
Key Points
-
Peripheral neuropathy is a serious but often neglected complication of diabetes mellitus
-
Schwann cells support the structural and functional integrity of nerves, so their damage as a result of the metabolic consequences of diabetes adversely affects axons
-
High polyol pathway flux, oxidative stress and inflammation are the main pathways activated in Schwann cells during diabetic neuropathy
-
Disruption of Schwann cell metabolism by hyperglycaemia and/or dyslipidaemia results in accumulation of neurotoxic intermediates that confer axonal and vascular vulnerability to injury
-
Microvascular changes within the endoneurium create a hypoxic environment that has the potential to disrupt Schwann cell function, promoting activation of inflammatory cascades that lead to neurodegeneration
Abstract
The prevalence of diabetes worldwide is at pandemic levels, with the number of patients increasing by 5% annually. The most common complication of diabetes is peripheral neuropathy, which has a prevalence as high as 50% and is characterized by damage to neurons, Schwann cells and blood vessels within the nerve. The pathogenic mechanisms of diabetic neuropathy remain poorly understood, impeding the development of targeted therapies to treat nerve degeneration and its most disruptive consequences of sensory loss and neuropathic pain. Involvement of Schwann cells has long been proposed, and new research techniques are beginning to unravel a complex interplay between these cells, axons and microvessels that is compromised during the development of diabetic neuropathy. In this Review, we discuss the evolving concept of Schwannopathy as an integral factor in the pathogenesis of diabetic neuropathy, and how disruption of the interactions between Schwann cells, axons and microvessels contribute to the disease.
Similar content being viewed by others


Main
The incidence of diabetes mellitus has increased dramatically over the past two to three decades. According to the International Diabetes Federation Diabetes Atlas, 415 million people worldwide had diabetes in 2015, and this number is expected to grow by 5% annually, predominantly as a result of increasing prevalence of type 2 diabetes. Furthermore, an estimated 100 million Europeans and 80 million Americans have impaired glucose tolerance or prediabetes. Few people die from acute diabetes in countries with comprehensive health care systems, but the disease is inflicting a huge medical, social and economic burden on society owing to the need for lifelong treatment of its systemic consequences and the insidious development of multi-organ damage.
Peripheral neuropathy (Boxes 1,2) is a common but often neglected complication of long-term diabetes, and the lack of treatment options reflects an incomplete understanding of the pathogenic mechanisms. Hyperglycaemia is generally accepted as the primary pathogenic insult in type 1 and type 2 diabetic neuropathy, although roles are emerging for other factors, such as impaired insulin signalling, hypertension and dyslipidaemia (particularly for type 2 diabetes), which might precede overt hyperglycaemia1. Many preclinical studies, and occasional clinical studies, have indicated that diabetic neuropathy — like diabetic nephropathy and retinopathy — results from microvascular disease, with a focus on axonal degeneration as a consequence of ischaemia and/or hypoxia. This mechanism, however, is likely to be only one aspect of a more complex pathogenesis.
The earliest descriptions of pathology in diabetic neuropathy indicated that Schwannopathy accompanied axonal degeneration. The majority of clinical and basic research in diabetic neuropathy since then has focused on the effects on neurons. However, accumulating data from research into the development and regeneration of the PNS has identified Schwann cells as equally indispensable components that maintain neuronal structure and function, nourish axons, and promote survival and growth upon injury. The early reports from 1979 that demonstrated morphological changes in Schwann cells in human diabetic neuropathy2 are now supported by an increased awareness of molecular alterations in Schwann cells during diabetes3. Schwann cells express a wide range of receptors and, when they sense insults or danger signals, they upregulate synthesis and secretion of factors that stimulate neuroprotection, regrowth and remyelination, or factors that aggravate disease phenotypes4. The most recent studies have demonstrated that Schwann cells regulate many aspects of axonal function, so that disruption of their metabolism by diabetes results in the accumulation of neurotoxic intermediates and compromises production of neuronal support factors, contributing to axonal degeneration, endothelial dysfunction and diabetic neuropathy.
Here, we review the interactions between Schwann cells, axons and microvessels that are known to contribute to the pathogenesis of diabetic neuropathy, with an emphasis on the mechanisms by which Schwannopathy might make axons and vessels vulnerable to injury.

Schwann cells and their interactions
Schwann cells are the most abundant cells in the PNS, and include two broad categories: myelinating and nonmyelinating Schwann cells. Together, the two types ensheath all axons of peripheral nerves. Myelinating Schwann cells individually wrap large-calibre axons (>1 μm diameter), leaving unmyelinated gaps (nodes of Ranvier) between adjacent Schwann cells that facilitate saltatory axonal conduction. Schwann cell myelin comprises various lipids and proteins compacted into a multilayer structure that provides electrical insulation. Nonmyelinating Schwann cells do not wrap axons, but small sensory axons (<1 μm diameter) are embedded in grooves in their membranes. Several small nociceptive axons can be embedded into the same Schwann cell, forming a Remak bundle3.
Schwann cells derive from neural crest cells that first develop into Schwann cell precursors (SCPs) that associate with the first compact columns of peripheral axons. These proto-nerves are devoid of all other structural elements that characterize mature nerves, such as connective tissue and blood supply, and the SCPs connect with each other to envelop large numbers of axons5. During embryonic development, Schwann cells facilitate neuronal survival via neurotrophin secretion; to migrate and form myelin. Schwann cells require axonal secretion of neurotrophins and must express neurotrophin receptors6,7. Schwann cell proliferation is stimulated by secretion of vascular endothelial growth factor (VEGF) from ganglion neurons, which also stimulates axonal outgrowth and formation of microvessels8. The development of peripheral nerves, therefore, depends on signalling between Schwann cells, the axon and microvessels in a complex temporal and spatial manner.
During Schwann cell maturation and alignment, multiple receptors, ligands and adhesion molecules orchestrate neuronal sprouting and targeting. Among these factors, the neurotrophins β-nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), NT-3 and NT-4/5 signal via two structurally unrelated types of receptors: the low-affinity neurotrophin receptor p75NTR and the tropomyosin-related kinase receptors TrkA, TrkB and TrkC. In the PNS, neurotrophins and their receptors are expressed by both Schwann cells and sensory neurons. Activation of the two receptor types can elicit opposing effects — survival and differentiation via Trks, or apoptosis via p75NTR9,10. Adding to the complexity, TrkA and TrkC can act as dependence receptors that elicit apoptosis when unstimulated11, and the biological effect of secreted proneurotrophins might depend on the expression ratio of Trk receptors and p75NTR12,13 and on expression of p75NTR co-receptors14,15.
Physical or metabolic damage to adult peripheral nerves induces rapid and robust changes in the synthesis of neurotrophins in neurons and Schwann cells to guide and support regeneration16,17. For example, hypoxia upregulates VEGF production after nerve injury, initiating migration of Schwann cells18. VEGF also stimulates formation of endoneurial blood vessels into hypoxic areas, directing Schwann cells to bridge the gap between the proximal and distal nerve stumps18. However, studies have shown that diabetes reduces expression of the Schwann-cell-derived neurotrophic factors ciliary neuronotrophic factor (CNTF)19 and sonic hedgehog20, which act on neurons and endothelial cells, respectively. Moreover, Schwann cells that have been removed from mice with diabetes and maintained under hyperglycaemic conditions in vitro to model nerve injury also exhibit impaired production of NGF and NT-3 (Ref. 21). The fact that Schwann cells ensheath the vast majority of axonal plasma membranes means that changes in their production of signalling molecules are likely to influence axons. Schwann cells are also in close contact with the basal lamina and the endoneurium, and receptor-mediated interactions between Schwann cells and these structures are known to affect several Schwann cell functions, including radial sorting and myelination22. Overall, these complex molecular interactions suggest that disruption of Schwann cell functions, including those triggered by diabetes, could have widespread consequences for peripheral nerve structure and function.
Schwann cells in diabetic neuropathy
Morphological changes. Nerve biopsy samples from patients with diabetes reveal overall fibre loss; degenerating fibres and clusters of regenerating axons are both present. Reduced axonal diameters, which suggest impaired maturation or atrophy, are frequently reported in rat models of diabetes, but are not a notable feature in many mouse models or humans with diabetes23,24. Whether axonopathy or Schwannopathy develops first in diabetic neuropathy25 has long been debated, and a single nerve biopsy sample is not sufficient to determine whether Schwann cells undergo independent structural damage during diabetes or are responding to axonal degeneration. However, teased fibre sural nerve preparations from humans, cats and rodents with diabetes indicate morphological changes in the myelin sheath — ranging from thin myelin that suggests cycles of demyelination and remyelination, to full segmental demyelination — in the presence of an apparently normal axon, indicating that Schwannopathy can develop independently of axonopathy2,3,26,27,28 (Fig. 1). Teased fibres from the same nerves can, however, exhibit predominantly axonal pathology, indicating that diabetes affects both cell types independently, as well as disrupting their interactions. Degenerative changes in nerve fibres are accompanied by the presence of enlarged mitochondria with effaced cristae and numerous vacuoles in the Schwann cells29, an observation that is consistent with emerging evidence of mitochondrial dysfunction and damage in axons and Schwann cells in animal models of diabetes30. Other diverse pathological markers, ranging from nonspecific signs of cellular stress through to overt cellular degeneration, are also present in Schwann cells from both animal models of diabetes and human nerve biopsy samples. Nonspecific Schwann cell changes include glycogen inclusions, lysosomal inclusions, accumulation of lipid droplets in the cytoplasm, cytoplasmic expansion, and an increased number of plasmalemmal vesicles3,31. The Schwann cell basal lamina also reduplicates and thickens3,31. In one study in a rat model of type 1 diabetes, levels of heparan sulfate in the basement membranes of Schwann cells in the dorsal root ganglia gradually decreased with increasing disease duration, with a reciprocal increase in deposited laminin. The authors of that study speculated that the early increase in heparan sulfate can exacerbate propagation of pain, whereas in advanced stages of the disease, basement membrane composition is maintained by overproduction of laminin in the Schwann cells, thereby blocking pain transmission and thermal neuronal stimuli; this hypothesis helps to explain the bimodal pain profile in diabetic neuropathy32.

Diabetic neuropathy has a complex pathogenesis involving interaction of axonopathy, schwannopathy and microvasculopathy. The figure shows the anatomical organization of myelinated and unmyelinated axons within nerve fascicles. Their nutrient supply is secured via endoneurial capillaries which, together with the perineurial membrane, form the blood–nerve barrier. a | Human skin biopsy samples immunostained with PGP9.5 to show progression of peripheral nerves from the dermis into the epidermis, where they exist as small unmyelinated C-fibres (scale bar 40 μm). Left panel shows loss of fibres in a patient with diabetic neuropathy and right panel shows fibres in a healthy individual. b | Changes in axons and myelin in diabetic neuropathy, showing degeneration of Schwann cells and nerve fibres, culminating in nerve and intraepidermal fibre loss. c | Endoneurial capillaries from patients with diabetes. Top panel shows a capillary from a patient without diabetic neuropathy, and bottom panel shows a capillary from a patient with neuropathy, in which endothelial cell hyperplasia and basement membrane thickening have reduced the size of the capillary lumen. d | Narrowing of individual capillaries might not prevent blood from passing through the endoneurial capillary bed per se, but the resulting increase in velocity of blood through endoneurial functional shunts or epineurial arteriovenous shunts prevents efficient oxygen extraction, causing hypoxia. Panel a courtesy of Dr Páll Karlsson, Danish Pain Research Center, Department of Clinical Medicine, Aarhus University, Denmark.
Polyol pathway flux and aldose reductase. Over the past 50 years, circumstantial33 and experimental34 evidence has established hyperglycaemia-driven increases in flux through the polyol pathway as the best-understood pathogenic mechanism of diabetic neuropathy. Numerous preclinical studies have demonstrated that inhibition of aldose reductase (the first enzyme in the polyol pathway) prevents almost all manifestations of neuropathy. Studies in which expression of enzymes involved in the polyol pathway has been manipulated have placed a particular focus on the metabolic consequences of excessive metabolism of glucose to sorbitol owing to high activity of aldose reductase35,36,37,38.
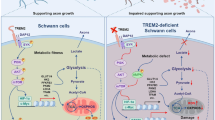
On the basis of the involvement of the polyol pathway, the perception of diabetic neuropathy as a microvascular complication of diabetes has been underpinned by immunocytochemical studies that showed localization of aldose reductase to the endothelial cells of epineurial and dorsal root ganglion blood vessels and to perivascular sympathetic axons39,40,41. Indeed, recent data implicates aldose reductase activity in the production of endothelial proinflammatory and prothrombotic signals42, a pathogenic cascade that is currently receiving close attention in the context of diabetic neuropathy42 and is discussed in detail below (see the section 'Microvascular changes in Schwann cells'). However, within the endoneurium, aldose reductase is restricted to the myelinating Schwann cells of somatic nerves and to satellite glial cells in the dorsal root ganglia39,40,41 (Box 3). This localization implies that aldose reductase dysfunction does not only alter vascular function, although this observation has led to something of a conundrum with regard to the pathogenic role of the enzyme: demyelination can be a feature of human diabetic neuropathy28,43, but diabetic rodents rarely exhibit marked Schwann cell pathology or demyelination in peripheral nerves until long after nerve dysfunction is detected44,45, unless neuropathy is accompanied by other stressors, such as hypertension46. Nevertheless, many rodent models of diabetes exhibit indicators of Schwann cell dysfunction, including reduced expression of myelin-associated proteins47 and Schwann cell-derived trophic factors, such as CNTF19 and desert hedgehog20,48. These perturbations have the potential to affect both neuronal49 and vascular48 function, and changes in CNTF expression are prevented by aldose reductase inhibition50. In addition, studies in cultured Schwann cells have suggested that increased flux through the polyol pathway drives Schwann cells towards an immature phenotype51. Furthermore, galactose intoxication, which increases polyol pathway activity52, can produce Schwann cell pathology that parallels the pathology seen in human diabetic neuropathy29,53, and can be prevented by inhibition of aldose reductase. This observation illustrates the cytotoxic capacity of increased flux through the aldose reductase component of the polyol pathway, and the fact that extreme stress is required to damage Schwann cells but does not necessarily evoke demyelination. Hyperglycaemia-induced increases in polyol pathway flux in Schwann cells might, therefore, be a primary pathogenic mechanism that impairs their structural, metabolic and trophic support of axons and blood vessels before (or without) precipitating frank demyelination (Fig. 2). Despite being highly controversial as a model for diabetic neuropathy, galactose intoxication elicits extreme osmotic forces and oedema, thereby leading to compression of the microvasculature and highlighting the importance of endoneurial hypoxia54.

Hyperglycaemia and dyslipidaemia ultimately lead to reduction of neuronal support from Schwann cells and microvessels. In Schwann cells, RAGE (receptor for advanced glycosylation end products) signalling leads to increased glucose metabolism by aldose reductase, which generates local oxidative damage, causes inflammation and drives cells to an immature phenotype. It also affects mitochondrial function, which increases oxygen consumption, and reduces production of desert hedgehog (DHH), which affects endothelial cell function. Endothelial cells also express aldose reductase, and increased polyol pathway flux activates proinflammatory and prothrombotic pathways that reduce nerve blood flow. Disruption of neuronal support by Schwann cells and the vascular system contributes to neuropathy, in conjunction with the direct effects of diabetes on neurons themselves.
Aldose reductase inhibitors (ARIs) are highly effective in animal models of diabetic neuropathy55,56, but their clinical development as a therapy in humans has largely stalled as a result of multiple inconclusive or negative studies57. Poor trial design and choices of end points, and limited drug potency have all been suggested as contributing factors in this translational failure58. Nevertheless, the regulatory approval of the ARI epalrestat in Japan remains the only example of a drug being licensed to treat diabetic neuropathy, and a recent trial of an ARI in people with mild to moderate neuropathy has re-emphasized earlier findings59 that this therapeutic approach can mitigate against some manifestations of diabetic neuropathy60. In the section 'Microvascular changes and Schwann cell function' below, we discuss polyol pathway activity and ARIs from the perspective of substrate availability for nerve energy metabolism.
Oxidative stress and mitochondrial dysfunction. A contribution of oxidative stress and mitochondrial disorders in Schwann cells to neuronal dysfunction during diabetes is becoming increasingly evident. Chronic hyperglycaemia is widely accepted as a trigger of excessive production of reactive oxygen species (ROS) in all cells, as is the fact that this toxic process is exacerbated by a concomitant reduction in endogenous antioxidant defences61. For example, genetic variations and polymorphisms in endogenous antioxidant enzymes have been associated with an increased susceptibility to diabetic neuropathy62. Potential sources of ROS in peripheral nerves include the polyol pathway, the mitochondrial electron transport chain, RAGE (receptor for advanced glycosylation end products) signalling, and NADPH oxidases and nitric oxide synthases63,64,65. Schwann cells are increasingly being recognized as an important site of ROS production, which, in this context, affects Schwann cell function and their interactions with other cell types within the nerve trunk.
Several lines of evidence indicate that oxidative stress in Schwann cells contributes to nerve damage in diabetic neuropathy. Reports that ARI treatment prevents oxidative damage to lipids and DNA in nerves of rats with diabetes66,67 indicate that increased flux through the polyol pathway contributes to oxidative stress, and implicates Schwann cells in causing this oxidative stress, as they express aldose reductase. Moreover, increased flux through the polyol pathway depletes cytosolic NADPH and subsequently reduces levels of glutathione (an important antioxidant), leaving neurons and Schwann cells vulnerable to toxic oxygen free radicals and peroxides61. Altered RAGE signalling in Schwann cells is another proposed mechanism of hyperglycaemia-induced free radical-mediated nerve damage. Excess formation of advanced glycation end-products (AGE) as a result of excess glucose activates RAGE and precipitates ROS formation68 (Fig. 2). Sural nerve biopsy samples from patients with diabetes have revealed overexpression of RAGE in Schwann cells69, and RAGE signalling has been implicated in several complications of diabetes70. Furthermore, AGE-induced modifications to key proteins (such as collagen), lipids and nucleic acids have the potential to alter the structure and function of Schwann cells, with detrimental effects on the axons they ensheath, leading to potentiation of diabetic neuropathy71. Finally, Schwann cells have been identified as the focus of injury induced by free radicals and oxidants and mediated by activation of poly(ADP-ribose) polymerase, a downstream effector of oxidative and nitrosative stress that also contributes to formation of superoxide anion radicals and peroxynitrite in diabetic peripheral nerves72. Hyperglycaemia can increase production of peroxynitrite in all nerve cells, and preoxynitrite in turn nitrates tyrosine residues to produce 3-nitrotyrosine, a fingerprint of nitrosative stress; levels of 3-nitrotyrosine and inducible nitric oxide synthase have been shown to be increased in Schwann cells73,74,75.
Impaired mitochondrial function has been implicated in many complications of diabetes. Initial studies in cultured endothelial cells indicated that hyperglycaemia increased flux through the electron transport chain, leading to ROS formation76, but these observations have been superseded by descriptions of impaired mitochondrial activity and bioenergetics in cultures of adult sensory neurons and in animal models of chronic diabetic neuropathy77 and nephropathy78. Mitochondria do not seem to be a source of excess ROS in peripheral axons79, but several findings have shifted the focus to Schwann cell mitochondria and their impact on axonal function. Exposure of Schwann cells to high glucose levels in vitro causes oxidative stress that is accompanied by overactivation of caspase-9 and apoptosis regulator BAX, and decreased levels of apoptosis regulator Bcl-2 (Ref. 80), changes that indicate mitochondrial internal stress. In an important study published in 2011, targeted disruption of Schwann cell mitochondria in a transgenic mouse model critically affected neuronal survival and produced a peripheral neuropathy resembling that caused by diabetes81. Subsequent work demonstrated that mitochondrial deficits activated a maladaptive stress response mediated by haem-regulated inhibitor kinase, and that acylcarnitines that were released from Schwann cells on mitochondrial disruption induced axonal degeneration82, emphasizing the importance of Schwann cell mitochondrial homeostasis in axon–glia interactions83.
Hyperglycaemia has also been shown to drive remodelling of the Schwann cell mitochondrial proteome that leads to increased expression of the α and β subunits of ATP synthase, and to cause suboptimal respiratory capacity by increasing the overall rate of oxygen consumption, suggesting that high glucose levels contribute to mitochondrial dysfunction and decrease the efficiency of oxidative phosphorylation in Schwann cells84. In a study published in 2016, mitochondrial damage, with upregulation of multiple subunits of complexes I, III, IV, V and of mitochondrial Rho GTPase 1, was described in a rodent model of type 1 diabetes85. These changes were observed only in peripheral nerves and not in the sensory or trigeminal ganglia, suggesting that they reflect disrupted Schwann cell metabolism.
Lipid metabolism and inflammation. Diabetes is associated with alterations in lipid levels in the circulation and nerves, and Schwann cell dysfunction has been linked to altered lipid metabolism and downstream inflammatory consequences. Accumulation of triglycerides, cholesterol and free fatty acids in blood plasma in diabetes seems to drive lipid-mediated neuropathology via mechanisms that are incompletely understood but might involve oxidative and inflammatory pathways in Schwann cells86. In rodents, a high-fat diet causes accumulation of oxidized lipids and activation of lipoxygenases in peripheral nerves, suggestive of a prediabetic condition24. Moreover, accumulation of oxidized LDLs in peripheral nerves promotes oxidative stress that has been associated with a low nerve conduction velocity and sensory deficits87. Cell culture studies suggest that Schwann cells are susceptible to lipotoxicity from free fatty acids, possibly as a result of lysosomal dysfunction88,89. Furthermore, evidence from a mouse model of peripheral neuropathy secondary to Schwann cell mitochondrial dysfunction suggests that disruption of Schwann cell mitochondria causes lipid metabolism to shift away from fatty acid synthesis toward lipid oxidation, resulting in early depletion of myelin lipid components and accumulation of acylcarnitine lipid intermediates, leading to axonal degeneration and neuropathy83. Perturbation of lipid metabolism has also been found in the peripheral nerves of mice with streptozotocin (STZ)-induced diabetes; these perturbations include a reduction in short-chain triacylglycerols, changes in major structural and/or membrane lipids, and diminished levels of palmitic, stearic and eicosanoic fatty acids85,90. Exposure of human Schwann cells to high extracellular glucose levels reduced their synthesis of phospholipids, and this effect was counteracted by an ARI, implicating high rates of glucose metabolism via the polyol pathway in dysregulation of Schwann cell lipid metabolism91.
In diabetes, lipoproteins circulating in the plasma are exposed to an oxidizing environment and can be modified by consequent glycation. Schwann cells express several Toll-like receptors (TLRs) and RAGE92,93, to which the modified LDLs can bind and drive an inflammatory response94. After an injury, such as those triggered by hyperglycaemia or modified LDLs, intracellular cascades can be activated in Schwann cells, including nuclear translocation of NF-κB and consequent expression of various cytokines and chemokines, similar to the scenario after traumatic nerve damage. These events contribute to Wallerian degeneration and emphasize the potential involvement of Schwann cells in the subclinical inflammation that is observed in patients with type 2 diabetes and is detectable as increased circulating levels of C-reactive protein and IL-6, which are consistently associated with polyneuropathy95. Furthermore, via production of cytokines and chemokines, Schwann cells might contribute to immune cell recruitment4,96 and nerve inflammation in diabetes.
Macrophage infiltration and proliferation have been observed in nerves from mouse models of diabetes97,98, and upregulation of proinflammatory genes during activation of inflammatory pathways in the PNS99,100,101 further implicates involvement of RAGE-mediated Schwann cell involvement in the inflammatory response. One upregulated gene encodes the S100 calcium binding protein A8/A9 (Ref. 101), which is overexpressed in patients with type 2 diabetes102. This protein has been shown to stimulate a local inflammatory response via interaction with RAGE103, leading to activation of mitogen-activated protein kinases (MAPKs), NF-κB and apoptosis104,105. Similarly, MAPKs (such as p38 MAPK, MAPK1, MAPK3, MAPK8 and MAPK10) are activated by high glucose levels in human Schwann cell cultures106; p38 is of particular interest, as it is a negative regulator of Schwann cell differentiation and myelination107.
Some evidence also suggests that Schwann cells and T cells have reciprocal effects on each other in diabetes. A study published in 2013 suggested that recruitment of T cells into the nerves of patients with diabetic neuropathy is induced by production of the chemokines CXCL-9, CXCL-10 and CXCL-11 by glucose-stimulated Schwann cells108. Conversely, infiltrating CD8+ T cells have been shown to mediate Schwann cell cytotoxicity by activating apoptosis, thus contributing to progression of neuropathy108. Furthermore, as T cells found in diabetic nerves express high levels of CXCR3, the authors of this study suggest that this receptor is important for migration of CD8+ T cells into the peripheral nerve and, thus, offer a novel potential target for therapy.
Additional factors in diabetes that promote immune cell infiltration of nerves and activation of Schwann cells are accumulation of AGE and the modification of myelin antigenicity via glycosylation of myelin proteins. Activation of Schwann cells in turn promotes their secretion of proinflammatory cytokines, thereby producing a positive feedback mechanism that perpetuates injury109,110. Inflammatory cytokines such as IL-1, tumour necrosis factor (TNF) and IL-17 can be produced by many cell types, including Schwann cells, and these factors sensitize Aδ-fibres and C-fibres, triggering neuropathic pain111,112,113. Indeed, blocking the TNF signalling pathway with a recombinant human TNF receptor–antibody fusion protein has been shown to be beneficial in animal models of diabetic neuropathy, leading to recovery of nerve conduction velocity and increased expression of myelin basic protein109.
Schwann cells in the peripheral nerve express a variety of TLRs, including TLR4 (Ref. 92), which might have a role in the pathogenesis of metabolic-induced retinal impairment and nephropathy114,115. Whether TLR4 in Schwann cells has a role in diabetic neuropathy, however, remains to be elucidated. Interestingly, an interrelationship between TLR4 and the p75NTR receptor has been demonstrated in immune cells. In lipopolysaccharide-stimulated dendritic cells, expression of p75NTR was increased via a TLR4-dependent mechanism. In TLR4-activated dendritic cells, NGF promotes secretion of cytokines, but silencing of p75NTR with small interfering RNA blocks this process116. These observations suggest that the increased expression of p75NTR in myelin sheaths around fibres that are susceptible to axonal degeneration in diabetic neuropathy117 might be related to TLR4 signalling.
Microvascular changes and Schwann cells. Microvascular damage in diabetic neuropathy is likely to affect Schwann cell function by promoting inflammatory cascades such as those described above, and by disrupting their access to oxygen and glucose. The morphology and function of microvessels is altered in nerve biopsy samples from patients with diabetic neuropathy118,119 (Fig. 1); these microvascular changes are particularly severe in endoneurial capillaries, and include capillary basement membrane thickening, loss of pericyte coverage, and endothelial hyperplasia120. Furthermore, microangiopathy can precede development of diabetic neuropathy120,121, and the degree of microvascular changes correlates with the clinical severity of diabetic neuropathy119,120.
Microvascular changes have been thought to elicit nerve damage by limiting nerve blood supply (ischaemia), but evidence now shows that endoneurial hypoxia can be caused by inefficient oxygen extraction alone via capillary dysfunction122. Tissue hypoxia, in turn, can activate the same cellular signalling pathways that inflammation activates123. These pathways can be activated in all cell types that are subjected to hypoxia, including Schwann cells. Low tissue oxygen tension upregulates the expression of hypoxia-inducible factor 1-α (HIF-1α) and NF-κB123, a proinflammatory transcription factor that is present at high levels in peripheral nerves and dorsal root ganglia in experimental diabetic neuropathy124. HIF-1α causes upregulation of NADPH oxidase 2 levels125, a major source of ROS in vessel walls126. NADPH-derived superoxides react with the vasodilator nitric oxide to produce peroxynitrite127, which causes severe nitrosative tissue damage and inactivates tissue plasminogen activator (tPA)128, creating a highly prothrombotic environment. Importantly, elevated tPA levels increase the formation of BDNF from proBDNF, and proBDNF is associated with apoptosis129; this observation is consistent with reports that the levels of BDNF in distal muscles negatively correlate with the severity of neuropathy in diabetic patients130. These mechanisms suggest that microvascular changes are likely to create a hypoxic endoneurial environment that exacerbates oxidative stress and inflammation, and leads to a loss of trophic support for Schwann cells and neurons.
Capillary dysfunction affects the uptake of glucose and oxygen into tissue131, and breakdown of glucose under hypoxic conditions generates far less ATP than does oxidative phosphorylation. When access to oxygen is impaired, therefore, Schwann cells and axons are likely to depend on alternative substrates or metabolic pathways to cover their metabolic needs. Glucose breakdown via the polyol pathway — despite its detrimental long-term effects — is an efficient source of ATP in these conditions. As a result, aldose reductase inhibitors could be detrimental in nerve fibres with severely affected microvessels, as they will limit this alternative source of energy122.
The evidence discussed indicates that in diabetic neuropathy, capillary dysfunction disrupts the function of Schwann cells and axons — and vice versa — by altering the endoneurial microenvironment. In light of this hypothesis, creation of animal models of diabetes that include microvascular damage46, or use of cell culture systems that mimic the diabetic endoneurial microenvironment, might increase the success of translational research.
Nodal disruption and ion channels. The tight association between Schwann cells and axons means that disturbances in Schwann cell function are likely to influence the excitability of large and small fibres and the propagation of action potentials. The resting membrane potential, depolarization and repolarization of axons are mediated by a series of voltage-gated sodium (NaV) channels, potassium (KV) channels, calcium channels and various ligand-gated channels (for example, transient receptor potential channels)132. In myelinated axons, the ion channels are clustered at the nodes of Ranvier to facilitate saltatory conduction, whereas in nonmyelinated axons, the channels are distributed diffusely and almost homogeneously along the axon133,134. Schwann cells are not excitable themselves, but do express ion channels that are involved in forming the nodal and paranodal regions where the neuronal ion channels are clustered, giving them the potential to influence axonal excitability.
The effects of Schwann cell dysfunction on neuronal ion channel function are not known and may be indirect. Impaired mitochondrial function and oxidative stress in Schwann cells (see above) could lead to changes in the distribution of NaV and KV channels at the nodes of Ranvier and the paranodal and juxataparanodal regions, contributing to axonal damage. Mitochondrial dysfunction may also be a consequence of increased intracellular calcium135. A detailed discussion of the role of ion channels in diabetic neuropathy is beyond the scope of this Review, but abnormalities such as paranodal swelling and axon–glial disjunction have been described in diabetic neuropathy in humans, and were proposed as a cause of nodal ion channel redistribution136. NaV channel expression in diabetes can be modified by various mechanisms, including neurotrophic factors137 and methylglyoxal, a metabolite that is upregulated as a result of hyperglycaemia, and has been shown to change the distribution of NaV1.8 (Ref. 138). Together with changes in ion channel function and energy metabolism in the axon itself, such as impaired Na+–K+ ATPase activity in nodal regions of the axon, Schwann cell dysfunction could — owing to the normal function of these cells in saltatory conduction — contribute to the reduction in action potential propagation seen in diabetic neuropathy. In a study published in 2016, mathematical modelling was used to provide evidence that abnormalities in the excitability of sensory and motor axons in patients with type 1 diabetes were the result of reduced nodal Na+ currents, reduced nodal and paranodal K+ conductance, and Na+–K+ ATPase dysfunction139. Evidence also suggests that altered expression of NaV channels, specifically NaV1.7, NaV1.8 and NaV1.9, contributes to the neuronal hyperexcitability and allodynia seen in experimental and clinical diabetic neuropathy52,53,54,55.
Impact of schwannopathy on neurons
As discussed above, hyperglycaemia causes chronic Schwann cell dysfunction, and further evidence suggests that consequent transcriptional changes lead to persistent increases in glycolysis, ROS formation, cellular NADH depletion and altered DNA methylation that contribute to diabetic neuropathy140. This Schwann cell dysfunction has direct effects on neuronal function as a result of myelin disruption, demyelination, changes in axonal conduction, and impaired regeneration.
Myelination of nerve fibres by Schwann cells is essential for rapid saltatory conduction, and insults to the myelin sheath can lead to motor and sensory nerve dysfunction. Evidence indicates that the effects of hyperglycaemia on Schwann cells lead to disruption of the myelin sheath, which contributes to diabetic neuropathy. In one study, hyperglycaemia induced a progressive decrease of caveolin-1 in Schwann cells in culture and in STZ-induced type 1 diabetes in mice141. Caveolin-1 is a structural protein found in specialized sphingolipid–cholesterol microdomains called caveolae, which are thought to be important for Schwann cell physiology because cholesterol comprises ∼25% of the total lipid content of myelin142. In this study, the reduction of caveolin-1 prolonged the kinetics of ErbB2 phosphorylation and enhanced the mitogenic response of Schwann cells to neuregulin1-β1; subsequent studies have demonstrated that changes in caveolin-1 expression in Schwann cells co-cultured with neurons leads to neuregulin-induced demyelination, and that interactions between caveolin-1 and ErbB2 signalling contribute to peripheral neuropathy in rodents with diabetes143,144.
Studies have suggested that expression of the major myelin components myelin glycoprotein P0, myelin-associated glycoprotein and early growth response protein 2 in Schwann cells is reduced in diabetic neuropathy, and that these deficits might contribute to disorganization and loss of myelin145,146,147. In the case of P0, in vitro studies using glucose-stimulated primary Schwann cells showed that the reduced expression was apparently dependent on upregulated MAPK signalling, and was partially rescued by stimulation of Schwann cells with 10 nM insulin147,148. The importance of the MAPK pathway for nerve regeneration has been well documented149, but evidence indicates that prolonged MAPK signalling also maintains Schwann cells in a dedifferentiated state, as myelinating Schwann cells in transgenic mice did not respond to pro-differentiation signals from axons while MAPK activation was maintained150. Studies have also shown that the MAPK p38 is similarly upregulated in diabetes and, together with increased flux in the polyol pathway, contributes to slowing of nerve conduction and generation of pain35,151. These observations emphasize the need for an appropriate signalling balance to ensure optimal cell proliferation and nerve fibre maintenance.
Deficits in large fibre conduction velocity are generally considered to be among the earliest functional markers of glucose neurotoxicity, as they typically present before decreases in axonal diameter or structural disruption of the myelin in rodents152. In the absence of structural pathology, impaired nodal biophysical properties, such as ion flux and current densities, that might involve Schwann cell dysfunction have been implicated152,153. A study of rats with STZ-induced diabetes has shown that reduced motor conduction velocity is associated with upregulation of the Rho–Rho-kinase signalling pathway and consequent misexpression and aberrant distribution of the myelin sheath adhesion molecules p120 catenin and epithelial cadherin, which are crucial for normal myelin formation to allow rapid propagation of action potentials154.
Some evidence suggests that impaired Schwann cell function also affects peripheral axonal regeneration after injury has occurred. After traumatic injury to a nerve, reciprocal signalling normally occurs between the axon and glia, and this signalling is necessary for the reformation of the myelin–axon unit during regeneration. Compromised Schwann cell production of the neurotrophic factors NGF and NT-3 that are essential to nerve structure and function is linked to the loss of glia–axon associations and decreased neurite outgrowth, suggesting that reduced production of these factors by Schwann cells has an important role in impaired axonal regeneration21,155,156. Indeed, after experimental axotomy, Schwann cell growth was robust and extended into the superficial dermis in people without diabetic neuropathy, whereas people with injury as a result of diabetes exhibited atrophic Schwann tubes that were limited to the mid-dermis, and limited regenerative axonal sprouting157.
Schwann cell proliferation after physical nerve injury can be studied in culture to investigate discrete pathogenic mechanisms. In this context, exposure to high levels of glucose reduces the number of Schwann cells, and cell loss occurs by apoptosis158. The remaining cells look thin and short, and fail to adequately extend processes75,147, and chromatin becomes increasingly condensed with shrinkage of the Schwann cell cytosol158,159. When co-cultured with neurons, hyperglycaemia-stressed Schwann cells are dysfunctional143, and a study from 2014 suggests that the stress causes them to produce VEGF, which subsequently impairs neurite outgrowth from co-cultured sensory neurons160. These observations indicate that impaired nerve regeneration after injury in diabetes is, at least in part, secondary to disruption of Schwann cell metabolism.
Several therapeutic strategies are being pursued to manipulate nerve plasticity and neurite outgrowth in diabetes, with the aim of encouraging axonal repair161,162,163. A study published in 2016 identified p75NTR as a novel therapeutic target in type 2 diabetes, on the basis of results using an adipocyte-specific conditional null mouse model shown to be resilient to high-fat-diet-induced obesity and insulin resistance164. p75NTR signalling increased adipocyte lipolysis via cyclic AMP and the protein kinase A pathway, thereby regulating energy balance164. The findings highlight the importance of this neurotrophin receptor for obesity and the metabolic syndrome. In patients with diabetes, high p75NTR expression has been observed in myelin sheaths around fibres that are susceptible to axonal degeneration, indicating a role for this receptor in axonopathy117. Furthermore, results obtained from rodent models of peripheral nerve injury indicate that p75NTR is upregulated in neurons and Schwann cells, and can induce growth cone collapse, dependent on its interactions with myelin- associated proteins, or regrowth, by forming neurotrophin chemoattractant gradients165,166. In vitro studies have demonstrated that interactions between p75NTR and NGF inhibit Schwann cell apoptosis159. These observations suggest that neuronal or glial synthesis of p75NTR might have an important role in the progression of diabetic neuropathy, but further investigation is required to determine the precise role of p75NTR.
Towards a new understanding
The fact that primary axonopathy is a major pathological feature of diabetic neuropathy is inescapable. Indeed, diabetic neuropathy is usually identified by clinical and neurophysiological examinations to detect indications of axonopathy, namely, reduced conduction velocity and amplitude in large fibres, and abnormal thresholds to sensory stimuli. The focus on axonopathy is highlighted by a growing interest in the detection of early degeneration of small sensory fibres by measuring intraepidermal nerve fibre density in skin biopsy samples or corneal nerve density by confocal microscopy167 (Fig. 1). Nevertheless, we have presented a case that the direct effects of diabetes on Schwann cells can in turn affect the vasculature and axons, and distinguishing the precise contribution of Schwann cell dysfunction to that of axonopathy could provide valuable new insights.
One appealing approach to separating the roles of axonopathy and schwannopathy in diabetic neuropathy is to identify differences in the primary pathogenic mechanisms. In vitro, Schwann cells are damaged by hyperglycaemia158 and, as the sole site of the polyol pathway in the endoneurium, are a source of glucotoxicity. Glia require relatively low energy under normal conditions; they derive sufficient ATP from glycolysis and supply excess pyruvate to axons168. Schwann cells might not, therefore, be well adapted to metabolize excess glucose. By contrast, neurons consume high amounts of energy and use the full capacity of oxidative phosphorylation, such that primary sensory neurons in culture thrive in conditions that mimic physiological hyperglycaemia. Consequently, hyperglycaemia is perhaps less of a direct threat to axons, suggesting that axonopathy in diabetes reflects other primary pathogenic insults, such as loss of trophic support169.
The hypothesis that axonopathy in diabetic neuropathy results from a loss of trophic support is reinforced by the effects of insulin. Adult sensory neurons express insulin receptors, and addition of insulin or insulin-like growth factor 1 (IGF-1) to cultured adult sensory neurons drives a dose-dependent increase in neurite outgrowth170. Similarly, direct local injections of insulin into the skin at concentrations that do not modulate systemic glycaemia promote axonal growth in the epidermis171. Moreover, in animal models of type 2 diabetes, sensory neurons develop impaired insulin signalling172. Thus, it is becoming increasingly recognized that diabetes-associated loss of trophic support from insulin, and perhaps IGF-1 and C-peptide, represents a primary insult to neurons in both type 1 and type 2 diabetes173.
Differences between the effects of diabetes on mitochondria in Schwann cells and neurons might also provide insight into the pathogenic mechanisms. Evidence indicates that in neurons, diabetes decreases the expression of mitochondrial proteins174, the inner membrane potential175 and the spare respiratory capacity77 of otherwise functional mitochondria as a result of disruption to the AMPK–PGC1 pathway, which serves as a nutrient sensor and mitochondrial regulator176. These effects leave the neuron energetically viable but restricted in its capacity to respond to the increased energy demand of regrowth after physical injury, and to survive disease-mediated stress. This suppression of neuronal mitochondrial function in diabetes is in stark contrast to the impact on Schwann cell mitochondria, in which hyperglycaemia increases expression of proteins involved in the tricarboxylic acid cycle and oxidative phosphorylation, and increases overall oxygen consumption while decreasing coupled respiration84. The fundamental differences between Schwann cells and neurons in normal glucose metabolism168 and the handling of hyperglycaemia might underlie the complex pathogenesis of diabetic neuropathy.
Conclusions
Primary Schwann cell damage is a largely forgotten aspect of diabetic neuropathy, partly as a result of the major historical focus on microvascular disease and axonopathy. Studies showing that injury to Schwann cells affects both the vasculature and axons have reignited interest in Schwannopathy as a primary cause of diabetic neuropathy. In light of these findings, now is the time to rethink therapeutic strategies that account for both primary axonopathy and primary Schwannopathy in the setting of ongoing stress that arises from vascular hypoxia, hyperglycaemia, impaired trophic support, dyslipidaemia, mitochondrial dysfunction, oxidative stress, and activation of inflammatory pathways.
An increased mechanistic understanding of the Schwann cell response to diabetes could unravel novel molecular mechanisms that can ultimately be targeted to ameliorate disease. A particularly pertinent approach might be to expand on the recent development of conditional knockout mouse models to understand the specific role of genes expressed by Schwann cells in the pathophysiology of diabetic neuropathy, and the relative contributions of axonopathy and Schwannopathy. Our current understanding that a diverse range of signalling pathways are disrupted in diabetic neuropathy presents substantial challenges for drug development. Nevertheless, we believe that innovative drug delivery systems, improvements in clinical trial design and the generation of new biomarkers will combine to generate a multimodal approach that ultimately proves useful for treating diabetic neuropathy.
References
Callaghan, B. C., Hur, J. & Feldman, E. L. Diabetic neuropathy: one disease or two? Curr. Opin. Neurol. 25, 536–541 (2012).
Yagihashi, S. & Matsunaga, M. Ultrastructural pathology of peripheral nerves in patients with diabetic neuropathy. Tohoku J. Exp. Med. 129, 357–366 (1979).
Mizisin, A. P. Mechanisms of diabetic neuropathy: Schwann cells. Handb. Clin. Neurol. 126, 401–428 (2014). A detailed review describing ultrastructural Schwann cell changes in diabetic patients and animal models.
Ydens, E. et al. The neuroinflammatory role of Schwann cells in disease. Neurobiol. Dis. 55, 95–103 (2013).
Jessen, K. R. & Mirsky, R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6, 671–682 (2005). A comprehensive description of Schwann cell biology during development of peripheral nerves.
Yamauchi, J., Chan, J. R. & Shooter, E. M. Neurotrophin 3 activation of TrkC induces Schwann cell migration through the c-Jun N-terminal kinase pathway. Proc. Natl Acad. Sci. USA 100, 14421–14426 (2003).
Cosgaya, J. M., Chan, J. R. & Shooter, E. M. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 298, 1245–1248 (2002).
Sondell, M., Lundborg, G. & Kanje, M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J. Neurosci. 19, 5731–5740 (1999).
Chao, M. V. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309 (2003).
Reichardt, L. F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1545–1564 (2006).
Nikoletopoulou, V. et al. Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature 467, 59–63 (2010).
Masoudi, R. et al. Biological activity of nerve growth factor precursor is dependent upon relative levels of its receptors. J. Biol. Chem. 284, 18424–18433 (2009).
Lee, R., Kermani, P., Teng, K. K. & Hempstead, B. L. Regulation of cell survival by secreted proneurotrophins. Science 294, 1945–1948 (2001).
Nykjaer, A. et al. Sortilin is essential for proNGF-induced neuronal cell death. Nature 427, 843–848 (2004).
Deinhardt, K. et al. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci. Signal. 4, ra82 (2011).
Richner, M. et al. Peripheral nerve injury modulates neurotrophin signaling in the peripheral and central nervous system. Mol. Neurobiol. 50, 945–970 (2014).
Leinninger, G. M., Vincent, A. M. & Feldman, E. L. The role of growth factors in diabetic peripheral neuropathy. J. Peripher. Nerv. Syst. 9, 26–53 (2004).
Cattin, A.-L. et al. Macrophage-induced blood vessels guide Schwann cell-mediated regeneration of peripheral nerves. Cell 162, 1127–1139 (2015).
Calcutt, N. A., Muir, D., Powell, H. C. & Mizisin, A. P. Reduced ciliary neuronotrophic factor-like activity in nerves from diabetic or galactose-fed rats. Brain Res. 575, 320–324 (1992).
Calcutt, N. A. et al. Therapeutic efficacy of sonic hedgehog protein in experimental diabetic neuropathy. J. Clin. Invest. 111, 507–514 (2003).
Dey, I. et al. Diabetic Schwann cells suffer from nerve growth factor and neurotrophin-3 underproduction and poor associability with axons. Glia 61, 1990–1999 (2013).
Court, F. A., Wrabetz, L. & Feltri, M. L. Basal lamina: Schwann cells wrap to the rhythm of space-time. Curr. Opin. Neurobiol. 16, 501–507 (2006).
Engelstad, J. K., Davies, J. L., Giannini, C., O'Brien, P. C. & Dyck, P. J. No evidence for axonal atrophy in human diabetic polyneuropathy. J. Neuropathol. Exp. Neurol. 56, 255–262 (1997).
Obrosova, I. G. et al. High-fat diet induced neuropathy of pre-diabetes and obesity: effects of 'healthy' diet and aldose reductase inhibition. Diabetes 56, 2598–2608 (2007).
Behse, F., Buchthal, F. & Carlsen, F. Nerve biopsy and conduction studies in diabetic neuropathy. J. Neurol. Neurosurg. Psychiatry 40, 1072–1082 (1977).
Dyck, P. J. et al. Human diabetic endoneurial sorbitol, fructose, and myo-inositol related to sural nerve morphometry. Ann. Neurol. 8, 590–596 (1980).
Mizisin, A. P., Shelton, G. D., Wagner, S., Rusbridge, C. & Powell, H. C. Myelin splitting, Schwann cell injury and demyelination in feline diabetic neuropathy. Acta Neuropathol. 95, 171–174 (1998). Paper describing reactive and degenerative Schwann cell changes and ballooning of the myelin sheath in a feline model of diabetes.
Malik, R. A. et al. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia 48, 578–585 (2005).
Kalichman, M. W., Powell, H. C. & Mizisin, A. P. Reactive, degenerative, and proliferative Schwann cell responses in experimental galactose and human diabetic neuropathy. Acta Neuropathol. 95, 47–56 (1998).
Chowdhury, S. K., Smith, D. R. & Fernyhough, P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol. Dis. 51, 56–65 (2013). A summary of the major features of mitochondrial dysfunction in neurons and Schwann cells in patients with diabetes and in experimental animal models.
Lennertz, R. C., Medler, K. A., Bain, J. L., Wright, D. E. & Stucky, C. L. Impaired sensory nerve function and axon morphology in mice with diabetic neuropathy. J. Neurophysiol. 106, 905–914 (2011).
Becker, M., Benromano, T., Shahar, A., Nevo, Z. & Pick, C. G. Changes in the basal membrane of dorsal root ganglia Schwann cells explain the biphasic pattern of the peripheral neuropathy in streptozotocin-induced diabetic rats. J. Mol. Neurosci. 54, 704–713 (2014).
Gabbay, K. H., Merola, L. O. & Field, R. A. Sorbitol pathway: presence in nerve and cord with substrate accumulation in diabetes. Science 151, 209–210 (1966). An original key paper describing elevated levels of glucose and sorbitol in diabetic nervous tissue.
Tomlinson, D. R., Holmes, P. R. & Mayer, J. H. Reversal, by treatment with an aldose reductase inhibitor, of impaired axonal transport and motor nerve conduction velocity in experimental diabetes mellitus. Neurosci. Lett. 31, 189–193 (1982).
Ho, E. C. et al. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes 55, 1946–1953 (2006). An important study that used aldose reductase-deficient mice to demonstrate that increased polyol pathway flux through aldose reductase in Schwann cells is a major contributing factor to the early signs of diabetic neuropathy.
Ng, T. F. et al. Effects of sorbitol dehydrogenase deficiency on nerve conduction in experimental diabetic mice. Diabetes 47, 961–966 (1998).
Song, Z. et al. Transgenic mice overexpressing aldose reductase in Schwann cells show more severe nerve conduction velocity deficit and oxidative stress under hyperglycemic stress. Mol. Cell. Neurosci. 23, 638–647 (2003).
Uehara, K., Yamagishi, S.-I., Otsuki, S., Chin, S. & Yagihashi, S. Effects of polyol pathway hyperactivity on protein kinase C activity, nociceptive peptide expression, and neuronal structure in dorsal root ganglia in diabetic mice. Diabetes 53, 3239–3247 (2004).
Jiang, Y., Calcutt, N. A., Ramos, K. M., Rames, K. M. & Mizisin, A. P. Novel sites of aldose reductase immunolocalization in normal and streptozotocin-diabetic rats. J. Peripher. Nerv. Syst. 11, 274–285 (2006).
Ludvigson, M. A. & Sorenson, R. L. Immunohistochemical localization of aldose reductase. I. Enzyme purification and antibody preparation — localization in peripheral nerve, artery, and testis. Diabetes 29, 438–449 (1980).
Powell, H. C., Garrett, R. S., Kador, P. F. & Mizisin, A. P. Fine-structural localization of aldose reductase and ouabain-sensitive, K+-dependent p-nitro-phenylphosphatase in rat peripheral nerve. Acta Neuropathol. 81, 529–539 (1991).
Vedantham, S. et al. Aldose reductase drives hyperacetylation of Egr-1 in hyperglycemia and consequent upregulation of proinflammatory and prothrombotic signals. Diabetes 63, 761–774 (2014).
Sharma, K. R. Demyelinating neuropathy in diabetes mellitus. Arch. Neurol. 59, 758–765 (2002).
Kato, N., Mizuno, K., Makino, M., Suzuki, T. & Yagihashi, S. Effects of 15-month aldose reductase inhibition with fidarestat on the experimental diabetic neuropathy in rats. Diabetes Res. Clin. Pract. 50, 77–85 (2000).
Powell, H. et al. Alloxan diabetic neuropathy: electron microscopic studies. Neurology 27, 60–66 (1977).
Gregory, J. A., Jolivalt, C. G., Goor, J., Mizisin, A. P. & Calcutt, N. A. Hypertension-induced peripheral neuropathy and the combined effects of hypertension and diabetes on nerve structure and function in rats. Acta Neuropathol. 124, 561–573 (2012).
Kawashima, R. et al. Alterations in mRNA expression of myelin proteins in the sciatic nerves and brains of streptozotocin-induced diabetic rats. Neurochem. Res. 32, 1002–1010 (2007).
Chapouly, C. et al. Impaired Hedgehog signalling-induced endothelial dysfunction is sufficient to induce neuropathy: implication in diabetes. Cardiovasc. Res. 109, 217–227 (2016).
Mizisin, A. P., Vu, Y., Shuff, M. & Calcutt, N. A. Ciliary neurotrophic factor improves nerve conduction and ameliorates regeneration deficits in diabetic rats. Diabetes 53, 1807–1812 (2004).
Mizisin, A. P., Calcutt, N. A., DiStefano, P. S., Acheson, A. & Longo, F. M. Aldose reductase inhibition increases CNTF-like bioactivity and protein in sciatic nerves from galactose-fed and normal rats. Diabetes 46, 647–652 (1997).
Hao, W. et al. Hyperglycemia promotes Schwann cell de-differentiation and de-myelination via sorbitol accumulation and Igf1 protein down-regulation. J. Biol. Chem. 290, 17106–17115 (2015).
Yagihashi, S. et al. Galactosemic neuropathy in transgenic mice for human aldose reductase. Diabetes 45, 56–59 (1996).
Mizisin, A. P. & Powell, H. C. Schwann cell injury is attenuated by aldose reductase inhibition in galactose intoxication. J. Neuropathol. Exp. Neurol. 52, 78–86 (1993).
Myers, R. R. & Powell, H. C. Galactose neuropathy: impact of chronic endoneurial edema on nerve blood flow. Ann. Neurol. 16, 587–594 (1984).
Mizisin, A. P., Powell, H. C. & Myers, R. R. Edema and increased endoneurial sodium in galactose neuropathy. Reversal with an aldose reductase inhibitor. J. Neurol. Sci. 74, 35–43 (1986).
Stavniichuk, R., Shevalye, H., Hirooka, H., Nadler, J. L. & Obrosova, I. G. Interplay of sorbitol pathway of glucose metabolism, 12/15-lipoxygenase, and mitogen-activated protein kinases in the pathogenesis of diabetic peripheral neuropathy. Biochem. Pharmacol. 83, 932–940 (2012).
Tentolouris, N. et al. Standard and emerging treatment options for diabetic neuropathy. Curr. Pharm. Des. 20, 3689–3704 (2014). A recent review of current treatment options for diabetic neuropathy.
Fernyhough, P. & Calcutt, N. A. New directions in diabetic neuropathy: evolution or extinction? Int. Rev. Neurobiol. 127, 229–234 (2016).
Greene, D. A., Arezzo, J. C. & Brown, M. B. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Zenarestat Study Group. Neurology 53, 580–591 (1999).
Polydefkis, M. et al. Safety and efficacy of ranirestat in patients with mild-to-moderate diabetic sensorimotor polyneuropathy. J. Peripher. Nerv. Syst. 20, 363–371 (2015).
Vincent, A. M., Russell, J. W., Low, P. & Feldman, E. L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 25, 612–628 (2004). An examination of animal and cell culture models of diabetes, as well as clinical trials of antioxidants, describing how hyperglycaemia induces oxidative stress in diabetic neuropathy.
Babizhayev, M. A. et al. The role of oxidative stress in diabetic neuropathy: generation of free radical species in the glycation reaction and gene polymorphisms encoding antioxidant enzymes to genetic susceptibility to diabetic neuropathy in population of type I diabetic patients. Cell Biochem. Biophys. 71, 1425–1443 (2014).
Fidanboylu, M., Griffiths, L. A. & Flatters, S. J. Global inhibition of reactive oxygen species (ROS) inhibits paclitaxel-induced painful peripheral neuropathy. PLoS ONE 6, e25212 (2011).
Kallenborn-Gerhardt, W. et al. NADPH oxidase-4 maintains neuropathic pain after peripheral nerve injury. J. Neurosci. 32, 10136–10145 (2012).
Uttara, B., Singh, A. V., Zamboni, P. & Mahajan, R. T. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 7, 65–74 (2009).
Cunha, J. M. et al. Elevated lipid peroxidation and DNA oxidation in nerve from diabetic rats: effects of aldose reductase inhibition, insulin, and neurotrophic factors. Metab. Clin. Exp. 57, 873–881 (2008).
Obrosova, I. G. et al. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes 54, 234–242 (2005).
Vincent, A. M. et al. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology 148, 548–558 (2007).
Haslbeck, K. M. et al. Activation of the RAGE pathway: a general mechanism in the pathogenesis of polyneuropathies? Neurol. Res. 29, 103–110 (2007).
Schmidt, A. M. & Stern, D. M. RAGE: a new target for the prevention and treatment of the vascular and inflammatory complications of diabetes. Trends Endocrinol. Metab. 11, 368–375 (2000).
Yagihashi, S., Kamijo, M., Baba, M., Yagihashi, N. & Nagai, K. Effect of aminoguanidine on functional and structural abnormalities in peripheral nerve of STZ-induced diabetic rats. Diabetes 41, 47–52 (1992).
Obrosova, I. G. et al. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes 54, 3435–3441 (2005).
Askwith, T., Zeng, W., Eggo, M. C. & Stevens, M. J. Taurine reduces nitrosative stress and nitric oxide synthase expression in high glucose-exposed human Schwann cells. Exp. Neurol. 233, 154–162 (2012).
Drel, V. R. et al. New therapeutic and biomarker discovery for peripheral diabetic neuropathy: PARP inhibitor, nitrotyrosine, and tumor necrosis factor-α. Endocrinology 151, 2547–2555 (2010).
Gadau, S. D. Nitrosative stress induces proliferation and viability changes in high glucose-exposed rat Schwannoma cells. Neuro Endocrinol. Lett. 33, 279–284 (2012).
Giacco, F. et al. GLP-1 cleavage product reverses persistent ROS generation after transient hyperglycemia by disrupting an ROS-generating feedback loop. Diabetes 64, 3273–3284 (2015).
Roy Chowdhury, S. K. et al. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 135, 1751–1766 (2012).
Sharma, V. & Sharma, P. L. Role of different molecular pathways in the development of diabetes-induced nephropathy. J. Diabetes Metab. http://dx.doi.org/10.4172/2155-6156.S9-004 (2013).
Chowdhury, S. K. et al. Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 59, 1082–1091 (2010).
Han, J. et al. Fuzi attenuates diabetic neuropathy in rats and protects schwann cells from apoptosis induced by high glucose. PLoS ONE 9, e86539 (2014).
Viader, A. et al. Schwann cell mitochondrial metabolism supports long-term axonal survival and peripheral nerve function. J. Neurosci. 31, 10128–10140 (2011). This paper examines how Schwann cell mitochondrial dysfunction affects axonal survival and contributes to the decline of peripheral nerve function.
Hinder, L. M. et al. Long-chain acyl coenzyme A synthetase 1 overexpression in primary cultured Schwann cells prevents long chain fatty acid-induced oxidative stress and mitochondrial dysfunction. Antioxid. Redox Signal. 21, 588–600 (2014).
Viader, A. et al. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 77, 886–898 (2013). An important study showing how Schwann cells with mitochondrial dysfunction release acylcarnitines and induce axonal degeneration.
Zhang, L. et al. Hyperglycemia alters the schwann cell mitochondrial proteome and decreases coupled respiration in the absence of superoxide production. J. Proteome Res. 9, 458–471 (2010).
Freeman, O. J. et al. Metabolic dysfunction is restricted to the sciatic nerve in experimental diabetic neuropathy. Diabetes 65, 228–238 (2016).
Hiukka, A., Maranghi, M., Matikainen, N. & Taskinen, M.-R. PPARalpha: an emerging therapeutic target in diabetic microvascular damage. Nat. Rev. Endocrinol. 6, 454–463 (2010).
Vincent, A. M. et al. Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. Diabetes 58, 2376–2385 (2009).
Almaguel, F. G. et al. Lipotoxicity-mediated cell dysfunction and death involve lysosomal membrane permeabilization and cathepsin L activity. Brain Res. 1318, 133–143 (2010).
Padilla, A., Descorbeth, M., Almeyda, A. L., Payne, K. & De Leon, M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Res. 1370, 64–79 (2011).
Beirowski, B. et al. Metabolic regulator LKB1 is crucial for Schwann cell-mediated axon maintenance. Nat. Neurosci. 17, 1351–1361 (2014). This paper demonstrates how impairment of Schwann cell metabolism results in degeneration of primarily unmyelinated small sensory fibres while motor axons were comparatively spared.
Kuruvilla, R. & Eichberg, J. Depletion of phospholipid arachidonoyl-containing molecular species in a human Schwann cell line grown in elevated glucose and their restoration by an aldose reductase inhibitor. J. Neurochem. 71, 775–783 (1998).
Goethals, S., Ydens, E., Timmerman, V. & Janssens, S. Toll-like receptor expression in the peripheral nerve. Glia 58, 1701–1709 (2010).
Sbai, O. et al. RAGE–TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J. Cell Sci. 123, 4332–4339 (2010).
Vincent, A. M., Callaghan, B. C., Smith, A. L. & Feldman, E. L. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat. Rev. Neurol. 7, 573–583 (2011).
Herder, C. et al. Subclinical inflammation and diabetic polyneuropathy: MONICA/KORA Survey F3 (Augsburg, Germany). Diabetes Care 32, 680–682 (2009).
Scheib, J. & Höke, A. Advances in peripheral nerve regeneration. Nat. Rev. Neurol. 9, 668–676 (2013).
Nukada, H., McMorran, P. D., Baba, M., Ogasawara, S. & Yagihashi, S. Increased susceptibility to ischemia and macrophage activation in STZ-diabetic rat nerve. Brain Res. 1373, 172–182 (2011).
Conti, G. et al. Macrophage infiltration and death in the nerve during the early phases of experimental diabetic neuropathy: a process concomitant with endoneurial induction of IL-1beta and p75NTR. J. Neurol. Sci. 195, 35–40 (2002).
Ma, J., Pan, P., Anyika, M., Blagg, B. S. & Dobrowsky, R. T. Modulating molecular chaperones improves mitochondrial bioenergetics and decreases the inflammatory transcriptome in diabetic sensory neurons. ACS Chem. Neurosci. 6, 1637–1648 (2015).
O'Brien, P. D. et al. BTBR ob/ob mice as a novel diabetic neuropathy model: neurological characterization and gene expression analyses. Neurobiol. Dis. 73, 348–355 (2015).
O'Brien, P. D. et al. Gender-specific differences in diabetic neuropathy in BTBR ob/ob mice. J. Diabetes Complicat. 30, 30–37 (2016).
Krisp, C. et al. Proteome analysis reveals antiangiogenic environments in chronic wounds of diabetes mellitus type 2 patients. Proteomics 13, 2670–2681 (2013).
Narumi, K. et al. Proinflammatory proteins S100A8/S100A9 activate NK cells via interaction with RAGE. J. Immunol. 194, 5539–5548 (2015).
Hermani, A., De Servi, B., Medunjanin, S., Tessier, P. A. & Mayer, D. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp. Cell Res. 312, 184–197 (2006).
Ghavami, S. et al. S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. 20, 314–331 (2010).
Stavniichuk, R. et al. 12/15-Lipoxygenase inhibition counteracts MAPK phosphorylation in mouse and cell culture models of diabetic peripheral neuropathy. J. Diabetes Mellitus 3, 101–110 (2013).
Yang, D. P. et al. p38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. J. Neurosci. 32, 7158–7168 (2012).
Tang, W. et al. CD8+ T cell-mediated cytotoxicity toward Schwann cells promotes diabetic peripheral neuropathy. Cell. Physiol. Biochem. 32, 827–837 (2013).
Shi, X., Chen, Y., Nadeem, L. & Xu, G. Beneficial effect of TNF-α inhibition on diabetic peripheral neuropathy. J. Neuroinflammation 10, 69 (2013).
King, R. H. The role of glycation in the pathogenesis of diabetic polyneuropathy. Mol. Pathol. 54, 400–408 (2001).
Gabay, E., Wolf, G., Shavit, Y., Yirmiya, R. & Tal, M. Chronic blockade of interleukin-1 (IL-1) prevents and attenuates neuropathic pain behavior and spontaneous ectopic neuronal activity following nerve injury. Eur. J. Pain 15, 242–248 (2011).
Kim, C. F. & Moalem-Taylor, G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. J. Pain 12, 370–383 (2011).
Li, Y. et al. Curcumin attenuates diabetic neuropathic pain by downregulating TNF-α in a rat model. Int. J. Med. Sci. 10, 377–381 (2013).
Ma, J. et al. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS ONE 9, e97985 (2014).
Lee, J.-J. et al. High-fat diet induces toll-like receptor 4-dependent macrophage/microglial cell activation and retinal impairment. Invest. Ophthalmol. Vis. Sci. 56, 3041–3050 (2015).
Jiang, Y. et al. TLR4 signaling induces functional nerve growth factor receptor p75NTR on mouse dendritic cells via p38MAPK and NF-kappa B pathways. Mol. Immunol. 45, 1557–1566 (2008).
Scarpini, E. et al. Induction of p75NGFR in human diabetic neuropathy. J. Neurol. Sci. 135, 55–62 (1996).
Yagihashi, S., Mizukami, H. & Sugimoto, K. Mechanism of diabetic neuropathy: where are we now and where to go? J. Diabetes Investig. 2, 18–32 (2011).
Cameron, N. E., Eaton, S. E., Cotter, M. A. & Tesfaye, S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia 44, 1973–1988 (2001). An important review of studies in humans and animal models underlining the importance of vascular dysfunction, driven by metabolic change, as a cause of diabetic neuropathy.
Giannini, C. & Dyck, P. J. Basement membrane reduplication and pericyte degeneration precede development of diabetic polyneuropathy and are associated with its severity. Ann. Neurol. 37, 498–504 (1995).
Thrainsdottir, S. et al. Endoneurial capillary abnormalities presage deterioration of glucose tolerance and accompany peripheral neuropathy in man. Diabetes 52, 2615–2622 (2003).
Østergaard, L. et al. The effects of capillary dysfunction on oxygen and glucose extraction in diabetic neuropathy. Diabetologia 58, 666–677 (2015).
Schwartz, R. S., Eltzschig, H. K. & Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 364, 656–665 (2011).
Toth, C. et al. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes 57, 1002–1017 (2008).
Yuan, G. et al. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell. Physiol. 226, 2925–2933 (2011).
Pop-Busui, R., Sima, A. & Stevens, M. Diabetic neuropathy and oxidative stress. Diabetes Metab. Res. Rev. 22, 257–273 (2006).
Dawson, V. L. & Dawson, T. M. Nitric oxide neurotoxicity. J. Chem. Neuroanat. 10, 179–190 (1996).
Hafer-Macko, C. E., Ivey, F. M., Sorkin, J. D. & Macko, R. F. Microvascular tissue plasminogen activator is reduced in diabetic neuropathy. Neurology 69, 268–274 (2007).
Teng, H. K. et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 25, 5455–5463 (2005).
Andreassen, C. S., Jakobsen, J., Flyvbjerg, A. & Andersen, H. Expression of neurotrophic factors in diabetic muscle — relation to neuropathy and muscle strength. Brain 132, 2724–2733 (2009).
Angleys, H., Jespersen, S. N. & Østergaard, L. The effects of capillary transit time heterogeneity (CTH) on the cerebral uptake of glucose and glucose analogs: application to FDG and comparison to oxygen uptake. Front. Comput. Neurosci. 10, 97 (2016).
Waxman, S. G. & Zamponi, G. W. Regulating excitability of peripheral afferents: emerging ion channel targets. Nat. Neurosci. 17, 153–163 (2014).
Krishnan, A. V., Lin, C. S., Park, S. B. & Kiernan, M. C. Axonal ion channels from bench to bedside: a translational neuroscience perspective. Prog. Neurobiol. 89, 288–313 (2009).
Zenker, J., Ziegler, D. & Chrast, R. Novel pathogenic pathways in diabetic neuropathy. Trends Neurosci. 36, 439–449 (2013). This review takes a novel approach by focusing on nodal regions, areas of intense interactions between Schwann cells and axons, and how they may be particularly sensitive to diabetes-induced nerve degeneration.
Fernyhough, P. & Calcutt, N. A. Abnormal calcium homeostasis in peripheral neuropathies. Cell Calcium 47, 130–139 (2010).
Cherian, P. V., Kamijo, M., Angelides, K. J. & Sima, A. A. Nodal Na+-channel displacement is associated with nerve-conduction slowing in the chronically diabetic BB/W rat: prevention by aldose reductase inhibition. J. Diabetes Complicat. 10, 192–200 (1996).
Leffler, A. et al. GDNF and NGF reverse changes in repriming of TTX-sensitive Na+ currents following axotomy of dorsal root ganglion neurons. J. Neurophysiol. 88, 650–658 (2002).
Bierhaus, A. et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 18, 926–933 (2012).
Kwai, N. C. et al. In vivo evidence of reduced nodal and paranodal conductances in type 1 diabetes. Clin. Neurophysiol. 127, 1700–1706 (2016).
Kim, E. S., Isoda, F., Kurland, I. & Mobbs, C. V. Glucose-induced metabolic memory in Schwann cells: prevention by PPAR agonists. Endocrinology 154, 3054–3066 (2013).
Tan, W. et al. Nerve growth factor blocks the glucose-induced down-regulation of caveolin-1 expression in Schwann cells via p75 neurotrophin receptor signaling. J. Biol. Chem. 278, 23151–23162 (2003).
Parton, R. G. & Simons, K. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8, 185–194 (2007).
Yu, C., Rouen, S. & Dobrowsky, R. T. Hyperglycemia and downregulation of caveolin-1 enhance neuregulin-induced demyelination. Glia 56, 877–887 (2008).
McGuire, J. F., Rouen, S., Siegfreid, E., Wright, D. E. & Dobrowsky, R. T. Caveolin-1 and altered neuregulin signaling contribute to the pathophysiological progression of diabetic peripheral neuropathy. Diabetes 58, 2677–2686 (2009).
Cermenati, G. et al. Diabetes-induced myelin abnormalities are associated with an altered lipid pattern: protective effects of LXR activation. J. Lipid Res. 53, 300–310 (2012).
Rachana, K. S., Manu, M. S. & Advirao, G. M. Insulin influenced expression of myelin proteins in diabetic peripheral neuropathy. Neurosci. Lett. 629, 110–115 (2016).
Liu, D., Liang, X. & Zhang, H. Effects of high glucose on cell viability and differentiation in primary cultured Schwann cells: potential role of ERK signaling pathway. Neurochem. Res. 41, 1281–1290 (2016).
Shettar, A. & Muttagi, G. Developmental regulation of insulin receptor gene in sciatic nerves and role of insulin on glycoprotein P0 in the Schwann cells. Peptides 36, 46–53 (2012).
Agthong, S., Kaewsema, A., Tanomsridejchai, N. & Chentanez, V. Activation of MAPK ERK in peripheral nerve after injury. BMC Neurosci. 7, 45 (2006).
Napoli, I. et al. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron 73, 729–742 (2012). This study demonstrates how prolonged MAPK activation in Schwann cells may underlie reduced expression of myelin components.
Cheng, H. T., Dauch, J. R., Hayes, J. M., Yanik, B. M. & Feldman, E. L. Nerve growth factor/p38 signaling increases intraepidermal nerve fiber densities in painful neuropathy of type 2 diabetes. Neurobiol. Dis. 45, 280–287 (2012).
Tomlinson, D. R. & Gardiner, N. J. Glucose neurotoxicity. Nat. Rev. Neurosci. 9, 36–45 (2008).
Tan, A. M., Samad, O. A., Dib-Hajj, S. D. & Waxman, S. G. Virus-mediated knockdown of Nav1.3 in dorsal root ganglia of STZ-induced diabetic rats alleviates tactile allodynia. Mol. Med. 21, 544–552 (2015).
Kanazawa, Y. et al. The Rho-kinase inhibitor fasudil restores normal motor nerve conduction velocity in diabetic rats by assuring the proper localization of adhesion-related molecules in myelinating Schwann cells. Exp. Neurol. 247, 438–446 (2013).
Suzuki, T., Sekido, H., Kato, N., Nakayama, Y. & Yabe-Nishimura, C. Neurotrophin-3-induced production of nerve growth factor is suppressed in Schwann cells exposed to high glucose: involvement of the polyol pathway. J. Neurochem. 91, 1430–1438 (2004).
Tosaki, T. et al. Reduced NGF secretion by Schwann cells under the high glucose condition decreases neurite outgrowth of DRG neurons. Exp. Neurol. 213, 381–387 (2008).
Ebenezer, G. J. et al. Impaired neurovascular repair in subjects with diabetes following experimental intracutaneous axotomy. Brain 134, 1853–1863 (2011). A study investigating the interplay between blood vessels, axons and Schwann cells during regeneration, and how Schwann cell regrowth seemed to be atrophic in patients with diabetes.
Delaney, C. L., Russell, J. W., Cheng, H. L. & Feldman, E. L. Insulin-like growth factor-I and over-expression of Bcl-xL prevent glucose-mediated apoptosis in Schwann cells. J. Neuropathol. Exp. Neurol. 60, 147–160 (2001). Study describing the mechanism underlying Schwann cell death on hyperglycaemia.
Vincent, A. M., Brownlee, M. & Russell, J. W. Oxidative stress and programmed cell death in diabetic neuropathy. Ann. N. Y. Acad. Sci. 959, 368–383 (2002).
Taiana, M. M. et al. Neutralization of schwann cell-secreted VEGF is protective to in vitro and in vivo experimental diabetic neuropathy. PLoS ONE 9, e108403 (2014).
Habash, T., Saleh, A., Roy Chowdhury, S. K., Smith, D. R. & Fernyhough, P. The proinflammatory cytokine, interleukin-17A, augments mitochondrial function and neurite outgrowth of cultured adult sensory neurons derived from normal and diabetic rats. Exp. Neurol. 273, 177–189 (2015).
Singh, B. et al. Peripheral neuron plasticity is enhanced by brief electrical stimulation and overrides attenuated regrowth in experimental diabetes. Neurobiol. Dis. 83, 134–151 (2015).
Han, J. W., Choi, D., Lee, M. Y., Huh, Y. H. & Yoon, Y.-S. Bone marrow-derived mesenchymal stem cells improve diabetic neuropathy by direct modulation of both angiogenesis and myelination in peripheral nerves. Cell Transplant. 25, 313–326 (2016).
Baeza-Raja, B. et al. p75 Neurotrophin receptor regulates energy balance in obesity. Cell Rep. 14, 255–268 (2016).
Meeker, R. B. & Williams, K. S. The p75 neurotrophin receptor: at the crossroad of neural repair and death. Neural Regen. Res. 10, 721–725 (2015).
Zhou, X.-F. & Li, H.-Y. Roles of glial p75NTR in axonal regeneration. J. Neurosci. Res. 85, 1601–1605 (2007).
Quattrini, C. et al. Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes 56, 2148–2154 (2007).
Halim, N. D. et al. Phosphorylation status of pyruvate dehydrogenase distinguishes metabolic phenotypes of cultured rat brain astrocytes and neurons. Glia 58, 1168–1176 (2010).
Zochodne, D. W. Sensory neurodegeneration in diabetes: beyond glucotoxicity. Int. Rev. Neurobiol. 127, 151–180 (2016).
Fernyhough, P., Willars, G. B., Lindsay, R. M. & Tomlinson, D. R. Insulin and insulin-like growth factor I enhance regeneration in cultured adult rat sensory neurones. Brain Res. 607, 117–124 (1993).
Guo, G., Kan, M., Martinez, J. A. & Zochodne, D. W. Local insulin and the rapid regrowth of diabetic epidermal axons. Neurobiol. Dis. 43, 414–421 (2011).
Grote, C. W. et al. Peripheral nervous system insulin resistance in ob/ob mice. Acta Neuropathol. Commun. 1, 15 (2013).
Zochodne, D. W. Diabetes and the plasticity of sensory neurons. Neurosci. Lett. 596, 60–65 (2015).
Akude, E. et al. Diminished superoxide generation is associated with respiratory chain dysfunction and changes in the mitochondrial proteome of sensory neurons from diabetic rats. Diabetes 60, 288–297 (2010).
Huang, T. J. et al. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes 52, 2129–2136 (2003).
Fernyhough, P. Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Curr. Diab. Rep. 15, 89–10 (2015).
Jensen, T. S. & Baron, R. Translation of symptoms and signs into mechanisms in neuropathic pain. Pain 102, 1–8 (2003).
Jensen, T. S. & Finnerup, N. B. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol. 13, 924–935 (2014).
Truini, A. et al. Peripheral nociceptor sensitization mediates allodynia in patients with distal symmetric polyneuropathy. J. Neurol. 260, 761–766 (2013).
Hoeijmakers, J. G., Faber, C. G., Merkies, I. S. & Waxman, S. G. Channelopathies, painful neuropathy, and diabetes: which way does the causal arrow point? Trends Mol. Med. 20, 544–550 (2014).
Tesfaye, S., Boulton, A. J. & Dickenson, A. H. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care 36, 2456–2465 (2013).
Truini, A. et al. Does the epidermal nerve fibre density measured by skin biopsy in patients with peripheral neuropathies correlate with neuropathic pain? Pain 155, 828–832 (2014).
Truini, A., Garcia-Larrea, L. & Cruccu, G. Reappraising neuropathic pain in humans — how symptoms help disclose mechanisms. Nat. Rev. Neurol. 9, 572–582 (2013).
Peltier, A., Goutman, S. A. & Callaghan, B. C. Painful diabetic neuropathy. BMJ 348, g1799 (2014).
Callaghan, B. C., Cheng, H. T., Stables, C. L., Smith, A. L. & Feldman, E. L. Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurol. 11, 521–534 (2012).
Vinik, A. I., Nevoret, M., Casellini, C. & Parson, H. Neurovascular function and sudorimetry in health and disease. Curr. Diab. Rep. 13, 517–532 (2013).
Pop-Busui, R., Lu, J., Lopes, N. & Jones, T. L. BARI 2D Investigators. Prevalence of diabetic peripheral neuropathy and relation to glycemic control therapies at baseline in the BARI 2D cohort. J. Peripher. Nerv. Syst. 14, 1–13 (2009).
Abbott, C. A., Malik, R. A., van Ross, E. R. E., Kulkarni, J. & Boulton, A. J. M. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 34, 2220–2224 (2011).
Bouhassira, D., Letanoux, M. & Hartemann, A. Chronic pain with neuropathic characteristics in diabetic patients: a French cross-sectional study. PLoS ONE 8, e74195 (2013).
Daousi, C. et al. Chronic painful peripheral neuropathy in an urban community: a controlled comparison of people with and without diabetes. Diabet. Med. 21, 976–982 (2004).
Dublin, P. & Hanani, M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav. Immun. 21, 592–598 (2007).
Huang, L.-Y. M., Gu, Y. & Chen, Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 61, 1571–1581 (2013).
Gardiner, N. J. et al. Expression of hexokinase isoforms in the dorsal root ganglion of the adult rat and effect of experimental diabetes. Brain Res. 1175, 143–154 (2007).
Berti-Mattera, L. N., Larkin, B., Hourmouzis, Z., Kern, T. S. & Siegel, R. E. NF-κB subunits are differentially distributed in cells of lumbar dorsal root ganglia in naïve and diabetic rats. Neurosci. Lett. 490, 41–45 (2011).
Hanani, M., Blum, E., Liu, S., Peng, L. & Liang, S. Satellite glial cells in dorsal root ganglia are activated in streptozotocin-treated rodents. J. Cell. Mol. Med. 18, 2367–2371 (2014).
Liu, S. et al. LncRNA NONRATT021972 siRNA regulates neuropathic pain behaviors in type 2 diabetic rats through the P2X7 receptor in dorsal root ganglia. Mol. Brain 9, 44 (2016).
Acknowledgements
The authors gratefully acknowledge Dr Páll Karlsson from the Danish Pain Research Center, Department of Clinical Medicine, Aarhus University, for providing the pictures of unmyelinated fibres from the human skin biopsy samples. We would also like to thank Ken Kragsfeldt from the Clinical Institute, Aarhus University, for the graphical design. This work has been made possible thanks to a challenge grant from the Novo Nordisk Foundation (NNF14OC0011633) and an NIH grant (NS081082).
Author information
Authors and Affiliations
Contributions
N.P.G., C.B.V, L.Ø., N.A.C. and T.S.J. researched data for the article and wrote the article. N.P.G., C.B.V, H.A., N.A.C. and T.S.J. made substantial contributions to discussion of the content of the article. All authors reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
FURTHER INFORMATION
PowerPoint slides
Glossary
- Prediabetes
-
A condition in which blood glucose levels are higher than normal but not high enough to be considered as type 2 diabetes; individuals with prediabetes often develop diabetic neuropathy.
- Dependence receptors
-
Proteins that mediate apoptosis by monitoring the absence of certain trophic factors.
- Basal lamina
-
Sheets of extracellular matrix that are secreted by Schwann cells and surround a nerve.
- Endoneurium
-
A layer of interstitial connective tissue that surrounds all axons, thereby separating individual nerve fibres.
- Radial sorting
-
The process that underlies selection of one axon for myelination by a Schwann cell during development.
- Capillary dysfunction
-
A state of dysregulated capillary blood flow patterns, in which oxygen, glucose and other diffusible molecules cannot be extracted efficiently by the tissue; uptake of these molecules can become critically impaired, although reduced tissue blood supply is not obvious.
- C-peptide
-
A short polypeptide that is cleaved from proinsulin in the production of insulin, and can be measured in the blood.
- Coupled respiration
-
A process in which oxygen uptake is dependent on the presence of ADP and phosphate.
Rights and permissions
About this article

Cite this article
Gonçalves, N., Vægter, C., Andersen, H. et al. Schwann cell interactions with axons and microvessels in diabetic neuropathy. Nat Rev Neurol 13, 135–147 (2017). https://doi.org/10.1038/nrneurol.2016.201
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneurol.2016.201
This article is cited by
-
Comparison of therapeutic effects of N-Acetylcysteine with pregabalin in improving the clinical symptoms of painful diabetic neuropathy: a randomized, double-blind clinical trial
Clinical Diabetes and Endocrinology (2024)
-
Rab32 facilitates Schwann cell pyroptosis in rats following peripheral nerve injury by elevating ROS levels
Journal of Translational Medicine (2024)
-
Chitosan as a promising materials for the construction of nanocarriers for diabetic retinopathy: an updated review
Journal of Biological Engineering (2024)
-
A muscarinic receptor antagonist reverses multiple indices of diabetic peripheral neuropathy: preclinical and clinical studies using oxybutynin
Acta Neuropathologica (2024)
-
Glial cell alterations in diabetes-induced neurodegeneration
Cellular and Molecular Life Sciences (2024)
