiotic treatment for the prevention and/or anti-inflammatory therapeutic effect against radiation induced intestinal mucositis. A review. Recent Pat Inflamm Allergy Drug Discov 2009; 3: 195–200.
Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998; 9: 143–150.
Acknowledgements
We would like to thank Professor Peter Dockery and Mr Mark Canney of Anatomy Department in National University of Ireland, Galway for their assistance in histology and members of our laboratory for helpful comments on this article. MyD88 knockout mice were generously provided by Dr Padraic Fallon, Institute for Molecular Medicine, St James’ Hospital, Dublin. This work was funded by Health Research Board and Science Foundation Ireland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lai, X., Egan, L. Suppression of radiation-induced DNA double-strand break repair by MyD88 is accompanied by apoptosis and crypt loss in mouse colon. Oncogenesis 2, e62 (2013). https://doi.org/10.1038/oncsis.2013.22
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2013.22
Keywords
This article is cited by
-
Radioprotective potential of probiotics against gastrointestinal and neuronal toxicity: a preclinical study
Clinical and Translational Oncology (2023)
-
Hydrogen-water ameliorates radiation-induced gastrointestinal toxicity via MyD88’s effects on the gut microbiota
Experimental & Molecular Medicine (2018)
-
Blockade of TLR3 protects mice from lethal radiation-induced gastrointestinal syndrome
Nature Communications (2014)
