
Abstract
Structure-based drug design is frequently used to accelerate the development of small-molecule therapeutics. Although substantial progress has been made in X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, the availability of high-resolution structures is limited owing to the frequent inability to crystallize or obtain sufficient NMR restraints for large or flexible proteins. Computational methods can be used to both predict unknown protein structures and model ligand interactions when experimental data are unavailable. This paper describes a comprehensive and detailed protocol using the Rosetta modeling suite to dock small-molecule ligands into comparative models. In the protocol presented here, we review the comparative modeling process, including sequence alignment, threading and loop building. Next, we cover docking a small-molecule ligand into the protein comparative model. In addition, we discuss criteria that can improve ligand docking into comparative models. Finally, and importantly, we present a strategy for assessing model quality. The entire protocol is presented on a single example selected solely for didactic purposes. The results are therefore not representative and do not replace benchmarks published elsewhere. We also provide an additional tutorial so that the user can gain hands-on experience in using Rosetta. The protocol should take 5–7 h, with additional time allocated for computer generation of models.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

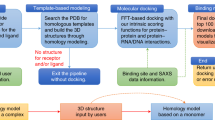
References
Rohl, C.A., Strauss, C.E.M., Misura, K.M.S. & Baker, D. Protein structure prediction using Rosetta. Methods Enzymol. 383, 66–93 (2004).
Siegel, J.B. et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 329, 309–313 (2010).
Kuhlman, B. et al. Design of a novel globular protein fold with atomic-level accuracy. Science 302, 1364–1368 (2003).
Davis, I.W. & Baker, D. RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 385, 381–392 (2009).
Misura, K.M.S., Chivian, D., Rohl, C.A., Kim, D.E. & Baker, D. Physically realistic homology models built with ROSETTA can be more accurate than their templates. Proc. Natl. Acad. Sci. USA 103, 5361–5366 (2006).
Davis, I.W., Raha, K., Head, M.S. & Baker, D. Blind docking of pharmaceutically relevant compounds using RosettaLigand. Protein Sci. 18, 1998–2002 (2009).
Das, R. & Baker, D. Macromolecular modeling with Rosetta. Annu. Rev. Biochem. 77, 363–382 (2008).
Kaufmann, K.W., Lemmon, G.H., Deluca, S.L., Sheehan, J.H. & Meiler, J. Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry 49, 2987–2998 (2010).
Rohl, C.A., Strauss, C.E.M., Chivian, D. & Baker, D. Modeling structurally variable regions in homologous proteins with Rosetta. Proteins 55, 656–677 (2004).
Meiler, J. & Baker, D. Coupled prediction of protein secondary and tertiary structure. Proc. Natl. Acad. Sci. USA 100, 12105–12110 (2003).
Yarov-Yarovoy, V., Schonbrun, J. & Baker, D. Multipass membrane protein structure prediction using Rosetta. Proteins 62, 1010–1025 (2006).
Bradley, P. et al. Free modeling with Rosetta in CASP6. Proteins 61 (suppl. 7), 128–134 (2005).
Bradley, P., Misura, K.M.S. & Baker, D. Toward high-resolution de novo structure prediction for small proteins. Science 309, 1868–1871 (2005).
Das, R. et al. Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home. Proteins 69 (suppl. 8), 118–128 (2007).
Rohl, C.A. Protein structure estimation from minimal restraints using Rosetta. Methods Enzymol. 394, 244–260 (2005).
Lange, O.F. et al. Determination of solution structures of proteins up to 40 kDa using CS-Rosetta with sparse NMR data from deuterated samples. Proc. Natl. Acad. Sci. USA 109, 10873–10878 (2012).
Lange, O.F. & Baker, D. Resolution-adapted recombination of structural features significantly improves sampling in restraint-guided structure calculation. Proteins 80, 884–895 (2012).
Kaufmann, K.W. et al. Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins 74, 630–642 (2009).
Lees-Miller, J.P. et al. Interactions of H562 in the S5 helix with T618 and S621 in the pore helix are important determinants of hERG1 potassium channel structure and function. Biophys. J. 96, 3600–3610 (2009).
Keeble, A.H. et al. Experimental and computational analyses of the energetic basis for dual recognition of immunity proteins by colicin endonucleases. J. Mol. Biol. 379, 745–759 (2008).
Fortenberry, C. et al. Exploring symmetry as an avenue to the computational design of large protein domains. J. Am. Chem. Soc. 133, 18026–18029 (2011).
Eswar, N., Eramian, D., Webb, B., Shen, M.-Y. & Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 (2008).
Perola, E., Walters, W.P. & Charifson, P.S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 56, 235–249 (2004).
Carlsson, J. et al. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol. 7, 769–778 (2011).
Ballester, P.J., Westwood, I., Laurieri, N., Sim, E. & Richards, W.G. Prospective virtual screening with Ultrafast Shape Recognition: the identification of novel inhibitors of arylamine N-acetyltransferases. J. R. Soc. Interface 7, 335–342 (2010).
Schneider, G. & Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 4, 649–663 (2005).
Schneider, G. et al. Voyages to the (un)known: adaptive design of bioactive compounds. Trends Biotechnol. 27, 18–26 (2009).
Meiler, J. & Baker, D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins 65, 538–548 (2006).
Lemmon, G. & Meiler, J. Rosetta Ligand docking with flexible XML protocols. Methods Mol. Biol. 819, 143–155 (2012).
Laskowski, R.A. SURFNET: a program for visualizing molecular surfaces, cavities, and intermolecular interactions. J. Mol. Graph 13, 323–330, (1995).
Huang, B. & Schroeder, M. LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct. Biol. 6, 19 (2006).
Kalidas, Y. & Chandra, N. PocketDepth: a new depth-based algorithm for identification of ligand binding sites in proteins. J. Struct. Biol. 161, 31–42 (2008).
Dunbrack, R.L. & Karplus, M. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J. Mol. Biol. 230, 543–574 (1993).
Li, Z. & Scheraga, H. A. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. USA 84, 6611–6615 (1987).
Simons, K.T., Kooperberg, C., Huang, E. & Baker, D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 268, 209–225 (1997).
Metropolis, N., Rosenbluth, A.W., Rosenbluth, M.N., Teller, A.H. & Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 21, 1087 (1953).
Rarey, M., Kramer, B., Lengauer, T. & Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 261, 470–489 (1996).
Verdonk, M.L., Cole, J.C., Hartshorn, M.J., Murray, C.W. & Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 52, 609–623 (2003).
Friesner, R.A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Ewing, T.J., Makino, S., Skillman, A.G. & Kuntz, I.D. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 15, 411–428 (2001).
Gohlke, H., Hendlich, M. & Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 295, 337–356 (2000).
Kaufmann, K.W. & Meiler, J. Using RosettaLigand for small molecule docking into comparative models. PLoS ONE 7, e50769 (2012).
Mobley, D.L. et al. Predicting absolute ligand binding free energies to a simple model site. J. Mol. Biol. 371, 1118–1134 (2007).
Mooers, B.H.M. & Matthews, B.W. Extension to 2268 atoms of direct methods in the ab initio determination of the unknown structure of bacteriophage P22 lysozyme. Acta Crystallogr. D Biol. Crystallogr. 62, 165–176 (2006).
Altschul, S.F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Zhang, Z. et al. Protein sequence similarity searches using patterns as seeds. Nucleic Acids Res. 26, 3986–3990 (1998).
Söding, J., Biegert, A. & Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005).
Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009).
Eddy, S.R. Accelerated profile HMM searches. PloS Comput. Biol. 7, e1002195 (2011).
Kelley, L.A. & Sternberg, M.J.E. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 (2009).
Canutescu, A.A. & Dunbrack, R.L. Cyclic coordinate descent: a robotics algorithm for protein loop closure. Protein Sci. 12, 963–972 (2003).
Mandell, D.J., Coutsias, E.A. & Kortemme, T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 6, 551–552 (2009).
Coutsias, E.A., Seok, C., Jacobson, M.P. & Dill, K.A. A kinematic view of loop closure. J. Comput. Chem. 25, 510–528 (2004).
Gront, D., Kulp, D.W., Vernon, R.M., Strauss, C.E.M. & Baker, D. Generalized fragment picking in Rosetta: design, protocols and applications. PLoS ONE 6, e23294 (2011).
Wang, G. & Dunbrack, R.L. PISCES: a protein sequence culling server. Bioinformatics 19, 1589–1591 (2003).
Leaver-Fay, A., Kuhlman, B. & Snoeyink, J. Rotamer-pair energy calculations using a trie data structure. Lect. Notes Comput. Sci. 3692, 389–400 (2005).
Jones, D., Taylor, W. & Thorton, J. A New approach to protein fold recognition. Nature 358, 86–89 (1992).
Raman, S. et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 77, 89–99 (2009).
Hirst, S.J., Alexander, N., McHaourab, H.S. & Meiler, J. RosettaEPR: an integrated tool for protein structure determination from sparse EPR data. J. Struct. Biol. 173, 506–514 (2011).
Meiler, J. & Baker, D. The fumarate sensor DcuS: progress in rapid protein fold elucidation by combining protein structure prediction methods with NMR spectroscopy. J. Magn. Reson. 173, 310–316 (2005).
Alexander, N., Bortolus, M., Al-Mestarihi, A., Mchaourab, H. & Meiler, J. De novo high-resolution protein structure determination from sparse spin-labeling EPR data. Structure 16, 181–195 (2008).
DiMaio, F. et al. Improved molecular replacement by density- and energy-guided protein structure optimization. Nature 473, 540–543 (2011).
Shen, Y. et al. De novo structure generation using chemical shifts for proteins with high-sequence identity but different folds. Protein Sci. 19, 349–356 (2010).
Herzog, F. et al. Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352 (2012).
Grishaev, A., Guo, L., Irving, T. & Bax, A. Improved fitting of solution X-ray scattering data to macromolecular structures and structural ensembles by explicit water modeling. J. Am. Chem. Soc. 132, 15484–15486 (2010).
Pandit, D. et al. Mapping of discontinuous conformational epitopes by amide hydrogen/deuterium exchange mass spectrometry and computational docking. J. Mol. Recognit. 25, 114–124 (2012).
Rathmann, D. et al. Ligand-mimicking receptor variant discloses binding and activation mode of prolactin-releasing peptide. J. Biol. Chem. 287, 32181–32194 (2012).
Combs, S., Kaufmann, K., Field, J.R., Bakely, R.D. & Meiler, J. Y95 and E444 interaction required for high-affinity S-citalopram binding in the human serotonin transporter. ACS Chem. Neurosci. 2, 75–81 (2011).
Nannemann, D.P., Kaufmann, K.W., Meiler, J. & Bachmann, B.O. Design and directed evolution of a dideoxy purine nucleoside phosphorylase. Protein Eng. Des. Sel. 23, 607–616 (2010).
Leach, A.R., Shoichet, B.K. & Peishoff, C.E. Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J. Med. Chem. 49, 5851–5855 (2006).
Smith, J.A., Vanoye, C.G., George, A.L., Meiler, J. & Sanders, C.R. Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry 46, 14141–14152 (2007).
Larkin, M.A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Pettersen, E.F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Ward, J.J., McGuffin, L.J., Buxton, B.F. & Jones, D.T. Secondary structure prediction with support vector machines. Bioinformatics 19, 1650–1655 (2003).
Leman, J.K., Koehler, J., Mueller, R., Karakas, M., Woetzel, N. & Meiler, J. Simultaneous prediction of protein secondary structure and transmembrane spans. Proteins http://dx.doi.org/10.1002/prot.24258 (10 April 2013).
Wang, C., Bradley, P. & Baker, D. Protein-protein docking with backbone flexibility. J. Mol. Biol. 373, 503–519 (2007).
Qian, B. et al. High-resolution structure prediction and the crystallographic phase problem. Nature 450, 259–264 (2007).
Lemmon, G., Kaufmann, K. & Meiler, J. Prediction of HIV-1 protease/inhibitor affinity using RosettaLigand. Chem. Biol. Drug Des. 79, 888–896 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38 (1996).
Wass, M.N., Kelley, L.A. & Sternberg, M.J.E. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 38, W469–W473 (2010).
Morris, G.M. et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Case, D.A. et al. The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005).
Alexander, N., Woetzel, N. & Meiler, J. Bcl::Cluster: A method for clustering biological molecules coupled with visualization in the Pymol molecular graphics system. Computational Advances in Bio and Medical Sciences (ICCABS), 2011 IEEE 1st International Conference on, 13–18 (2011).
Dunbrack, R.L. & Cohen, F.E. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 6, 1661–1681 (1997).
Simons, K.T. et al. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins 34, 82–95 (1999).
Kuhlman, B. & Baker, D. Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. USA 97, 10383–10388 (2000).
Neria, E., Fischer, S. & Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 105, 1902–1921 (1996).
Lazaridis, T. & Karplus, M. Effective energy function for proteins in solution. Proteins 35, 133–152 (1999).
Gordon, D.B., Marshall, S.A. & Mayo, S.L. Energy functions for protein design. Curr. Opin. Struct. Biol. 9, 509–513 (1999).
Wedemeyer, W.J. & Baker, D. Efficient minimization of angle-dependent potentials for polypeptides in internal coordinates. Proteins 53, 262–272 (2003).
Ramachandran, G.N., Ramakrishnan, C. & Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99 (1963).
Fleishman, S.J. et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 6, e20161 (2011).
Acknowledgements
We thank J. Smith, as well as R. Levinson, for testing the protocol and for their helpful comments. Work in the Meiler laboratory is supported through grants from the National Institutes of Health (R01 GM080403, R01 MH090192) and the National Science Foundation (Career 0742762).
Author information
Authors and Affiliations
Contributions
All authors contributed equally to this work. All authors wrote substantial portions of the main text, the figures and the supplementary information. S.A.C. proposed the composition of the work for the benefit of the scientific community, tested the presented protocol and managed submission. S.L.D. wrote instructions on how to install the software, generated the comparative models, wrote data-processing scripts and managed references. S.H.D. wrote the supplementary glossary and was responsible for overall editing of the work. G.H.L. wrote the RosettaLigand program in its present form. D.P.N. carefully read through the manuscript for consistency and accuracy and helped in the analysis of the generated models. E.D.N. also generated comparative models, performed all of the ligand docking and performed the data analysis. J.R.W. contributed several figures, data-processing scripts, specialty movers, wrote large sections of the tutorial and managed references. J.H.S. tested the protocol, wrote the Troubleshooting section and edited the manuscript for clarity. J.M. helped define the scope of the work and guided the process of establishing the protocol.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Discussion
Discussion of clustering using Rosetta, Using Constraints as Filters in RosettaScripts, Installing Rosetta 3.4, Testing Rosetta, and Glossary. (PDF 676 kb)
Supplementary Data
A zipped file that contains example input files, command lines, and output files that the user can download. This tutorial is provided to allow the user to gain handson experience with docking small molecules into comparative models with Rosetta. (ZIP 16433 kb)
Rights and permissions
About this article
Cite this article
Combs, S., DeLuca, S., DeLuca, S. et al. Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc 8, 1277–1298 (2013). https://doi.org/10.1038/nprot.2013.074
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2013.074
This article is cited by
-
The pyruvate decarboxylase activity of IpdC is a limitation for isobutanol production by Klebsiella pneumoniae
Biotechnology for Biofuels and Bioproducts (2022)
-
Engineering of a thermophilic dihydroxy-acid dehydratase toward glycerate dehydration for in vitro biosystems
Applied Microbiology and Biotechnology (2022)
-
A structure-based computational workflow to predict liability and binding modes of small molecules to hERG
Scientific Reports (2020)
-
Laboratory evolution of an alcohol dehydrogenase towards enantioselective reduction of difficult-to-reduce ketones
Bioresources and Bioprocessing (2019)
-
Biochemical and structural features of diverse bacterial glucuronoyl esterases facilitating recalcitrant biomass conversion
Biotechnology for Biofuels (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
