
Abstract
Aberrant expression of neuroendocrine markers is extremely rare in endothelial neoplasms, with only a single report describing three cases. Although originally classified as conventional angiosarcoma, further assessment of these tumors revealed a strikingly composite morphology composed of retiform and epithelioid elements reminiscent of composite hemangioendothelioma, a rare subtype of hemangioendothelioma. To further investigate these findings, available materials from 11 morphologically distinctive endothelial tumors showing neuroendocrine marker expression were retrieved from our archives. Immunohistochemistry for CD31, CD34, FLI-1, synaptophysin, chromogranin, D2-40, ERG, keratin (OSCAR), and CAMTA1 was performed. Total RNA from five cases were extracted and subjected to whole transcriptome sequencing. Clinical follow-up was obtained. These tumors were found to arise in five males and six females in patients from 9 to 55 years in age (median 47 years). They arose both in superficial (wrist, ankle, scalp, hip, and foot) and deep (periaortic tissues, C5 vertebra, pulmonary vein, and liver) locations. All contained elongated, retiform vascular channels lined by hyperchromatic ‘hobnail’ endothelial cells and a solid growth of uniform epithelioid cells reminiscent of epithelioid hemangioendothelioma. Hemangioma-like foci also lined by hobnail endothelial cells were frequently present. Mitotic activity was typically <1/10 HPF, and necrosis or areas of conventional angiosarcoma was absent. The results of immunohistochemistry were: CD31 (10/10), FLI-1 (10/10), ERG (9/9), CD34 (5/10), D2-40 (7/10), synaptophysin (11/11), chromogranin A (1/11), CD56 (5/11), keratin (0/11), and CAMTA1 (0/6). Sequencing analysis showed one case with PTBP1-MAML2 and one case with EPC1-PHC2 fusion transcripts; fusion transcripts were not identified in the remaining cases. Follow-up (8 cases) revealed local recurrence in one patient and metastatic spread in four individuals (bone, lung, liver, and brain). One person died of disease. Although the morphological features of these tumors are characteristic of composite hemangioendothelioma, this distinctive subset with neuroendocrine differentiation more often involves deep locations and displays more aggressive behavior than typically described in other cases of composite hemangioendothelioma.
Similar content being viewed by others

Main
Composite hemangioendothelioma is an extraordinarily rare endothelial tumor of intermediate (borderline) malignancy, first formally described by Nayler et al.1 Composite hemangioendothelioma was first included in the World Health Organization (WHO) classification of Tumors of Soft Tissue and Bone in 2002, defined as a ‘locally aggressive, rarely metastasizing neoplasm with vascular differentiation, containing an admixture of histologically benign, intermediate and malignant components’ (emphasis added).2 However, the definition of composite hemangioendothelioma was modified in the more recent 2013 WHO classification to ‘locally aggressive, rarely metastasizing vascular neoplasm, containing an admixture of histologically distinct components’ (emphasis added).3 This latter definition places more emphasis on the presence of multiple histologically distinct components and not on whether the components are benign, intermediate, or malignant. Altogether, fewer than 40 cases of composite hemangioendothelioma have been reported.1, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 Composite hemangioendothelioma occur chiefly in adults, with very rare pediatric or congenital cases, and predominantly involve the skin and superficial soft tissues, with rare cases reported in lymph nodes, spleen, kidney and other deep locations.6 They have been reported to occur in association with vascular malformations, in a patient with Mafucci syndrome and in the setting of prior irradiation and lymphedema. In general, the prognosis for patients with composite hemangioendothelioma has been reported to be favorable, with a high rate of local recurrence (~50%), but a low risk of lymph node (6%) or distant (<1%) metastases.1, 6, 23
Over the past several years, we have encountered a small number of unusual vascular neoplasms showing neuroendocrine marker expression, most often synaptophysin. Although we initially regarded these neoplasms as ‘angiosarcoma,’28 we have more recently come to appreciate the ‘composite’ features of these tumors, with a distinctive admixture of retiform, nested, and solid/epithelioid areas, somewhat reminiscent of epithelioid hemangioendothelioma. We therefore undertook the present study to more completely elucidate the clinicopathological, immunohistochemical, and molecular genetic features of these and similar endothelial neoplasms from our collective archives.
Materials and methods
Approval for this study was granted by the Institutional Review Board of Mayo Clinic, Rochester, Minnesota. All available routinely stained and immunohistochemistry slides from 15 tumors previously diagnosed as ‘angiosarcoma showing neuroendocrine marker expression’ (three cases), ‘hemangioendothelioma with neuroendocrine marker expression’ (nine cases), and ‘composite hemangioendothelioma’ (three cases) were retrieved from our institutional and consultation archives and re-reviewed. The ‘angiosarcomas showing neuroendocrine marker expression’ had been previously reported by one of us (ALF);28 of these three cases, one was included in the present study (Case 1 from this prior study, with additional clinical information), and two were felt to show different morphological features and were excluded. Two cases previously reported by one of us (ALF) as ‘composite hemangioendothelioma’24 lacked available tissue blocks and were excluded. The final study population thus consisted of 11 cases, all with available formalin-fixed, paraffin-embedded tissue blocks or unstained slides.
Clinical information, including patient age and sex, tumor site and size, surgical interventions, adjuvant therapy, local recurrences, distant metastases, and current status was obtained from the clinical records, pathology reports, and contributing pathologists and clinicians. Pathologic features, including the presence of a precursor lesion, tumor growth patterns, cytologic features, mitotic activity, and the presence or absence of necrosis were recorded.
For immunohistochemistry, formalin-fixed paraffin-embedded sections of the tumor were immunostained at the Mayo Clinic using heat-induced epitope retrieval, the Ventana Ultraview detection system (Ventana Medical Systems, Tucson, AZ, USA) and commercially available antibodies directed against CD31 (JC/70a, 1/350; Dako Corp.), CD34 (PWS44, 1/100; Leica Biosystems, Buffalo Grove, IL, USA), FLI-1 (G146-254,1/50; BD Pharmingen, Franklin Lakes, NJ, USA), chromogranin A (LK2H10, prediluted; Ventana Medical Systems), D2-40 (D2-40, 1/200; Covance, Princeton, NJ, USA), CD56 (123C3, 1/100; Dako), ERG (9FY, 1/25; Biocare Medical, Concord, CA, USA), synaptophysin (27G12, 1/50; Leica Biosystems), and wide-spectrum keratin (OSCAR, 1/100; Covance). Additionally, using heat-induced epitope retrieval and the Leica Bond III detection system (Leica Microsystems), immunohistochemistry for the epithelioid hemangioendothelioma-associated marker CAMTA1 (polyclonal, 1/200; Novus Biological) was performed at the University of California, San Francisco. Also, immunohistochemistry for synaptophysin and chromogranin A was performed on whole section and tissue microarray sections from 101 ‘control’ vascular tumors, including retiform hemangioendothelioma (3 cases), Dabska-type hemangioendothelioma (papillary intralymphatic angioendothelioma) (2 cases), conventional angiosarcoma (24 cases), epithelioid hemangioendothelioma (17 cases), hemangioma (44 cases), lymphangioma (7 cases), littoral cell angioma (3 cases), and glomeruloid hemangioma (1 case).
To assess for fusion transcripts, RNA was extracted from three cases using formalin-fixed paraffin-embedded tissue (Qiagen, Hilden, Germany). Hundred nanograms of RNAs from tumor tissue was used to generate libraries using TruSeq RNA Access library prep protocol (Illumina, San Diego, CA, USA). Sequencing was performed using a HiSeq 2000 instrument with a 101 cycle paired-end read. FASTQ formatted raw files were mapped and aligned to hg19 using MAP-RSeq v.2.0.0 workflow.29 Fusion transcripts were identified with Tophat-Fusion,30 and the quality of the RNA-seq was assessed with Fusion Sense.31
To validate the fusion transcripts found by sequencing, fluorescent in situ hybridization studies were used using lab-developed break-apart probes targeted to MAML2 and EPC1 genes. The probe sets were constructed with bacterial artificial chromosomes selected using the University of California Santa Cruz Biotechnology Genome Browser and Database (http://genome.ucsc.edu), genome assembly hg19, and then obtained from Invitrogen (Carlsbad, CA, USA). DNA was isolated from bacterial cultures by a Plasmid Maxi Kit (Qiagen, Valencia, CA, USA) and fluorescently labeled via nick translation (Abbott Molecular, Des Plaines, IL, USA). Clone specificity was verified by PCR and by FISH on metaphases from normal male blood specimens. Once individually validated, the clones were combined for use as two break-apart strategy probe sets. The 3′ portion of EPC1 was detected by RP11-108P17, CTD-2309O18, and CTD-2334D16 labeled in Spectrum Green, and the 5′ portion of EPC1 was found by RP11-104F5 labeled in Spectrum Orange. The 3′ portion of MAML2 was detected by CTD-2252L1, RP11-1123F20, RP11-936C10, RP11-7D4, and CTD-254417 labeled in Spectrum Green, and the 5′ portion of MAML2 was covered by RP11-1056O10, CTD-2325K3 and RP11-8N17 labeled in Spectrum Orange. Following hybridization, these tumors were then scored as either positive (>15%) or negative (<15%).
Results
Clinical Findings Including Patient Follow-Up
The Table 1 summarizes the clinicopathological, immunohistochemical, and genetic features of the 11 studied cases. The tumors occurred in five males and six females, ranging in age from 9 to 55 years (median 47 years of age). Most were relatively small (median 3.0 cm, range 0.4–9.5 cm). They occurred both in the skin and superficial soft tissues (finger, wrist, arm, cheek, ear, hip, ankle, and foot) and in deeply situated structures (periaortic soft tissues, C5 vertebra, pulmonary vein, and liver). In one patient (case 6), the lesion rapidly arose behind the ear at a prior insertion site of an implantable hearing device. Another tumor (case 11) presented as a new, rapidly growing mass within a long-standing lymphatic vascular malformation of the cheek.
Clinical follow-up information was available for 8 of 11 patients (range 6–28 months, median 10 months). Local recurrence of the tumor was seen in two patients. Distant metastatic disease occurred in four patients, with metastatic sites including the bone, lung, liver, and brain. At the time of last follow-up, four patients are alive without disease, three are alive with persistent/metastatic disease, and one is dead of disease.
Morphological Findings
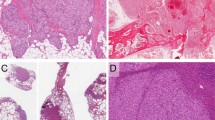
The morphological and immunohistochemical features of five representative cases are illustrated in Figures 1,2,3,4, and 5. Microscopically, all tumors grew in an infiltrative manner into the surrounding soft tissues, although some were relatively well-circumscribed and others much more infiltrative, with perineurial and perivascular invasion. The most notable feature of all of the tumors was the presence of three distinct but intimately admixed growth patterns having retiform, nested, and solid/epithelioid features. The retiform portions of these lesions were composed of elongated, branching vascular channels essentially identical to those seen in retiform hemangioendothelioma,29 lined by monotonous, ‘hobnail’ endothelial cells, with high nuclear to cytoplasmic ratios, hyperchromatic nuclei, and low mitotic activity. Occasionally the vascular channels lined by hobnail endothelial cells were dilated, creating a more ‘hemangioma-like’ appearance. The retiform zones of these tumors were typically juxtaposed to vaguely ‘neuroendocrine-appearing’ nests of epithelioid to slightly spindled endothelial cells with minimal pleomorphism, evenly dispersed chromatin, small nucleoli, and low mitotic activity. Both the retiform and nested endothelial cell formations were typically surrounded by fascicles of myoid-appearing spindled cells, which lacked expression of endothelial markers. The solid component of these lesions was composed of a sheet-like proliferation of uniform, small, epithelioid endothelial cells with well-defined cell borders, often containing individual intracytoplasmic vacuoles of the type typically seen in epithelioid hemangioendothelioma30 and epithelioid hemangioma.31 These epithelioid areas, however, lacked the distinctive myxochondroid to hyalinized matrix seen in epithelioid hemangioendothelioma or the capillary sized vessels and eosinophils that typify epithelioid hemangioma. In one tumor, abnormally configured thick-walled vessels resembling lymphatics and veins were present immediately adjacent to tumor showing typical solid/epithelioid, nested, and retiform morphology. No tumor contained foci resembling conventional angiosarcoma. Overall, mitotic activity was very low (<1 mitotic figure per 10 high-powered fields) and necrosis was absent.


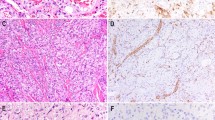
Composite hemangioendothelioma with neuroendocrine marker expression (Case 3), presenting as a small, well-circumscribed, para-aortic mass (arrow) (a). Portions of this tumor showed a predominantly solid growth pattern (b), and consisted of small nests of vaguely ‘neuroendocrine-appearing,’ short spindled to epithelioid cells with evenly dispersed chromatin, inapparent nucleoli, and absent mitotic activity and pleomorphism (c). Other area were composed of elongated, branching vascular channels surrounded by myoid-appearing spindled cells (d) and lined by hyperchromatic, ‘hobnail’ endothelial cells (e). This tumor was diffusely positive for CD31 (f), ERG protein (g), and synaptophysin (h). Whole transcriptome sequencing detected a PTBP1-MAML2 fusion in this case and FISH showed disruption/rearrangement of both copies of the MAML2 break-apart FISH probe resulting in a signal pattern of two 3′MAML2 Spectrum Green signals, two 5′MAML2 Spectrum Orange signals and no intact MAML2 fusion signals (i). This tumor metastasized to the sacrum.

Pulmonary metastasis from a primary composite hemangioendothelioma with neuroendocrine marker expression of the vertebra (Case 4) showing both nested and retiform features (a). Higher power view of retiform vascular channels lined by hobnail endothelial cells (b). In addition, this tumor contained hemangioma-like vascular channels lined by hobnail endothelial cells (c). CD31 (d) and synaptophysin (e) were diffusely positive in this tumor.

Composite hemangioendothelioma with neuroendocrine marker expression presenting as a cutaneous mass of the ankle (Case 3). This case showed an unusual admixture of dilated vascular channels, smaller channels lined by hobnail endothelial cells and solid areas (a). In other areas the solid cell nests were juxtaposed to elongated vascular channels (b). Solid and vaguely nested aggregates of epithelioid endothelial cells surrounded elongated channels lined by hobnail cells (c). Higher power view of uniform epithelioid endothelial cells, resembling those seen in epithelioid hemangioendothelioma. Myxochondroid matrix, however, is absent (d). This tumor was also diffusely positive for CD31 (e) and synaptophysin (e).

Composite hemangioendothelioma with neuroendocrine marker expression from the index finger of a child (Case 11). This tumor contained both retiform and solid/nested areas, although retiform vascular channels predominated (a). Higher power view of retiform vascular channels lined by hobnail endothelial cells, with prominent perivascular fibrosis (b). Solid/nested foci containing short spindled to epithelioid cells, adjacent to retiform channels (c). This tumor grew in a highly infiltrative fashion, with striking perineurial growth. Owing to multiple positive margins at the time of surgery, amputation of the finger was ultimately necessary to completely resect the tumor (d). In addition to other endothelial markers (not shown), this tumor was strongly positive for D2-40, suggesting lymphatic endothelial differentiation. Overall, D2-40 was expressed by 70% of cases (e). Synaptophysin expression was also diffusely present (f).

This composite hemangioendothelioma with neuroendocrine marker expression arose in association with a long-standing vascular malformation of the cheek (Case 10) (a). Dilated vascular channels lined by hobnailed cells, adjacent to solid masses of epithelioid endothelial cells (b). The retiform vascular channels in this tumor were often compressed, forming branching structures composed of a solid proliferation of darkly staining hobnail endothelial cells (c). The epithelioid areas showing striking intracytoplasmic vacuolization, as seen in other epithelioid endothelial cell tumors (d). Hemangioma-like structures lined by hobnail endothelial cells were also present (e). Synaptophysin was positive (f).
Immunohistochemical Findings
All studied cases were diffusely positive for CD31, FLI-1, and ERG. CD34 expression, in contrast, was much more limited, being focally present in 50% of tested cases and absent in the others. D2-40 expression was present in 70% of cases, including the tumor that arose in association with a lymphatic vascular malformation. Of the neuroendocrine markers, synaptophysin was strongly positive in all cases, whereas CD56 was expressed by just under 50% of cases; chromogranin A expression was seen in only one case. Synaptophysin expression was typically diffuse, and was not confined to areas of the tumors showing a particular morphological pattern. Keratin and CAMTA1 expression were absent.
Expression of synaptophysin was very limited in the ‘control’ endothelial neoplasms, present in only 3 of 101 (3%) tumors. Interestingly, all of these synaptophysin-positive cases were hemangioendotheliomas of ‘hobnail’ type, including two (of three) cases of retiform hemangioendothelioma and one (of two) cases of Dabska-type hemangioendothelioma (Figure 6). This Dabska-type hemangioendothelioma was additionally interesting, inasmuch as it had arisen in association with a lymphatic vascular malformation; synaptophysin expression in this tumor was seen in hobnail endothelial cells and intravascular papillations, but not in the lymphatic malformation. Chromogranin A expression was absent in all ‘control’ cases tested.

Dabska-type and retiform hemangioendothelioma (a and c, respectively), positive for synaptophysin (b and d, respectively). These were the only other types of vascular tumors that showed synaptophysin expression in the ‘control’ group.
Sequencing and FISH Findings
Whole transcriptome sequencing detected PTBP1-MAML2 and EPC1-PHC2 fusion transcripts in Cases 3 and 5, respectively. PTBP1-MAML2 is an in-frame fusion that joins exon 10 of PTB1 to exon 2 of MAML2, which is predicted to have the high mobility group box domain that binds to Notch receptors. The EPC1-PCH2 fusion extends from exon 6 to exon 1 of PHC2 and is predicted to drive the expression of PCH2. The remaining cases were negative for detectable fusion events. Validation studies using FISH break-apart probes targeted to MAML2 and EPC1 genes were positive in these same cases, confirming the sequencing findings. Six other tested cases were negative for MAML2 and EPC1 rearrangements by FISH.
Discussion
Herein, we have reported the clinicopathological and molecular genetic features of 11 cases of a distinctive endothelial neoplasm, characterized by an unusual admixture of solid/epithelioid, nested and retiform morphology and neuroendocrine marker expression, most often synaptophysin. Although the morphology of this tumor suggests that it is, for the time being at least, best considered a variant of composite hemangioendothelioma rather than a distinct entity, there are some differences between the clinicopathological features of the present cases and classical composite hemangioendothelioma, as will be discussed below.
The literature on composite hemangioendothelioma is difficult to critically evaluate, because composite hemangioendothelioma is a very difficult entity to precisely define. As originally described by Nayler et al.,1 composite hemangioendothelioma show a ‘remarkable admixture of epithelioid, retiform, and spindle cell hemangioendothelioma components,’ with some cases also showing ‘an angiosarcomatous or benign angiomatous component’. Although this description aptly summarizes the varied appearance of these unusual tumors, it arguably lacks both ‘minimal’ and ‘maximal’ criteria for diagnosis, and leaves individual pathologists considerable latitude in application. Additionally, the constituent elements of composite hemangioendothelioma (eg, retiform hemangioendothelioma, epithelioid hemangioendothelioma, spindle cell hemangioma, etc) are themselves quite rare and difficult for many pathologists to diagnose. Although a case-by-case review of previously reported examples of composite hemangioendothelioma is beyond the scope of the present study, we suspect that some cases of composite hemangioendothelioma might be better interpreted as representing other benign and malignant vascular tumors, including sinusoidal hemangioma,8, 32 anastomosing hemangioma,10, 33 epithelioid hemangioma with partial spindled growth,18, 34 and conventional angiosarcoma.9, 13, 16, 20 The distinction of angiosarcoma from composite hemangioendothelioma containing ‘foci resembling angiosarcoma’ is particularly problematic, as angiosarcoma is a notoriously protean and deceptive lesion that may show hemangioma-like, retiform, and epithelioid features in a single tumor.35 This issue is highlighted by a recent case of composite hemangioendothelioma reported by Leen et al,7 containing elements closely resembling conventional and epithelioid angiosarcoma. Although the (to date) benign clinical course of this patient would seem to support the authors’ classification of this particular tumor as composite hemangioendothelioma, we have little doubt that this case would engender significant debate, even among expert soft tissue pathologists.
Although composite hemangioendotheliomas have been described as showing any combination of vascular histologies, a subset of cases show a distinctive admixture of areas containing elongated, branching vascular channels lined by hobnail endothelial cells, closely resembling retiform hemangioendothelioma,29 and solid zones composed of epithelioid endothelial cells with intracytoplasmic vacuoles, as seen in epithelioid hemangioendothelioma.30 The myxochondroid matrix seen in ordinary epithelioid hemangioendothelioma is not, however, a feature of these cases. Composite hemangioendothelioma showing this unusual combination of features were first illustrated by Nayler et al1 and such areas were stressed as a possible ‘defining’ feature of composite hemangioendothelioma in a later report of five cases, by Fukunaga et al.24 Similar cases have also been reported by Requena et al,23 Bhat and Chowdappa,4 Liau et al,12 Cakir et al,19 and Fasolis et al.22 The 11 cases that comprise the present report also show this same distinctive combination of retiform and epithelioid features, in addition to containing areas with a striking nested appearance, a feature not previously noted in composite hemangioendothelioma.
In addition to ‘composite’ morphology, our cases were characterized by neuroendocrine marker expression, in particular synaptophysin. Neuroendocrine marker expression does not appear to have been previously evaluated in composite hemangioendotheliomas, perhaps not a surprising omission in an obviously endothelial neoplasm. We have specifically used the term ‘neuroendocrine marker expression,’ rather than ‘neuroendocrine differentiation’ to describe our cases, because they typically express only synaptophysin and to a lesser extent CD56, in the absence of chromogranin A. Synaptophysin is a transmembrane glycoprotein expressed by neural, endocrine, and neuroendocrine cells that participates in the storage and release of neurotransmitters and acts as a membrane channel protein.36 Expression of synaptophysin is not, however, limited to neural, neuroendocrine or endocrine tumors, and is well documented in alveolar rhabdomyosarcoma,37 melanoma,38 and extraskeletal myxoid chondrosarcoma,39 among others. Similarly, expression of CD56, a membrane-associated adhesion molecule expressed by a wide variety of cell types, is not at all specific for neural or neuroendocrine tumors.37, 40, 41, 42 In contrast, expression of chromogranin A, a calcium-binding granin protein present in the dense core granules of neural and neuroendocrine cells43, 44 seems to be limited in almost all instances to tumors truly showing neuroendocrine differentiation, and thus the absence of chromogranin expression in almost all of our cases would suggest that they do not show neuroendocrine differentiation.
Although the significance of synaptophysin expression in our cases is unclear, it does seem to be a phenomenon almost entirely restricted to the distinctive composite endothelial tumors that are the subject of this report. Intriguingly, the only other synaptophysin-positive endothelial tumors were retiform and Dabska-type hemangioendotheliomas, related lesions characterized by hobnail endothelial cells and lymphatic endothelial differentiation.45, 46, 47 This finding, along with the uniform presence of retiform hemangioendothelioma-like areas and frequent expression of D2-40, suggests the hypothesis that composite hemangioendothelioma with neuroendocrine marker expression is closely related to retiform and Dabska (‘hobnail’) hemangioendotheliomas. Given their more aggressive clinical behavior, as compared with ordinary retiform and Dabska-type hemangioendotheliomas, it is possible that our cases might represent a form of malignant progression in ‘hobnail’ hemangioendotheliomas.
A number of features suggest that our cases are not related to epithelioid hemangioendothelioma, despite the presence of similar-appearing areas. First, as noted above, these areas lack one of the cardinal morphological features of epithelioid hemangioendothelioma, myxochondroid matrix. Additionally, our cases were uniformly negative for CAMTA1, a marker expressed by >90% of epithelioid hemangioendotheliomas, reflecting the presence of the disease-defining WWTR1-CAMTA1 gene fusion.48, 49 We also did not find evidence of WWTR1-CAMTA1 rearrangement in one case tested by FISH (data not shown). Finally, true epithelioid hemangioendotheliomas are consistently synaptophysin-negative, unlike the epithelioid areas seen in our cases.
The genetic results in our studied cases are of uncertain significance. As noted above, we identified two individual cases showing PTBP1-MAML2 and EPC1-PHC2 fusion transcripts, respectively, and confirmed these findings with FISH. Rearrangements of the MAML2 gene are seen in mucoepidermoid carcinoma (MECT1-MAML2, CRTC1-MAML2).50 Rearrangements of the EPC1 gene are seen in ossifying fibromyxoid tumor and endometrial stromal sarcoma.51, 52 We are not aware of gene fusions involving the PTBP1 or PHC2 genes in human cancer. Study of additional cases of similar tumors, preferably with fresh/frozen tissue will be necessary to determine whether these transcripts are entity-defining or are merely unique to these two cases.
The behavior of composite hemangioendothelioma with neuroendocrine marker expression appears to be significantly more aggressive than that of previously reported composite hemangioendothelioma, with rapidly developing metastases in 50% of patients with follow-up. It is possible that this actually understates the malignant potential of these tumors, as our median follow-up duration was relatively short (10 months). We do not think that this reflects consultation bias, as only one of these cases was referred in consultation after the development of metastatic disease. Clinically aggressive behavior was seen in both superficially and deeply located tumors. Given the relatively small size of our series, we were not able to discern any relationship between other clinicopathological parameters (eg, patient age, tumor size, extent of synaptophysin expression) and outcome. Although the aggressive behavior of these tumors raises the question of whether they should be termed ‘angiosarcoma,’ rather than regarded as an aggressive variant of composite hemangioendothelioma, we believe the very distinctive morphology and immunophenotype of these lesions and the absence of features of conventional angiosarcoma (eg, high nuclear grade, brisk mitotic activity, necrosis) to argue in favor of our current nomenclature. Although the term ‘hemangioendothelioma’ is generally used to refer to lesions with a low metastatic risk, this term is also applied to epithelioid hemangioendothelioma, a tumor whose natural history is closer to that of a low-grade angiosarcoma.53
In summary, we have described the clinicopathological, immunohistochemical, and genetic features of an unusual endothelial neoplasm defined by ‘composite’ morphology, with retiform, nested, and solid/epithelioid features, and neuroendocrine marker expression. At present, we think it best to regard these lesions as representing a clinically aggressive variant of a WHO recognized entity, composite hemangioendothelioma, although it is also possible that they represent an unusual variant of retiform hemangioendothelioma, or even an altogether distinct entity. Study of additional cases of strictly defined composite hemangioendothelioma will be necessary to determine the exact relationship of those tumors showing neuroendocrine marker expression to those without this finding.
References
Nayler SJ, Rubin BP, Calonje E et al. Composite hemangioendothelioma: a complex, low-grade vascular lesion mimicking angiosarcoma. Am J Surg Pathol 2000;24:352–361.
Rubin BP. Composite haemangioendothelioma. In: Fletcher CD, Unni KK, Mertens F (eds). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone, 3rd edn. IARC Press: Lyon, France, 2002, pp 168–169.
Rubin BP. Composite haemangioendothelioma. In: Fletcher CD, Bridge JA, Pancras CW et al (eds). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone, 4th edn. IARC Press: Lyon, France, 2013, pp 149–150.
Bhat A, Chowdappa V . Composite hemangioendothelioma: report of a rare case. J Clin Diagn Res 2016;10:ED01–ED03.
Sakamoto A . Reconstruction with beta-tricalcium phosphate after navicular tumor resection. J Foot Ankle Surg 2017;56:98–102.
Shang Leen SL, Fisher C, Thway K . Composite hemangioendothelioma: clinical and histologic features of an enigmatic entity. Adv Anat Pathol 2015;22:254–259.
Leen SL, Clarke PM, Chapman J et al. Composite hemangioendothelioma of the submandibular region. Head Neck Pathol 2015;9:519–524.
Stojsic Z, Brasanac D, Stojanovic M et al. Cutaneous composite hemangioendothelioma: case report and review of published reports. Ann Saudi Med 2014;34:182–188.
Mahmoudizad R, Samrao A, Bentow JJ et al. Composite hemangioendothelioma: an unusual presentation of a rare vascular tumor. Am J Clin Pathol 2014;141:732–736.
Zhang J, Wu B, Zhou GQ et al. Composite hemangioendothelioma arising from the kidney: case report with review of the literature. Int J Clin Exp Pathol 2013;6:1935–1941.
Dong A, Bai Y, Wang Y et al. Bone scan, MRI, and FDG PET/CT findings in composite hemangioendothelioma of the manubrium sterni. Clin Nucl Med 2014;39:e180–e183.
Liau JY, Lee FY, Chiu CS et al. Composite hemangioendothelioma presenting as a scalp nodule with alopecia. J Am Acad Dermatol 2013;69:e98–e99.
McNab PM, Quigley BC, Glass LF et al. Composite hemangioendothelioma and its classification as a low-grade malignancy. Am J Dermatopathol 2013;35:517–522.
Chen YL, Chen WX, Wang J et al. Composite hemangioendothelioma on the neck. Kaohsiung J Med Sci 2012;28:564–565.
Tateishi J, Saeki H, Ito K et al. Cutaneous composite hemangioendothelioma on the nose treated with electron beam. Int J Dermatol 2013;52:1618–1619.
Yoda Y, Ohashi M . A case of composite hemangioendothelioma arising from the spleen. Jpn J Clin Oncol 2012;42:770.
Tsai JW, Huang HY, Lee JC et al. Composite haemangioendothelioma: report of four cases with emphasis on atypical clinical presentation. Pathology 2011;43:176–180.
Aydingoz IE, Demirkesen C, Serdar ZA et al. Composite haemangioendothelioma with lymph-node metastasis: an unusual presentation at an uncommon site. Clin Exp Dermatol 2009;34:e802–e806.
Cakir E, Demirag F, Gulhan E et al. Mediastinal composite hemangioendothelioma. A rare tumor at an unusual location. Tumori 2009;95:98–100.
Tejera-Vaquerizo A, Herrera-Ceballos E, Bosch-Garcia R et al. Composite cutaneous hemangioendothelioma on the back. Am J Dermatopathol 2008;30:262–264.
Utas S, Canoz O, Ferahbas A et al. Composite cutaneous haemangioendothelioma treated with interferon. J Eur Acad Dermatol Venereol 2008;22:503–505.
Fasolis M, Iaquinta C, Montesco MC et al. Composite hemangioendothelioma of the oral cavity: case report and review of the literature. Head Neck 2008;30:974–979.
Requena L, Luis Diaz J, Manzarbeitia F et al. Cutaneous composite hemangioendothelioma with satellitosis and lymph node metastases. J Cutan Pathol 2008;35:225–230.
Fukunaga M, Suzuki K, Saegusa N et al. Composite hemangioendothelioma: report of 5 cases including one with associated Maffucci syndrome. Am J Surg Pathol 2007;31:1567–1572.
Tronnier M, Vogelbruch M, Kutzner H . Spindle cell hemangioma and epithelioid hemangioendothelioma arising in an area of lymphedema. Am J Dermatopathol 2006;28:223–227.
Biagioli M, Sbano P, Miracco C et al. Composite cutaneous haemangioendothelioma: case report and review of the literature. Clin Exp Dermatol 2005;30:385–387.
Reis-Filho JS, Paiva ME, Lopes JM . Congenital composite hemangioendothelioma: case report and reappraisal of the hemangioendothelioma spectrum. J Cutan Pathol 2002;29:226–231.
Tessier Cloutier B, Costa FD, Tazelaar HD et al. Aberrant expression of neuroendocrine markers in angiosarcoma: a potential diagnostic pitfall. Hum Pathol 2014;45:1618–1624.
Calonje E, Fletcher CD, Wilson-Jones E et al. Retiform hemangioendothelioma. A distinctive form of low-grade angiosarcoma delineated in a series of 15 cases. Am J Surg Pathol 1994;18:115–125.
Weiss SW, Enzinger FM . Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer 1982;50:970–981.
Fetsch JF, Weiss SW . Observations concerning the pathogenesis of epithelioid hemangioma (angiolymphoid hyperplasia). Mod Pathol 1991;4:449–455.
Calonje E, Fletcher CD . Sinusoidal hemangioma. A distinctive benign vascular neoplasm within the group of cavernous hemangiomas. Am J Surg Pathol 1991;15:1130–1135.
Montgomery E, Epstein JI . Anastomosing hemangioma of the genitourinary tract: a lesion mimicking angiosarcoma. Am J Surg Pathol 2009;33:1364–1369.
Keel SB, Rosenberg AE . Hemorrhagic epithelioid and spindle cell hemangioma: a newly recognized, unique vascular tumor of bone. Cancer 1999;85:1966–1972.
Miettinen M . Hemangioendotheliomas, angiosarcomas, and Kaposi's sarcoma. In: Miettinen M (ed). Modern Soft Tissue Pathology Tumors and Non-Neoplastic Conditions, 1st edn. Cambridge University Press: NY, USA, 2010, pp 624–647.
Gould VE, Wiedenmann B, Lee I et al. Synaptophysin expression in neuroendocrine neoplasms as determined by immunocytochemistry. Am J Pathol 1987;126:243–257.
Bahrami A, Gown AM, Baird GS et al. Aberrant expression of epithelial and neuroendocrine markers in alveolar rhabdomyosarcoma: a potentially serious diagnostic pitfall. Mod Pathol 2008;21:795–806.
Romano RC, Carter JM, Folpe AL . Aberrant intermediate filament and synaptophysin expression is a frequent event in malignant melanoma: an immunohistochemical study of 73 cases. Mod Pathol 2015;28:1033–1042.
Goh YW, Spagnolo DV, Platten M et al. Extraskeletal myxoid chondrosarcoma: a light microscopic, immunohistochemical, ultrastructural and immuno-ultrastructural study indicating neuroendocrine differentiation. Histopathology 2001;39:514–524.
Shipley WR, Hammer RD, Lennington WJ et al. Paraffin immunohistochemical detection of CD56, a useful marker for neural cell adhesion molecule (NCAM), in normal and neoplastic fixed tissues. Appl Immunohistochem 1997;5:87–93.
Folpe AL, Schmidt RA, Chapman D et al. Poorly differentiated synovial sarcoma: immunohistochemical distinction from primitive neuroectodermal tumors and high-grade malignant peripheral nerve sheath tumors. Am J Surg Pathol 1998;22:673–682.
Graham RP, Dry S, Li X et al. Ossifying fibromyxoid tumor of soft parts: a clinicopathologic, proteomic, and genomic study. Am J Surg Pathol 2011;35:1615–1625.
Erickson LA, Lloyd RV . Practical markers used in the diagnosis of endocrine tumors. Adv Anat Pathol 2004;11:175–189.
Lloyd RV . Practical markers used in the diagnosis of neuroendocrine tumors. Endocr Pathol 2003;14:293–301.
Fukunaga M . Expression of D2-40 in lymphatic endothelium of normal tissues and in vascular tumours. Histopathology 2005;46:396–402.
Fanburg-Smith JC, Michal M, Partanen TA et al. Papillary intralymphatic angioendothelioma (PILA): a report of twelve cases of a distinctive vascular tumor with phenotypic features of lymphatic vessels. Am J Surg Pathol 1999;23:1004–1010.
Miettinen M, Wang ZF . Prox1 transcription factor as a marker for vascular tumors-evaluation of 314 vascular endothelial and 1086 nonvascular tumors. Am J Surg Pathol 2012;36:351–359.
Tanas MR, Sboner A, Oliveira AM et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med 2011;3:98ra82.
Doyle LA, Fletcher CD, Hornick JL . Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am J Surg Pathol 2016;40:94–102.
Behboudi A, Enlund F, Winnes M et al. Molecular classification of mucoepidermoid carcinomas-prognostic significance of the MECT1-MAML2 fusion oncogene. Genes Chromosomes Cancer 2006;45:470–481.
Antonescu CR, Sung YS, Chen CL et al. Novel ZC3H7B-BCOR, MEAF6-PHF1, and EPC1-PHF1 fusions in ossifying fibromyxoid tumors—molecular characterization shows genetic overlap with endometrial stromal sarcoma. Genes Chromosomes Cancer 2014;53:183–193.
Micci F, Panagopoulos I, Bjerkehagen B et al. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res 2006;66:107–112.
Goldblum JR, Folpe AL, Weiss SW . Hemangioendothelioma: vascular tumors of intermediate malignancy. In: Goldblum JR, Folpe AL, Weiss SW (eds). Enzinger and Weiss's Soft Tissue Tumors, 6th edn. Saunders Elsevier: Philadelphia, PA, USA, 2014, pp 681–702.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Perry, K., Al-lbraheemi, A., Rubin, B. et al. Composite hemangioendothelioma with neuroendocrine marker expression: an aggressive variant. Mod Pathol 30, 1589–1602 (2017). https://doi.org/10.1038/modpathol.2017.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.83
This article is cited by
-
Patient with composite haemangioendothelioma containing angiosarcoma-like areas in the setting of congenital lymphoedema mimicking Stewart-Treves syndrome: a case report
Diagnostic Pathology (2023)
-
Composite hemangioendothelioma in the cervical spine with kaposiform hemangioendothelioma features in an elderly patient: a case report
BMC Geriatrics (2022)
-
Thoracic composite hemangioendothelioma with neuroendocrine marker expression
Surgical Case Reports (2021)
-
Whole Exome Sequencing Identifies Somatic Variants in an Oral Composite Hemangioendothelioma Characterized by YAP1-MAML2 Fusion
Head and Neck Pathology (2021)
-
Genome-wide association study for circulating fibroblast growth factor 21 and 23
Scientific Reports (2020)
