
Abstract
Current treatment strategies for anxiety disorders are predominantly symptom-based. However, a third of anxiety patients remain unresponsive to anxiolytics highlighting the need for more effective, mechanism-based therapeutic approaches. We have previously compared high vs low anxiety mice and identified changes in mitochondrial pathways, including oxidative phosphorylation and oxidative stress. In this work, we show that selective pharmacological targeting of these mitochondrial pathways exerts anxiolytic effects in vivo. We treated high anxiety-related behavior (HAB) mice with MitoQ, an antioxidant that selectively targets mitochondria. MitoQ administration resulted in decreased anxiety-related behavior in HAB mice. This anxiolytic effect was specific for high anxiety as MitoQ treatment did not affect the anxiety phenotype of C57BL/6N and DBA/2J mouse strains. We furthermore investigated the molecular underpinnings of the MitoQ-driven anxiolytic effect and found that MitoQ treatment alters the brain metabolome and that the response to MitoQ treatment is characterized by distinct molecular signatures. These results indicate that a mechanism-driven approach based on selective mitochondrial targeting has the potential to attenuate the high anxiety phenotype in vivo, thus paving the way for translational implementation as long-term MitoQ administration is well-tolerated with no reported side effects in mice and humans.
Similar content being viewed by others


INTRODUCTION
High anxiety levels are responsible for a significant health-related life quality loss in the general population (Saarni et al, 2007). Anxiety disorders, the most common psychiatric conditions, affect approximately one out of five people worldwide, are highly co-morbid with other major psychiatric diseases (Achim et al, 2011), and pose an additional burden to patients with a negative impact on recovery and disease outcome (Braga et al, 2005). Owing to the lack of in-depth molecular understanding of anxiety pathobiology, current therapeutic approaches are mainly symptom- rather than mechanism-driven with a third of patients being unresponsive to anxiolytic treatment (Bystritsky, 2006).
To elucidate the molecular underpinnings of anxiety traits, we have compared mice selectively inbred for ∼45 generations for high (HAB) or low anxiety-related behavior (LAB) according to their performance on the elevated plus maze (Kromer et al, 2005). Using a hypothesis-free systems biology platform based on proteomics, metabolomics, and bioinformatics (Filiou and Turck, 2012; Frank et al, 2009; Haegler et al, 2009; Zhang et al, 2009), we identified alterations in multiple mitochondrial pathways in high anxiety. The most predominant change was the increased expression of more than 60 proteins across all electron transport chain subunits of oxidative phosphorylation resulting in increased oxidative stress and decreased antioxidant capacity in HAB mice (Filiou et al, 2011, 2014; Iris et al, 2014).
In this work, we examined whether we can manipulate the high anxiety phenotype by pharmacologically targeting the identified mitochondrial changes in vivo. We treated HAB mice with MitoQ, a mitochondria-targeted compound that enhances the mitochondrial protection against oxidative damage (Cocheme et al, 2007). MitoQ consists of a lipophilic cation moiety that allows mitochondria-specific accumulation and a ubiquinone that is converted to the antioxidant ubiquinol by the activity of complex II of the electron transport chain (Smith et al, 2003). MitoQ was selected based on a series of criteria. First of all, long-term MitoQ administration has been reported to be non-toxic and well-tolerated in mice (Smith and Murphy, 2010). MitoQ is water soluble and can be administered orally through the drinking water, thus circumventing invasive and more stressful ways of administration (eg injections) that may interfere with the anxiety phenotype. Furthermore, MitoQ can cross the blood–brain barrier, albeit slowly, as it is detected in the mouse brain after treatment (Rodriguez-Cuenca et al, 2010). MitoQ administration was shown to have protective effects in animal models of diseases characterized by mitochondrial oxidative damage (Adlam et al, 2005). In brain disorders, MitoQ administration prevented cognitive decline and neuropathological correlates of Alzheimer’s disease in a transgenic mouse model (McManus et al, 2011).
We analyzed the behavioral and molecular effects of this selective mitochondrial targeting and investigated differences between MitoQ treatment responders and non-responders. We focused our analyses on cortical and hippocampal areas due to their involvement in emotional processing (Tovote et al, 2015) and on plasma as it is the specimen of choice for translational implementation. To determine whether our findings are specific for high anxiety, we also assessed the behavioral effects of MitoQ treatment in DBA/2J and C57BL/6N inbred mouse strains. Here we report on a molecular mechanism-driven anxiolytic effect of selective mitochondrial targeting which is specific for high anxiety.
MATERIALS AND METHODS
Animals
Mouse experiments were approved by local authorities and conducted according to current regulations for animal experimentation in Germany and the European Union (European Communities Council Directive 2010/63/EU). HAB (6 weeks old), C57BL/6N (Martinsried, Germany), and DBA/2J (Charles River, Sulzfeld, Germany) male mice were group housed under standard conditions in the animal facility of the Max Planck Institute of Psychiatry as previously described (Frank et al, 2009).
Total Antioxidant Capacity
Prior to the assignment to the MitoQ treatment or control group, tail blood was collected in 1.5 ml EDTA-coated microcentrifuge tubes (Kabe Labortechnik, Nümbrecht-Elsenroth, Germany) without anesthesia by a small incision in the dorsal tail vein using a scalpel blade. Plasma was obtained by centrifugation for 10 min, at 1300 g, 4 °C. Tail blood sampling took place between 0900 and 1100 hours. Plasma total antioxidant capacity (TAC) representing the amount of small molecule and protein antioxidants was assessed by a TAC kit (K274-100; Biovision, Mountain View, CA) according to the manufacturer’s instructions.
MitoQ Treatment
After a week of habituation, HAB mice were randomly assigned to MitoQ treatment and control groups. The treatment group of HAB mice (n=13) received 400 μM MitoQ for 10 weeks (MitoQ was provided in the form of MS010 (1.2 g/l), a β-cyclodextrin complex of MitoQuninone mesylate, corresponding to 400 μM of MitoQ). MitoQ was dissolved in drinking water and was prepared fresh twice a week. The control group of HAB mice (n=13) received only drinking water. Drinking behavior was monitored twice a week and animal weight once a week during the treatment period. C57BL/6N and DBA/2J mice (n=15 for the treatment group and n=15 for the control group for each strain) were treated with MitoQ following the same protocol as used for HAB mice.
Behavioral Phenotyping
During the tenth week of MitoQ treatment a behavioral battery assessing anxiety-related (dark–light box (DL), open field (OF)) and depression-like (tail suspension test (TST)) behavior was performed. Behavioral tests were kept to a minimum to avoid learning/memory effects and reduce the burden of continuous behavioral testing that may hamper behavioral readouts in mice. Experimenters were blind to animal treatment for all behavioral tests.
Dark–light box
The set up for DL was as previously described (Kromer et al, 2005) using 10 and 300–400lux for the dark and illuminated areas, respectively. Each mouse was placed in the corner of the darkened compartment facing away from the aperture. During the 5 min test, data were scored using Eventlog 1.0 (EMCO Software, Reykjavik, Iceland). A mouse was defined to have entered the lit box when both front paws and shoulders were inside the lit compartment.
Open field
OF was performed 2 days after DL as previously described (Bunck et al, 2009) illuminating the arena with dim light of 50lux.
Tail suspension test
TST was performed 2 days after OF as previously described (Kromer et al, 2005).
Animal Sampling
Two days after TST, animals were killed, blood was collected by cardiac puncture, and plasma was obtained by centrifugation for 10 min, at 1300 g, 4 °C. Mice were perfused with 0.9% saline. The prefrontal cortex, hippocampus, and cerebellum were dissected according to the mouse brain atlas (Paxinos and Franklin, 2001) and stored at −80 °C. Hippocampi and plasma were used for metabolite-based analyses and cortices were used for protein-based analyses.
MitoQ Quantification in the Brain
Brain tissue from all MitoQ-treated HAB treated animals (∼50 mg wet weight per animal) was used to estimate the amount of MitoQ in the brain as previously described (Rodriguez-Cuenca et al, 2010). Briefly, the tissue was homogenized, spiked with a deuterated MitoQ internal standard, and then extracted into acetonitrile/formic acid. The extract was then analyzed for MitoQ content by LC-MS/MS relative to a MitoQ standard curve and MitoQ levels between responder and non-responder groups were compared by unpaired Student’s t-test.
Metabolomics Analysis
Hippocampi and plasma from MitoQ-treated and untreated HAB mice were prepared for LC-MS/MS-based targeted metabolite analysis as previously described (Filiou et al, 2014). LC-MS/MS metabolomics measurements were performed at the Mass Spectrometry Core of Beth Israel Deaconess Medical Center (Boston, MA) using a 5500 QTRAP triple quadrupole mass spectrometer (AB/SCIEX, Framingham, MA) coupled to a Prominence UFCL HPLC system (Shimadzu, Columbia, MD). A targeted single reaction monitoring (SRM)-based platform that quantifies the levels of 286 metabolites was used (Yuan et al, 2012). Following normal phase chromatography using a 4.6-mm i.d. × 10 cm Amide Xbridge HILIC column (Waters Corp., Milford, MA) samples were introduced to the mass spectrometer. Selected metabolites were targeted in both positive and negative modes by positive/negative polarity switching. All other measurement parameters were as previously described (Webhofer et al, 2013).
Catalase Activity Determination
Catalase activity in the cortex of MitoQ-treated HAB mice was measured as previously described (Filiou et al, 2012) using a catalase assay kit (707002; Cayman, Ann Arbor, MI).
Western Blot
Western blot analysis in the cortex of MitoQ-treated HAB mice was performed as previously described (Filiou et al, 2010) using the following primary antibodies: Slc25a22 (1 : 2000, ab60332; Abcam, Cambridge, UK), malate dehydrogenase 1 (1 : 1000, ARP48284_T100; Aviva Systems Biology, San Diego, CA), HK1 (1 : 600, HPA007044; Sigma Aldrich, Darmstadt, Germany), and Prdx3 (1 : 2000, ab16751; Abcam) (10 μg per lane). Equal total protein loading was ensured by staining of the transfer membrane with Coomasie Brilliant Blue R-250 (BioRad, Hercules, CA) and signal intensity comparison.
Statistical Analysis
Unless otherwise specified, animal data were analyzed by the non-parametric Mann–Whitney test to address non-normal data distributions and are presented as mean±SEM. Metabolite data were statistically analyzed using MetaboAnalyst (Xia et al, 2009). For metabolites measured both in positive and negative modes, only one of the two measurements was included for further analysis. Metabolites with >10% missing values were not considered for statistical analysis. Data were median-normalized (row-wise) and pareto-scaled (column-wise). To identify metabolite level alterations the significance analysis of microarrays (SAM) was used. SAM also follows non-parametric statistics and uses permutation-based analysis to determine statistically significant changes in metabolite levels, thus avoiding parametric assumptions about the distribution of individual metabolites unlike other approaches, which assume equal variance and/or independence of tests (Tusher et al, 2001). For SAM, significant differences with FDR<0.1 and q<0.1 were accepted. Metabolite data are presented in box and whisker plots of median normalized peak intensities. For metabolite pathway identification the KEGG database was used (http://www.genome.jp/kegg/genes.html).
RESULTS
MitoQ Treatment Exerts Anxiolytic Effects in HAB Mice
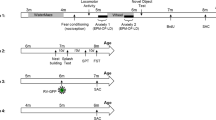
HAB mice were treated with MitoQ for 10 weeks (Figure 1). To address potential effects of intra-line heterogeneity, we compared TAC in tail plasma prior to the beginning of the MitoQ administration between treatment and control HAB groups and found no difference (Supplementary Figure S1A). MitoQ treatment did not result in morphological or body weight alterations in HAB mice (Supplementary Figure S1B). During the last week of treatment, mice were subjected to a behavioral battery of anxiety-related (OF, DL) and depression-like (TST) tests. We observed an anxiolytic effect in DL with MitoQ-treated HAB mice spending significantly more time in the lit compartment compared with untreated HAB mice (Figure 2a). No significant differences were observed in OF or TST (Supplementary Figure S1C and D).

Experimental set up of MitoQ treatment in HAB mice. DL, dark–light box; HAB, high anxiety-related behavior; OF, open field; TAC, total antioxidant capacity; TST, tail suspension test.

MitoQ treatment exerts anxiolytic effects in HAB mice. (a) HAB mice treated with 400 μM MitoQ (n=13) spent more time in the lit compartment of DL compared with untreated HAB mice (n=13) (p=0.0455). (b) Decreased levels of sarcosine (p=0.0009), AMP (p=0.001), and ascorbic acid (p=0.002) in hippocampi of MitoQ-treated compared with untreated HAB mice. CT, untreated HAB mice; DL, dark–light box; MitoQ, MitoQ-treated HAB mice. *p<0.05.
MitoQ Treatment Alters the Hippocampal Metabolome
To identify molecular correlates of the MitoQ-driven anxiolytic effect in DL, we compared the hippocampal metabolomes of MitoQ-treated vs untreated HAB mice by an SRM-based, targeted metabolomics platform (Yuan et al, 2012). After application of exclusion criteria, we quantified 222 metabolites in the hippocampus (Supplementary Table S1). SAM analysis revealed decreased levels of sarcosine, AMP, and ascorbic acid in MitoQ-treated compared with untreated HAB mice (Figure 2b).
MitoQ Anxiolytic Effects are Present Only in High Anxiety
To investigate whether the MitoQ-driven anxiolytic effects are specific for high anxiety, 400 μM MitoQ were administered for 10 weeks to mice from C57BL/6N and DBA/2J strains that are not inbred for anxiety traits following the same protocol for treatment and behavioral testing that was used for HAB mice. For both strains, no behavioral differences were observed in anxiety-related (DL, OF) and depression-like (TST) behavior between treated and untreated mice (Supplementary Figure S2). These results indicate that the anxiolytic effects of MitoQ are specific for HAB mice.
MitoQ DL Responders Show Altered Brain Metabolite and Protein Signatures Compared with Non-Responders
It has been recently reported that responders to psychiatric drug treatment show distinct molecular signatures compared with non-responders (Belzeaux et al, 2012). Based on these findings, we examined whether a similar phenomenon can also be observed for the MitoQ treatment response. It should be noted that although HAB mice are inbred for anxiety traits, variability of inbred strains in behavioral testing readouts is comparable to that of outbred strains. Based on DL behavioral data, MitoQ-treated mice were median separated to high responders (n=6) and low responders (n=7) according to their % time spent in the DL lit compartment (Figure 3a). For simplification purposes, these two groups will be referred to as responders and non-responders from now on. The group of DL MitoQ responders showed a significant increase in % time spent in the lit compartment compared with untreated HAB mice while DL MitoQ non-responders showed no difference in the % time spent in the lit compartment compared with untreated HAB mice (Supplementary Figure S3).

Altered molecular profiles between MitoQ responders and non-responders in DL. (a) Mice were classified into MitoQ responder and non-responder groups based on the median value of the % time spent in DL lit compartment. (b) No differences in brain MitoQ amount (measured as pmol/mg brain tissue) between MitoQ responders and non-responders in DL (p>0.05). (c) Increased levels of xanthosine 5-phosphate (p=0.0003) and UDP (p=0.0006) in MitoQ responders compared with non-responders. (d) Increased catalase activity (p=0.0221) in MitoQ responder compared with non-responder cortices. (e) Decreased expression levels of Slc25a22 (p=0.0303) and increased expression levels of MDH1 (p=0.0177) and HK1 (p=0.0101) in MitoQ responder vs non-responder cortices. HK1, hexokinase 1; MDH1, malate dehydrogenase 1; NR, non-responders; R, responders; UDP, uridine diphosphate. *p<0.05.
To ensure that the response to treatment was not a consequence of different MitoQ amounts in the brain, we determined MitoQ brain levels in all MitoQ-treated HAB mice by LC-MS/MS. MitoQ was present in all treated HAB brains, and no difference in MitoQ brain amounts was observed between DL responders and non-responders (Figure 3b). We then compared the brain metabolomes of the two groups. Of the 226 metabolites quantified in hippocampi (Supplementary Table S2), we found increased levels of xanthosine 5-phosphate and uridine diphosphate in DL responders compared with non-responders (Figure 3c). Based on identified differences in mitochondrial pathways between high and low anxiety, which included oxidative stress, metabolism, and mitochondrial import/transport (Filiou et al, 2011), we investigated the expression of a selected number of mitochondrial/energy metabolism proteins and oxidative stress markers in MitoQ-responder vs non-responder cortices. We observed an increased activity of the antioxidant catalase, which detoxifies reactive oxygen species (Figure 3d). In addition, we found decreased expression of the mitochondrial glutamate carrier Slc25a22 and increased expression of the enzyme malate dehydrogenase 1 and the glycolysis enzyme hexokinase (HK1) in DL MitoQ responders compared with non-responders (Figure 3e).
MitoQ OF Responders Show a Distinct Metabolite Signature in Plasma Compared with Non-Responders
To get further insights into the molecular correlates of MitoQ treatment response we also classified MitoQ-treated HAB mice according to their performance in OF. HAB mice that entered the OF inner zone (n=3) were classified as responders. HAB mice that did not enter the OF inner zone were classified as non-responders (n=10) (Figure 4a). MitoQ brain levels were not significantly different between the two groups (Figure 4b). We then quantified the levels of 246 metabolites in plasma (Supplementary Table S3) and found altered levels of 11 metabolites between MitoQ OF responders and non-responders (Table 1). These metabolites are primarily involved in secondary metabolite and amino acid metabolism. We also observed increased expression of peroxiredoxin-3 (Prdx3), a mitochondrial protein antioxidant, in MitoQ-responder compared with non-responder cortices (Figure 4c).

Molecular changes between MitoQ responders and non-responders in OF. (a) Mice were classified into MitoQ responder and non-responder groups based on whether they entered the inner zone in OF. (b) No differences in brain MitoQ amount (measured as pmol/mg brain tissue) between MitoQ responders and non-responders in OF (p>0.05). (c) Increased Prdx3 protein expression in MitoQ responders compared with non-responder cortices (p=0.0467). NR, non-responders; OF, open field; Prdx3, peroxiredoxin 3; R, responders. *p<0.05.
DISCUSSION
In this work we report that pharmacological manipulation of mitochondria exerts anxiolytic effects in vivo. This approach is based on previous, hypothesis-free data that revealed altered mitochondrial pathways in a high anxiety mouse strain (Filiou et al, 2011). Following up on these findings, we hypothesized that selective pharmacological targeting of these mitochondrial pathways may result in a mitigation of the high anxiety phenotype. HAB mice, which were treated with the mitochondria-specific antioxidant MitoQ, showed a decreased anxiety-related behavior in the DL, a test assessing the aversion of mice to lit environments (Bourin and Hascoet, 2003). Owing to selective inbreeding for the high anxiety traits for ∼45 generations, the HAB phenotype is extremely robust and barely amenable to any kind of pharmacological manipulation (Landgraf et al, 2007). Intriguingly, HAB mice, which largely remain unresponsive to chronic treatment with conventional anxiolytics or widely used non-specific antioxidants (Sah et al, 2012), responded to MitoQ that specifically targets mitochondria.
At the molecular level, MitoQ treatment resulted in altered levels of sarcosine, AMP, and ascorbic acid in the HAB hippocampus. Sarcosine (N-methylglycine) has been discussed in the context of psychiatric disorders as an inhibitor of the glycine transporter. Inhibition of glycine transporters has been suggested to both induce and attenuate anxiety-related behavior (Depoortere et al, 2005; Labrie et al, 2009). AMP is involved in purine metabolism, components of which have been associated with symptoms of major psychiatric disorders (Yao et al, 2012) whereas ascorbic acid is a known antioxidant.
In addition to comparing treated vs untreated HAB mice, we performed a molecular comparison between MitoQ responders vs non-responders in DL and OF anxiety-related tests. Ideally, anxiolytic response to MitoQ administration can be assessed at the level of individual animals by performing the anxiety-related tests before and after treatment. However, repeating the same behavioral test more than one time has been shown to compromise its validity due to strong memory effects of mice (Holmes et al, 2001). Therefore, we assessed the effect of treatment in responders vs the untreated control group and showed that responders have significantly lower anxiety-related behavior in the DL, unlike the non-responders, vs the control group (Supplementary Figure 3).
In our analysis, comparison of MitoQ responders vs non-responders revealed altered protein expression in the brain and a distinct metabolite signature in plasma of HAB mice that responded to treatment. Glucose metabolism was one of the predominantly implicated pathways as altered levels of glycolysis (HK1, fructose-6-phosphate) and glyconeogenesis (uridine diphosphate, hexose-phosphate) enzymes and metabolites were observed upon MitoQ treatment. Increased levels of citric acid cycle components (fumarate) implicate mitochondrial energy metabolism in response to MitoQ whereas several antioxidant-related readouts were affected, including increased catalase activity as well as altered Prdx3 and glutathione disulfide levels.
Although different metabolites were affected between MitoQ-treated vs untreated HAB mice and between MitoQ responders vs non-responders, the overall affected pathways are partially overlapping, with oxidative stress being the common denominator of the observed changes. Response to MitoQ may depend on the antioxidant status of each animal upon treatment, which is shaped by different antioxidants. Variations in the basal levels of these antioxidants across individual animals may interfere with the MitoQ-driven antioxidant effect and result into differences in MitoQ response. Based on our results, we suggest that by acting as a regulator of oxidative stress in mitochondria, MitoQ affects on the one hand the equilibrium of antioxidant defense and on the other mitochondrial energy metabolism. Oxidative stress has been linked to increased anxiety (Rammal et al, 2008) and MitoQ may complement the action of more generalized antioxidants (eg ascorbic acid). Alterations in the abundance of antioxidants such as catalase, the activity of which was found to be increased in MitoQ-responders, have been also shown to exert beneficial effects in anxiety-related behavior (Olsen et al, 2013), in accordance to our findings. Furthermore, prevention of oxidative damage to the mitochondria by MitoQ may alter mitochondrial energy metabolism pathways that have been also implicated in high anxiety (Szego et al, 2010). In a future set of experiments, tissue cultures from MitoQ-responder mice will be established to investigate in detail the effect of MitoQ on protein expression. Future work will also include assessment of MitoQ effects in a larger number of HAB mice as well as in other anxiety mouse models.
Interestingly, there is extensive overlap between the MitoQ-driven molecular alterations in HAB mice reported here and the molecular differences previously described between HAB and LAB mice (Filiou et al, 2011, 2014; Zhang et al, 2011). AMP levels were found to be altered both in MitoQ-treated vs untreated HAB brain and in HAB vs LAB brain (Filiou et al, 2014). We also observed a considerable overlap between the molecular alterations of MitoQ responders vs non-responders and HAB vs LAB mice. Both MitoQ responders and LAB mice (Zhang et al, 2011) have higher plasma myo-inositol levels. At the protein level, Prdx3 expression in the brain was higher in responders vs non-responders. Members of the peroxiredoxin protein family were also expressed at higher levels in LAB compared with HAB mice (Filiou et al, 2011). This was also the case for Slc25a22, and HK1 with altered expression levels in MitoQ responders vs non-responders as well as in HAB vs LAB mice. At the pathway level, amino acid metabolism was differentially affected in MitoQ responder vs non-responder plasma as well as in HAB vs LAB mouse plasma.
Our findings suggest that MitoQ treatment exerts its anxiolytic effects through molecular pathways, which are affected in anxiety-related behavior. The resulting anxiolytic response of mitochondrial pathways to MitoQ treatment indicates that a mechanism-driven pharmacological strategy to manipulate the high anxiety phenotype is viable.
Mitochondrial targeting has emerged as a therapeutic approach for several disorders involving mitochondrial oxidative stress (Edeas and Weissig, 2013; Fulda et al, 2010; Smith et al, 2012). Although accumulating evidence points towards mitochondrial dysfunction in psychiatric disorders (Manji et al, 2012), the therapeutic potential of mitochondrial targeting has only been recently highlighted for bipolar disorder (de Sousa et al, 2014) and autism (Ghanizadeh et al, 2013) and has never been tested in vivo. Our study is the first in vivo application of selective mitochondrial targeting in the context of psychiatric disorders. The anxiolytic effect of MitoQ in mice selectively bred for an extreme genetic predisposition towards high anxiety is indicative of the potential that selective mitochondrial targeting holds to manipulate behavioral phenotypes related to trait anxiety. The fact that the anxiolytic effect was only present in HAB mice and not in other mouse strains which are not inbred for high anxiety, underlines a specificity that could be of use for translational applications in patients with anxiety disorders. Importantly, long-term treatment with MitoQ is also well-tolerated in human cohorts and was administered in clinical trials for Parkinson’s disease (Snow et al, 2010) and chronic hepatitis C (Gane et al, 2010) with no adverse side effects.
In summary, we show that mitochondrial targeting exerts beneficial effects in high anxiety in vivo. This is a mechanism-driven approach to manipulate disease symptoms and is based on reported metabolic and mitochondrial alterations which have been linked to anxiety pathogenesis (Einat et al, 2005; Filiou et al, 2011). As advanced methodologies are now available for selective compound transport to mitochondria (Pathak et al, 2014; Smith et al, 2011), pharmacological manipulation of mitochondrial pathways may serve as the basis for a novel therapeutic path for translational applications in high anxiety and stress-related psychiatric disorders.
FUNDING AND DISCLOSURE
This work was funded by a grant from the Deutsche Forschungsgemeinschaft (FI 1895/1-1) to MDF, the Max Planck Society (MDF, CWT) and the UK MRC (MPM and AL). MPM holds IP related to MitoQ and consults for Antipodean Pharmaceuticals, who are commercializing MitoQ. All other authors declare no conflict of interest.
References
Achim AM, Maziade M, Raymond E, Olivier D, Merette C, Roy MA (2011). How prevalent are anxiety disorders in schizophrenia? A meta-analysis and critical review on a significant association. Schizophr Bull 37: 811–821.
Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP et al (2005). Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 19: 1088–1095.
Belzeaux R, Bergon A, Jeanjean V, Loriod B, Formisano-Treziny C, Verrier L et al (2012). Responder and nonresponder patients exhibit different peripheral transcriptional signatures during major depressive episode. Transl Psychiatry 2: e185.
Bourin M, Hascoet M (2003). The mouse light/dark box test. Eur J Pharmacol 463: 55–65.
Braga RJ, Mendlowicz MV, Marrocos RP, Figueira IL (2005). Anxiety disorders in outpatients with schizophrenia: prevalence and impact on the subjective quality of life. J Psychiatr Res 39: 409–414.
Bunck M, Czibere L, Horvath C, Graf C, Frank E, Kessler MS et al (2009). A hypomorphic vasopressin allele prevents anxiety-related behavior. PLoS One 4: e5129.
Bystritsky A (2006). Treatment-resistant anxiety disorders. Mol Psychiatry 11: 805–814.
Cocheme HM, Kelso GF, James AM, Ross MF, Trnka J, Mahendiran T et al (2007). Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion 7: S94–S102.
de Sousa RT, Machado-Vieira R, Zarate CA, Manji HK (2014). Targeting mitochondrially mediated plasticity to develop improved therapeutics for bipolar disorder. Expert Opin Ther Targets 18: 1131–1147.
Depoortere R, Dargazanli G, Estenne-Bouhtou G, Coste A, Lanneau C, Desvignes C et al (2005). Neurochemical, electrophysiological and pharmacological profiles of the selective inhibitor of the glycine transporter-I SSR504734, a potential new type of antipsychotic. Neuropsychopharmacology 30: 1963–1985.
Edeas M, Weissig V (2013). Targeting mitochondria: strategies, innovations and challenges: the future of medicine will come through mitochondria. Mitochondrion 13: 389–390.
Einat H, Yuan P, Manji HK (2005). Increased anxiety-like behaviors and mitochondrial dysfunction in mice with targeted mutation of the Bcl-2 gene: further support for the involvement of mitochondrial function in anxiety disorders. Behav Brain Res 165: 172–180.
Filiou MD, Asara JM, Nussbaumer M, Teplytska L, Landgraf R, Turck CW (2014). Behavioral extremes of trait anxiety in mice are characterized by distinct metabolic profiles. J Psychiatr Res 58: 115–122.
Filiou MD, Bisle B, Reckow S, Teplytska L, Maccarrone G, Turck CW (2010). Profiling of mouse synaptosome proteome and phosphoproteome by IEF. Electrophoresis 31: 1294–1301.
Filiou MD, Teplytska L, Otte DM, Zimmer A, Turck CW (2012). Myelination and oxidative stress alterations in the cerebellum of the G72/G30 transgenic schizophrenia mouse model. J Psychiatr Res 46: 1359–1365.
Filiou MD, Turck CW (2012). Psychiatric disorder biomarker discovery using quantitative proteomics. Methods Mol Biol 829: 531–539.
Filiou MD, Zhang YY, Teplytska L, Reckow S, Gormanns P, Maccarrone G et al (2011). Proteomics and metabolomics analysis of a trait anxiety mouse model reveals divergent mitochondrial pathways. Biol Psychiatry 70: 1074–1082.
Frank E, Kessler MS, Filiou MD, Zhang Y, Maccarrone G, Reckow S et al (2009). Stable isotope metabolic labeling with a novel N-enriched bacteria diet for improved proteomic analyses of mouse models for psychopathologies. PLoS One 4: e7821.
Fulda S, Galluzzi L, Kroemer G (2010). Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 9: 447–464.
Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM et al (2010). The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int 30: 1019–1026.
Ghanizadeh A, Berk M, Farrashbandi H, Shoushtari AA, Villagonzalo KA (2013). Targeting the mitochondrial electron transport chain in autism, a systematic review and synthesis of a novel therapeutic approach. Mitochondrion 13: 515–519.
Haegler K, Mueller NS, Maccarrone G, Hunyadi-Gulyas E, Webhofer C, Filiou MD et al (2009). QuantiSpec-quantitative mass spectrometry data analysis of 15N-metabolically labeled proteins. J Proteomics 71: 601–608.
Holmes A, Iles JP, Mayell SJ, Rodgers RJ (2001). Prior test experience compromises the anxiolytic efficacy of chlordiazepoxide in the mouse light/dark exploration test. Behave Brain Res 122: 159–167.
Iris F, Filiou M, Turck CW (2014). Differential proteomics analyses reveal anxiety-associated molecular and cellular mechanisms in cingulate cortex synapses. AJPN 2: 25–42.
Kromer SA, Kessler MS, Milfay D, Birg IN, Bunck M, Czibere L et al (2005). Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. J Neurosci 25: 4375–4384.
Labrie V, Clapcote SJ, Roder JC (2009). Mutant mice with reduced NMDA-NR1 glycine affinity or lack of D-amino acid oxidase function exhibit altered anxiety-like behaviors. Pharmacol Biochem Behav 91: 610–620.
Landgraf R, Kessler MS, Bunck M, Murgatroyd C, Spengler D, Zimbelmann M et al (2007). Candidate genes of anxiety-related behavior in HAB/LAB rats and mice: focus on vasopressin and glyoxalase-I. Neurosci Biobehav Rev 31: 89–102.
Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M, Chen G (2012). Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci 13: 293–307.
McManus MJ, Murphy MP, Franklin JL (2011). The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J Neurosci 31: 15703–15715.
Olsen RH, Johnson LA, Zuloaga DG, Limoli CL, Raber J (2013). Enhanced hippocampus-dependent memory and reduced anxiety in mice over-expressing human catalase in mitochondria. J Neurochem 125: 303–313.
Pathak RK, Marrache S, Harn DA, Dhar S (2014). Mito-DCA: a mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem Biol 9: 1178–1187.
Paxinos G, Franklin K (2001) The Mouse Brain in Stereotaxic Coordinates, 2nd edn. Academic Press: San Diego, CA.
Rammal H, Bouayed J, Younos C, Soulimani R (2008). Evidence that oxidative stress is linked to anxiety-related behaviour in mice. Brain Behav Immun 22: 1156–1159.
Rodriguez-Cuenca S, Cocheme HM, Logan A, Abakumova I, Prime TA, Rose C et al (2010). Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic Biol Med 48: 161–172.
Saarni SI, Suvisaari J, Sintonen H, Pirkola S, Koskinen S, Aromaa A et al (2007). Impact of psychiatric disorders on health-related quality of life: general population survey. Br J Psychiatry 190: 326–332.
Sah A, Schmuckermair C, Sartori SB, Gaburro S, Kandasamy M, Irschick R et al (2012). Anxiety-rather than depression-like behavior is associated with adult neurogenesis in a female mouse model of higher trait anxiety- and comorbid depression-like behavior. Transl Psychiatry 2: e171.
Smith RAJ, Hartley RC, Cocheme HM, Murphy MP (2012). Mitochondrial pharmacology. Trends Pharmacol Sci 33: 341–352.
Smith RAJ, Hartley RC, Murphy MP (2011). Mitochondria-targeted small molecule therapeutics and probes. Antioxid Redox Sign 15: 3021–3038.
Smith RAJ, Murphy MP (2010). Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci 1201: 96–103.
Smith RAJ, Porteous CM, Gane AM, Murphy MP (2003). Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci USA 100: 5407–5412.
Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V et al (2010). A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Movement Disord 25: 1670–1674.
Szego EM, Janáky T, Szabó Z, Csorba A, Kompagne H, Müller G et al (2010). A mouse model of anxiety molecularly characterized by altered protein networks in the brain proteome. Eur Neuropsychopharmacol 20: 96–111.
Tovote P, Fadok JP, Luthi A (2015). Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16: 317–331.
Tusher VG, Tibshirani R, Chu G (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121.
Webhofer C, Gormanns P, Reckow S, Lebar M, Maccarrone G, Ludwig T et al (2013). Proteomic and metabolomic profiling reveals time-dependent changes in hippocampal metabolism upon paroxetine treatment and biomarker candidates. J Psychiatr Res 47: 289–298.
Xia JG, Psychogios N, Young N, Wishart DS (2009). MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res 37: W652–W660.
Yao JK, Condray R, Dougherty GG, Keshavan MS, Montrose DM, Matson WR et al (2012). Associations between purine metabolites and clinical symptoms in schizophrenia. PLoS One 7: e42165.
Yuan M, Breitkopf SB, Yang X, Asara JM (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7: 872–881.
Zhang YY, Filiou MD, Reckow S, Gormanns P, Maccarrone G, Kessler MS et al (2011). Proteomic and metabolomic profiling of a trait anxiety mouse model implicate affected pathways. Mol Cell Proteomics 10: M111.008110.
Zhang YY, Webhofer C, Reckow S, Filiou MD, Maccarrone G, Turck CW (2009). A MS data search method for improved 15N-labeled protein identification. Proteomics 9: 4265–4270.
Acknowledgements
We thank Albin Varga and the personnel of the animal facility at the Max Planck Institute of Psychiatry for invaluable help with animal experiments. The authors also thank Rainer Landgraf and Carsten Wotjak for their insightful comments on the manuscript as well as Christiane Rewerts and Igor Stosic for expert assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
About this article

Cite this article
Nussbaumer, M., Asara, J., Teplytska, L. et al. Selective Mitochondrial Targeting Exerts Anxiolytic Effects In Vivo. Neuropsychopharmacol 41, 1751–1758 (2016). https://doi.org/10.1038/npp.2015.341
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.341
This article is cited by
-
Mitochondria as a therapeutic target for common pathologies
Nature Reviews Drug Discovery (2018)
