
Abstract
The carcinogenic role of high-risk human papillomavirus (HR-HPV) types in the increasing subset of vulvar intraepithelial neoplasia and vulvar cancer in young women has been established. However, the actual number of vulvar cancer cases attributed to HPV is still imprecisely defined. In an attempt to provide a more precise definition of HPV-driven vulvar cancer, we performed HPV-type-specific E6*I mRNA analyses available for 20 HR-/possible HR (pHR)-HPV types, on tissue samples from 447 cases of vulvar cancer. HPV DNA genotyping was performed using SPF10-LiPA25 assay due to its high sensitivity in formalin-fixed paraffin-embedded tissues. Data on p16INK4a expression was available for comparative analysis via kappa statistics. The use of highly sensitive assays covering the detection of HPV mRNA in a broad spectrum of mucosal HPV types resulted in the detection of viral transcripts in 87% of HPV DNA+ vulvar cancers. Overall concordance between HPV mRNA+ and p16INK4a upregulation (strong, diffuse immunostaining in >25% of tumor cells) was 92% (K=0.625, 95% confidence interval (CI)=0.531–0.719). Among these cases, 83% were concordant pairs of HPV mRNA+ and p16INK4a+ and 9% were concordant pairs of HPV mRNA− and p16INK4a−. Our data confirm the biological role of HR-/pHR-HPV types in the great majority of HPV DNA+ vulvar cancers, resulting in an HPV-attributable fraction of at least 21% worldwide. Most HPV DNA+ vulvar cancers were associated with HPV16 (85%), but a causative role for other, less frequently occurring mucosal HPV types (HPV26, 66, 67, 68, 70 and 73) was also confirmed at the mRNA level for the first time. These findings should be taken into consideration for future screening options as HPV-associated vulvar preneoplastic lesions have increased in incidence in younger women and require different treatment than vulvar lesions that develop from rare autoimmune-related mechanisms in older women.
Similar content being viewed by others

Main
With an annual incidence of 2 cases per 100 000 women, vulvar cancer is a rare malignancy.1, 2 The estimated global burden of vulvar cancer is 27 000 cases annually and it accounts for 3–5% of all gynecological cancers in developed countries.3 Most cases of vulvar cancer present as squamous cell carcinoma (>90%).1, 2 The International Agency for Research on Cancer and World Health Organization distinguish two pathways for the development of vulvar cancer. The first pathway is activated by underlying autoimmune-related processes while the second pathway is triggered by mucosal human papillomavirus (HPV) infection. Differentiated-type vulvar intraepithelial neoplasia is non-HPV associated and appears in older women (median age 70 years) who develop chronic inflammation of the anogenital area often diagnosed as lichen sclerosus or lichen planus.4 In 2–5% of cases, these lesions further develop into differentiated keratinizing squamous cell carcinoma of the vulva.5 Usual-type vulvar intraepithelial neoplasia is HPV associated and appears in younger women. Morphologically, it can be either basaloid, warty or mixed histology, and if untreated, 9–16% of lesions can progress to basaloid or warty squamous cell carcinoma of the vulva.5 In some countries, HPV-associated vulvar intraepithelial neoplasia increased four-fold between 1973 and 2000, particularly among younger women (median age 40 years).6, 7 Changes in sexual behavior, early onset of sexual activities and transmission of high-risk/possible high-risk HPV types (HR-/pHR-HPV), especially HPV16, are considered contributing factors to this observed increase in vulvar lesions.
Differentiating between HPV-associated and non-HPV-associated vulvar lesions has been recognized as an important distinction among clinicians as it has both therapeutic and postoperative management implications.8, 9, 10 Non-HPV-associated vulvar intraepithelial neoplasia is a rapidly progressive lesion requiring immediate excision and treatment.8 By contrast, HPV-associated vulvar intraepithelial neoplasia develops slowly and can spontaneously regress or regress postlocalized treatment (eg, with topical immune modulators).8, 10 However, patients with HPV-associated vulvar lesions are at increased risk of developing additional HPV-associated lesions within the anogenital tract. Consequently, careful examination and monitoring of the cervix and the perianal area is needed in these women.11
True estimation of the HPV-attributable fraction in vulvar cancer is still imprecise. World estimation of HPV DNA-positive (HPV DNA+) vulvar cancers stands currently at 43%.3 This estimation is based on a recent meta-analysis that assessed 63 epidemiological studies including the evaluation of 1873 vulvar cancer cases collected from across the globe.12 However, the mere presence of HPV DNA has shown to be insufficient to define HPV-driven mucosal cancers outside of the cervix.13, 14, 15, 16 Previously, we compared HPV DNA positivity alone and in combination with p16INK4a upregulation by assessing these markers directly in 1709 vulvar cancer tissue specimens collected from 39 countries worldwide.17 Our results showed a lower fraction of HPV DNA+ vulvar cancers compared with previous HPV DNA reports; 29%17 versus 43% based on a meta-analysis.3 In addition, adding p16INK4a upregulation data to the sole HPV DNA presence in tumor tissues lowered estimate of the HPV-attributable fraction of vulvar cancer from 29% to 25%.17
In the continuous effort to better define HPV-driven vulvar cancer, we focused on collecting biological evidence of HPV-transformed phenotype in the vulva by investigating the expression and concordance of HPV mRNA and p16INK4a in 447 HPV DNA+ cases of vulvar cancer. Making this distinction is particularly important toward the adequate assessment of potential clinical differences between HPV-driven and non-HPV-driven malignancies of the vulva.
Materials and methods
Ethics Statement
The samples obtained and analyzed here were selected from HPV VVAP (International Survey on HPV prevalence and type distribution in Vulvar, Vaginal, Anal, Penile neoplasias)17 for which the Institutional Review Board approval was received. All samples were anonymized. All protocols applied were approved by local and Catalan Institute of Oncology ethics committees.
Study Approach
All specimens analyzed were formalin fixed and paraffin embedded. Vulvar cancer cases with sufficient tissue quality and quantity that had been previously analyzed for the presence of HPV DNA and p16INK4a expression were eligible for further analyses. Based on these criteria, 447 HPV DNA+ histologically diagnosed vulvar cancers were analyzed for the expression of viral transcripts. HPV E6*I mRNA RT-PCR assays were available for 20 HR-/pHR-HPV types: HPV16, 18, 26, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 67, 68, 70, 73, and 82.
HPV mRNA Analysis
The mRNA extraction and mRNA detection from tissue ribbons were performed as previously described.18, 19 For all cases analyzed, hematoxylin and eosin stain before and after sectioning for RNA showed ≤10% tumor reduction. For each case, HPV-type-specific E6*I mRNA RT-PCR assays were performed for an HPV type(s) previously determined by genotyping and for a cellular ubiquitin C gene as a control for tissue quality. A second assay was performed to assess the presence of HPV16 E6*I mRNA in all cases, irrespective of HPV DNA result. Cases with HPV mRNA positive (HPV mRNA+) and/or ubiquitin C mRNA+ signal were considered ‘RNA valid’. All ‘RNA invalid’ samples, ie, cases that were HPV mRNA-negative (HPV mRNA−) and ubiquitin C mRNA−, were analyzed a second time, and according to signals obtained, classified as ‘RNA valid’ or ‘RNA invalid’. Of the 11 vulvar cancer cases initially classified as ‘RNA invalid’, six cases were reclassified as ‘RNA valid’ and five remained classified as ‘RNA invalid’ upon re-analyses.
Testing for HPV type mRNA in HPV DNA-negative (HPV DNA−) cases was not included as part of the primary scope of this work owing to the complexity of such analyses (Figure 1). However, a subset of 20 HPV DNA− cases were tested for HPV16 E6*I mRNA as a negative control (10 HPV DNA−/p16INK4a− and 10 HPV DNA−/p16INK4a+ cases). The 10 HPV DNA−/p16INK4a− cases were ‘RNA valid’ and also HPV16 mRNA−. From 10 HPV DNA−/p16INK4a+, 9 were ‘RNA valid’ and 3 of the 9 were HPV16 mRNA+ (33%). This observation had minimal impact on overall estimates owing to the small number of p16INKa+ cases among the HPV DNA− cases (9%; 103 of 1,194). Two out of the 10 HPV DNA−/HPV mRNA−/p16INK4a− cases showed partial basaloid features, whereas 2 out of the 3 HPV DNA−/HPV mRNA+/p16INK4a+ cases were basaloid.

Study algorithm. (1) HPV E6*I mRNA assays were developed for 12 HR-HPV: 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, and 59; and for 8 pHR-HPV: 26, 53, 66, 67, 68, 70, 73, and 82. (HPV-type classification was according to IARC classification.1) (2) In p16INK4a analysis: Tissue quality insufficient to analyze p16INK4a expression (n=12). In mRNA analysis: both HPV and ubiquitin C mRNA are negative and therefore invalid (n=5). (3) No splice site HPV mRNA assays available for the HPV types identified by SPF10-LiPA25 or sequencing; LR-HPV: HPV6 (n=5), HPV11 (n=2), HPV44 (n=3), HPV61 (n=2), HPV74 (n=3), HPV102 (n=1); pHR-HPV: HPV30 (n=1), HPV69 (n=2); HPV undetermined, ie, not identified by SPF10-LiPA25 or sequencing but positive with DEIA (n=20); not enough material (n=2; one HPV16 and one HPV6/16/39 or 68 or 73 case). (4) No result: In p16INK4a analysis: Insufficient tumor tissue in tissue blocks for p16INK4a analysis. (5) A random selection of 20 HPV DNA− vulvar cancer cases (10 HPV DNA−/p16INK4a− and 10 HPV DNA−/p16INK4a+ cases) were analyzed for HPV mRNA as control tissues. One case was invalid in mRNA analysis.
HPV Genotyping, p16INK4a Immunohistochemistry and Histological Assessment
HPV genotyping, p16INK4a immunohistochemistry and histological analyses were performed as previously described.17 For simplicity of reporting, we denote p16INK4a-positive (p16INK4a+) cases as cases that showed strong, diffuse immunostaining in >25% of tumor cells (p16INK4a upregulation) and p16INK4a negative (p16INK4a−) cases as cases with focal, weak immunostaining or immunostaining in ≤25% of tumor cells.16
Statistical Analysis
Information was available for ‘country’, ‘age at diagnosis’, ‘year of diagnosis’, ‘histopathological diagnosis’, ‘HPV DNA positivity’, ‘HPV type’ and ‘HPV mRNA and/or p16INK4a expression’. Chi-squared test and Student’s t-test were used to evaluate associations between variables and HPV DNA, HPV mRNA and p16INK4a positivity. Agreement between HPV DNA and p16INK4a and between HPV mRNA and p16INK4a was assessed by kappa score. The McNemar test was used for matched pair data to assess the unequal distribution of discordant results. The agreement was evaluated globally and by the different assays explored. All statistical inferences were based on two-sided tests. Results were statistically significant at P<0.05. Data analyses were performed using Statistical Package for the Social Sciences (SPSS) (Version 13.0, SPSS, Chicago, IL, USA) and with STATA (Version 10.0, Stata Corporation, Computing Resource Center, College Station, TX, USA).
Results
A total of 1709 vulvar cancer cases collected from 39 countries were HPV genotyped; 29% (488/1709) of these cases were HPV DNA+ (Figure 1).17 From 488 HPV DNA+ cases, 447 were analyzed in the current study for the expression of viral mRNA and 99% (442/447) were ‘RNA valid’ (HPV mRNA+ and/or ubiquitin C mRNA+) (Table 1). A subset of 433 HPV DNA+ vulvar cancers had available HPV mRNA and p16INK4a data (Table 1) included in the final statistical analysis of HPV mRNA and p16INK4a concordance (Table 2).
HPV E6*I mRNA and p16INK4a Expression in Vulvar Cancers Harboring Single or Multiple HPV Infections
Overall, 87% (384/442) of vulvar cancer cases were HPV DNA+/HPV mRNA+ (Table 1).
Of the 413 ‘RNA valid’ vulvar cancer cases harboring single HPV type DNA, 87% (360/413) were HPV mRNA+ (Table 1). Among 317 HPV16 single DNA+ cases, 88% (280/317) were HPV16 mRNA+. Among the 96 non-HPV16 DNA+ cases, 83% (80/96) were mRNA+ for the HPV type defined by SPF10-LiPA25 genotyping, including one HPV33 DNA+ case that expressed both HPV33 and HPV16 mRNA transcripts. We have identified nine vulvar cancer cases that were HPV DNA+ for a single pHR-HPV type (ie, HPV26, 53, 66, 67, 68, 70 and 73, respectively) (Supplementary Table S1). The biological activity of HPV26, 66, 67, 68, 70 and 73 in vulvar cancer was confirmed by the presence of HPV-type mRNA and upregulation of p16INK4a. Two HPV53 single DNA+ cases were HPV53 mRNA− and p16INK4a−.
Twenty-nine ‘RNA valid’ vulvar cancers harboring DNA of multiple HPV types were identified of which 83% (24/29) were HPV mRNA+ for at least one HPV type identified by genotyping (Table 1 and Table 3). From these 29 cases, 10 could be fully analyzed (ie, mRNA assays were available for all the types detected by genotyping). Overall, 40% (4/10) of these cases expressed transcripts of multiple HR-/pHR-HPV types (Table 3). Three of these four cases had a preneoplastic lesion adjacent to the invasive tumor.
Among vulvar cancer cases with HPV mRNA and p16INK4a data, 83% (360/433) were concordantly HPV mRNA+ and p16INK4a+ and 9% (37/433) were concordantly HPV mRNA− and p16INK4a−, yielding an overall concordance of 92% (K=0.625; 95% CI: (0.531–0.719); Table 4). A total of 36 cases (8%) showed discordant HPV mRNA and p16INK4a data: 19 HPV mRNA−/p16INK4a+ and 17 HPV mRNA+/p16INK4a cases (Table 4). McNemar tests (P=0.868) indicated that the discordant cases are equally distributed. Furthermore, there was no difference in kappa index concordance between histological diagnosis, region of origin or year of diagnosis (Table 2). However, differences in kappa index concordance were observed according to age group; agreement being higher among women aged >66 years compared with women aged <66 years (Table 2).
Cutoff for p16INK4a in HPV-Driven Vulvar Cancer
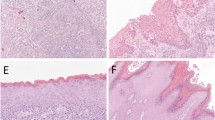
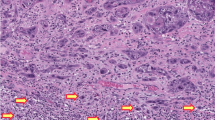
To define p16INK4a upregulation in vulvar cancer, we used the cutoff of >25% p16INK4a+ tumor cells, with strong staining intensity and in a diffuse pattern. As there is no current standardization for p16INK4a cutoff to define HPV-associated vulvar cancer, we challenged our >25% cutoff by different scenarios as shown in Tables 4 and 5. The >25% cutoff showed 83% (360/433) of HPV DNA+/HPV mRNA+ tumors to be p16INK4a+, yielding good agreement between p16INK4a and HPV mRNA positivity (Table 4). Increasing this cutoff to >50% or >75% resulted in a fair and poor agreement between p16INK4a and HPV mRNA positivity, respectively (Table 4). With >25% cutoff, 94% of HPV DNA+/HPV mRNA+ keratinizing tumors were defined as p16INK4a+ compared with only 44% when >75% cutoff was applied (Table 5). This difference was lower for non-keratinizing vulvar cancers where 95% of non-keratinizing HPV DNA+/HPV mRNA+ tumors were defined as p16INK4a+ with >25% cutoff against 71% with >75% cutoff (Table 5). Keratinizing vulvar cancers tend to present with a lower percentage of p16INK4a+ tumor cells, compared with non-keratinizing vulvar cancers (Table 5). Examples of vulvar cancer regarding histology, p16INK4a staining and HPV status are shown in Figure 2.



Examples of vulvar cancer cases regarding HPV status, p16INK4a immunostaining and the presence of keratinizing component. (a and b) HPV45 DNA+/HPV45 mRNA+ non-keratinizing vulvar cancer with >75% p16INK4a+ tumor cells (ID 20271). (c and d) HPV52 DNA+/HPV52 mRNA+ non-keratinizing vulvar cancer with >75% p16INK4a+ tumor cells (ID 20365). (e and f) HPV16 DNA+/HPV16 mRNA+ keratinizing vulvar cancer with 26–50% diffuse p16INK4a+ tumor cells (ID 22143). (g and h) HPV16 DNA+/HPV16 mRNA+ keratinizing vulvar cancer with 26–50% diffuse p16INK4a+ tumor cells (ID 22245). (i and j) HPV DNA−/HPV16 mRNA−/p16INK4a− keratinizing vulvar cancer (ID 20072). (k and l) HPV DNA−/HPV16 mRNA−/ p16INK4a− keratinizing vulvar cancer (ID 20462). × 10 magnification.
Patients’ Age and Tumor Histology, Origin and Year of Diagnosis
All discordant cases were equally distributed within each variable-dependent category (McNemar test yielded non-significant results). All vulvar cancers (100%) identified between 1980 and 1989 (n=3) and after 2010 (n=12) were HPV DNA+/HPV mRNA+/p16INK4a+ compared with 82% of such cases identified between 1990 and 2009 (n=418) (Table 2). Prevalence of HPV DNA+/HPV mRNA+/p16INK4a+ cases varied by geographic region with the highest prevalence observed in Oceania (91%) and the lowest in Africa (59%) (Table 2). In addition, the total number of vulvar cancer cases from Africa (n=17) was also low in comparison with Oceania (n=75) (Table 2). HPV DNA+/HPV mRNA+/p16INK4a+ vulvar cancers varied across five age groups and ranged between 78% (≥81 years) and 86% (<66 years) (Table 2).
Discussion
The etiological role of HPV in the development of vulvar cancer has been well recognized.1 However, the true attributable fraction of HPV in vulvar cancer remains unclear. It has become increasingly evident that a functional evidence of HPV activity or HPV transformation is necessary in addition to the HPV DNA presence, in order to define true HPV-driven tumors outside of the cervix uteri.20 In our study, we focused on obtaining evidence of HPV transcriptional activity, ie, the presence of HPV mRNA in HPV DNA+ vulvar cancer tissues. In an examination of 447 HPV DNA+ vulvar cancers, we combined HPV mRNA data with data on a well-established marker of HPV-transformed phenotype in mucosal cancers, p16INK4a. Viral mRNA was identified in 87% of the HPV DNA+ vulvar cancer cases. Among the 433 cases with both HPV mRNA and p16INK4a data available, 83% were concordant pairs of HPV mRNA+ and p16INK4a+. These data indicate that a proportion of HPV DNA+ cases (9%) does not express an additional marker of HPV activity, therefore questioning HPV attribution in that subset. Thus, in the absence of mechanistic data to define an HPV-driven cancer, markers of HPV activity and HPV-transformed phenotype demonstrated in addition to HPV DNA, should allow for more robust etiologic attribution.
To identify transcriptionally active HPV types in vulvar lesions, we applied HPV-type-specific and highly sensitive E6*I mRNA assays developed for 20 HR-/pHR-HPV types and validated for use in formalin-fixed paraffin-embedded tissues.18, 21 HPV transcripts are indicative of active virus but are not transformation specific.22, 23 But the expression of viral mRNA in mucosal tumors is a requirement for expression of E7 oncoprotein, which drives malignant transformation and induces p16INK4a upregulation. Our study is not the first to examine the expression of viral transcripts in vulvar lesions. However, in terms of the number of cases and range of HPV types analyzed, it is the broadest in scope. We identified eight earlier studies that provided data on viral E6/E7 transcripts in vulvar lesions, which focused on HPV16 and/or HPV18 transcripts only.24, 25, 26, 27, 28, 29, 30, 31 Our study also demonstrates that HR-HPV types other than HPV16 and 18, as well as a subset of pHR-HPV types, have an etiological role in the development of vulvar cancer.
Expression of p16INK4a protein in vulvar preneoplastic lesions and vulvar cancer has also been assiduously investigated.2, 27, 32, 33, 34, 35 In the context of HPV infection, p16INK4a upregulation is a result of a cellular defense mechanism referred to as ‘oncogene-induced senescence’.36 In cell lines of cervical cancer, an HPV-transformed tumor model, CDKN2A gene that encodes for p16INK4a protein is present as a wild-type, and p16INK4a upregulation is induced by HR-HPV E7 oncogene expression.36, 37, 38 In the vulva, up to 20% of cancers carry CDKN2A mutations but these are usually ‘silencing’ mutations primarily identified in HPV DNA− cancers and resulting in a loss of p16INK4a expression.39, 40 This confirms the value of p16INK4a upregulation as a marker of HPV-transforming activity in vulvar cancer in addition to HPV DNA and/or HPV mRNA or as a single marker once the precise cutoff has been defined.
Numerous immunohistochemical studies have confirmed p16INK4a upregulation as an excellent biomarker to define HR-HPV-associated lesions at different mucosal sites.13, 15, 21, 41, 42, 43, 44 In the vulva, p16INK4a also seems to be a superior marker to assess the effectiveness of imiquimod treatment suggested for HPV DNA+ vulvar intraepithelial neoplasia.10, 45 Our findings demonstrate p16INK4a upregulation in all HPV DNA+/HPV mRNA+ vulvar cancers harboring single pHR-HPV types 26, 66, 67, 70 or 73 (1 case each) and HPV DNA+ (no RNA assay available) pHR-HPV types 30 (1 case) or 69 (2 cases). These HPV types are not included in commercial HPV genotyping assays and might be missed when analyzing for HPV DNA only. However, the likelihood of identifying such cases is very low (<2%). The specificity of p16INK4a upregulation in support of HR-/pHR-HPV-type carcinogenicity is further substantiated by our findings that 89% (353/396) and 83% (10 of 12) of vulvar cancers harboring a single, active infection with HR-HPV or pHR-HPV type, respectively, showed p16INK4a upregulation. On the contrary, only 7% (1/15) of vulvar cancers harboring single LR-HPV type DNA showed p16INK4a upregulation (Supplementary Table S2). In this single LR-HPV6 DNA+/p16INK4a+ vulvar cancer case in our study (with low-intensity p16INK4a staining in >75% of tumor cells), HPV6 DNA was found in tumor cells, and not in the surrounding stroma, speculating that LR-HPV6 might be just less efficient in inducing p16INK4a upregulation.46 In such cases, as well as cases discordant for HPV mRNA and p16INK4a expression, investigation via CDKN2A gene sequencing should be explored in future studies. It is also possible that, in such cases, HPV DNA or HPV mRNA are present below detection limits of the applied assays. Or perhaps p16INK4a upregulation was a consequence of a CDKN2A gene mutation, although such CDKN2A mutations seem to be rare in vulvar malignancies.39, 40
The cutoff for upregulation of p16INK4a, as a biomarker to identify HPV-associated vulvar lesions, has not been specifically defined. The Lower Anogenital Squamous Terminology Standardization Project for HPV-Associated Lesions recommends when p16INK4a should be used in combination with histology to describe HPV-associated precancerous lesions.47, 48 The experts suggest that strong and diffuse, block-positive p16INK4a result should be used to support a categorization of precancerous disease.47, 48 However, no percentage of p16INK4a+ tumors cells has been specified as a guideline to help define HPV-associated vulvar cancer. In this study, we have applied a cutoff including >25% of p16INK4a+-stained tumor cells in a diffuse pattern and with high staining intensity. This cutoff was meticulously defined by a team of expert pathologists in a study on 321 cervical cancers.21 To test how the distribution of HPV-associated vulvar cancers would change if that cutoff would be increased, we have tested different scenarios (Tables 4 and 5). We found that keratinizing vulvar cancers, the most common histological subtype in this cancer series,17 were affected the most when cutoff for p16INK4a positivity was >25%. Application of >75% cutoff to define p16INK4a positivity resulted in 66% of HPV DNA+/HPV mRNA+ keratinizing tumors to be classified as p16INK4a−, compared with only 6% when >25% p16INK4a cutoff was applied. If p16INK4a positivity would be the only biomarker to assess HPV association, these cancers would be classified as non-HPV associated. Similar observation was made for non-keratinizing tumors but to a lesser extent—27% of HPV DNA+/HPV mRNA+ non-keratinizing tumors were to be classified as p16INK4a− with >75% cutoff, in contrast to 5% with >25% cutoff. As HPV DNA and HPV mRNA expression are considered to precede the upregulation of p16INK4a in HPV-associated mucosal tumors, the lack of correlation between high percentage of p16INK4a+ tumor cells and HPV mRNA positivity in this vulvar cancer series remains to be investigated. At present, we conclude that lower percentage of p16INK4a+-stained tumor cells is associated with a degree of keratinizing component, an observation already made in our series of 321 cervical cancers.21
A single study by Riethdorf et al27 also correlated the use of HPV mRNA and p16INK4a expression in vulvar lesions and found 90% (52 of the 58) of vulvar intraepithelial neoplasia and vulvar cancer cases to be HPV16 mRNA+/p16INK4a+. More specifically, the authors showed that 31% of vulvar cancers were HPV16 mRNA+ while 34% were p16INK4a+.27 In our study, the HPV mRNA+ and p16INK4a+ concordance among vulvar cancers yielded good agreement (K=0.625, 95% CI: (0.531–0.719)), with 83% concordant pairs of HPV mRNA+ and p16INK4a+ and 9% concordant pairs of HPV mRNA− and p16INK4a−. HPV DNA+/HPV mRNA+/p16INK4a+ vulvar cancers did not significantly vary among different age groups or different time periods. The highest fraction of HPV DNA+/HPV mRNA+/p16INK4a+ vulvar cancers was identified in Oceania (92%) and the lowest in Africa (59%), which was significantly different. However, we cannot exclude that this difference might be due to the number of cases available from these two continents (Africa: n=17 versus Oceania: n=82).
A clear definition of HPV-driven vulvar cancer is important for assessing potential clinical differences in HPV-associated vulvar cancers compared with those vulvar cancers that develop through autoimmune processes. Clinical studies have demonstrated better overall and disease-free survival for patients with HPV-associated head-and-neck15, 23, 41, 49 and anal cancers42 versus mutation-induced cancers at these anatomical sites. Several recent studies have also investigated whether survival of patients with vulvar cancer is associated with tumor HPV status or p16INK4a expression.32, 50, 51, 52 At present, there has not been a definitive indication of a need for different treatment or clinical management of HPV-associated and non-HPV-associated vulvar cancers. However, few recent studies demonstrated the association of p16INK4a positivity in vulvar cancer with prolonged overall survival rate,51, 52 lower in-field relapse51 and lower recurrence rate50 compared with vulvar cancers that were p16INK4a−. It remains to be seen whether classifying HPV-driven vulvar cancers through HPV mRNA positivity together with p16INK4a upregulation should further improve the classification and evaluation of HPV-driven versus non-HPV-driven vulvar cancers.
Although the set of vulvar cancer cases analyzed here represents the collection of tissue samples worldwide, our study is limited by the small number of cases originating from North America and Africa (<5% of all cases) as opposed to the other four continents. Other limitations include the absence of fully functional data for all of the samples tested as we did not analyze the transcriptional activity of pHR-HPV30 and 69 or LR-HPV types, for which no E6*I mRNA RT-PCR assays have been developed. In addition, as our study is not population based, there remains a question of potential selection bias. However, we believe that this is unlikely as the cases selected were obtained from large pathology laboratories some of which served as the unique national laboratory for the country. In addition, we requested that selection of consecutive cases should be based on an overall diagnosis of vulvar cancer or on the availability of tissues in a given period without any additional selection criteria, such as ‘histology’ or ‘age’.17
The biggest asset of the present study is the contribution of HPV mRNA data, in addition to the provision of data on p16INK4a expression in vulvar cancer; in this regard, the study undertakes the task of providing a more reliable description of vulvar cancers with HPV-transformed phenotype. The estimate of HPV attribution in vulvar cancer in our study is lower when compared with previous HPV DNA reports and meta-analyses.3 This might be due to the use of: (i) strict HPV protocols and different laboratories to avoid potential cross-contamination during regulated procedures (tissue sectioning, DNA and RNA extraction, PCR, RT-PCR and Luminex hybridization); (ii) control samples for assessing potential cross-contamination; and (iii) additional markers indicative of active virus in addition to the sole HPV DNA presence. Finally, a clear and accurate characterization of HPV-driven tumors is the essential starting point to define possibly required altered approaches in patient management or evaluation of therapeutic response.
HPV VVAP study group for vulvar site
Argentina: Myriam Perrotta, Ana Jaen (Hospital Italiano de Buenos Aires).
Australia: Suzanne M Garland, Sepher N Tabrizi (The Royal Women’s Hospital, The University of Melbourne); Gerard Vincent Wain, Catherine Jane Kennedy, Yoke-Eng Chiew (Gynaecological Oncology, Westmead Hospital); Raghwa Sharma (Department of Tissue Pathology and Diagnostic Oncology, University of Sydney and University of Western Sydney Westmead Hospital).
Austria: Elmar Armin Joura (University Hospital and Medical School); Josefine Stani and Reinhard Horvat, MD (Medical University and General Hospital Vienna, Austria).
Bangladesh: Ashrafun Nessa, AJE Nahar Rahman, Mohammed Kamal (Bangabandhu Sheikh Mujib Medical University – BSMMU); Faruk Ahmed (Dhaka Medical College Hospital).
Belarus: Halina Viarheichyk, Sitnikov Valeriy (Gomel State Medical University); Achynovich Searhei (Gomel Regional Clinical Oncological Hospital).
Bosnia Herzegovina: Ermina Iljazovic (University Clinical Center Tuzla BiH).
Brazil: Paula Maldonado, Gutemberg Leão Almeida, Isabel Val, Renata Fonseca, Roberto José Lima, Marcia Mannarino, Yara Furtado (Instituto de Ginecologia da Universidad Federal do Rio de Janeiro).
Chile: Rodrigo Prado, Carla Molina, Rosa Muñoz (Centro de Oncología Preventiva, Facultad de Medicina, Universidad de Chile); Ximena Rodriguez, Marisol Guerrero, Virginia Leiva, Elsa Olave, Claudia Ramis, Viviana Toro (Hospital de San José).
Colombia: Raúl Murillo, Gustavo Adolfo Hernández Suárez, Carlos Eduardo Pinzón, Nubia Muñoz (Instituto Nacional de Cancerología).
Czech Republic: Václav Mandys (3rd Faculty of Medicine and University Hospital King’s Wineyards, Prague); Jan Laco (Faculty Hospital Hradec Kralove).
Ecuador: Leopoldo Tinoco, Hospital Oncológico de Quito, Quito, Ecuador.
France: Christine Clavel, Philippe Birembaut, Veronique Dalstein (CHU de Reims, Laboratoire Pol Bouin/INSERM UMR-S 903, Reims); Christine Bergeron (Laboratoire Cerba, Department de Pathology, Cergy Pontoise); Massimo Tommasino (International Agency for Research on Cancer).
Germany: Monika Hampl, Prof. Baldus (Department of Obstetrics and Gynecology, Heinrich Heine University of Duesseldorf, Duesseldorf, Germany); Karl Ulrich Petry, Alexander Luyten (Klinikum Wolfsburg); Michael Pawlita, Gordana Halec, Daniela Hoefler, Dana Holzinger (German Cancer Research Center, Division of Molecular Diagnostics of Oncogenic Infections, Heidelberg, Germany).
Greece: Theodoros Agorastos (Aristotle University of Thessaloniki).
Guatemala: Luis Estuardo Lombardi, Edgar Kestler, Obdulia Salic, Sergio Marroquin, Victor Argueta (Centro de Investigación Epidemiológica en Salud Sexual y Reproductiva-CIESAR, Hospital General San Juan de Dios); Walter Guerra (Instituto Nacional del Cáncer); Hesler Morales (Instituto Guatemalteco de Seguridad Social, Instituto Nacional del Cáncer).
Honduras: Annabelle Ferrera (Escuela de Microbiología, Universidad Nacional Autónoma de Honduras); Odessa Henríquez and Silvia Portillo (Instituto Nacional Cardiopulmonar en Tegucigalpa).
India: Neerja Bhatla (All India Institute of Medical Sciences, New Delhi-110029, India).
Israel: Jacob Bornstein, Alejandro Livoff, Hector Itzhac Cohen (Western Galilee Hospital- Nahariya).
Italy: Luciano Mariani, Amina Vocaturo, Maria Benevolo, Fernando Marandino, Francesca Rollo (Regina Elena Cancer Istitute).
Korea-South: Hai-Rim Shin, Jin-Kyung Oh (National Cancer Center); Shin Gwang Kang (Asian Medical Center); Dong-chul Kim (Kangnam St. Mary’s Hospital).
Kuwait: Waleed Al-Jassar (Faculty of Medicine, Kuwait University), Rema’a Al-Safi (Maternity Hospital).
Lebanon: Muhieddine Seoud (The American University of Beirut Medical Center).
Mali: Bakarou Kamate, Cathy Ndiaye (Hospital National DU Point G).
Mexico: Isabel Alvarado-Cabrero (Instituto Mexicano del Seguro Social); Rubén López-Revilla, Claudia Magaña-León (Instituto Potosino de Investigación Científica y Tecnológica, AC); Cuauhtémoc Oros (Hospital Central Ignacio Morones Prieto, San Luis Potosí).
Mozambique: Carla Carrilho (Eduardo Mondlane University).
New Zealand: Susan M Bigby (Middlemore Hospital), Lois J Eva, Ronald W Jones, (Auckland City Hospital).
Nigeria: Adekunbiola AF Banjo, FB Abdulkareem, AO Daramola, CC Anunobi, RU Anorlu (Lagos University Teaching Hospital Idi-Araba); Sani Malami, Ali Bala Umar (Faculty of Medicine, Bayero University).
Paraguay: Elena Kasamatsu, Antonio Leopoldo Cubilla, Francisco Perrota (Instituto de Investigaciones en Ciencias de la Salud, Universidad Nacional de Asunción) Instituto de Patología e Investigación.
Philippines: Celia Ladines Llave, Jean Anne Toral (Cervical Cancer Prevention Center-CECAP, Cancer Institute (UP-CM-PGH)); Efren J Domingo, Maria Julieta V Germar, Jerico Thaddeus, P Luna, Arnold M Fernandez, Carolyn Zalameda Castro, Roslyn Balacuit (University of the Philippine College of Medicine General Hospital).
Poland: Andrzej Marcin Nowakowski (Medical University of Lublin); Robert Jach, Jolanta Orlowska-Heitzman, Monika Kabzinska-Turek, Paulina Przybylska, Marzena kula-Prykan (Jagiellonian University Medical College).
Portugal: Eugenia Cruz (Instituto Português de Oncologia de Coimbra, Francisco Gentil – Coimbra, Portugal); Ana Félix (Instituto Portugues de Oncologia de Lisboa Francisco Gentil).
Senegal: Cathy Ndiaye, Nafissatou Ndiaye Ba, Victorino Mendes (HOGGY stands for Hôpital Général de Grand Yoff; DANTEC – Hôpital A Le Dantec; FAC – Faculté de Médecine, Université Cheikh A Diop).
Spain: Maria Alejo, Ana Mulero (Hospital General d’Hospitalet); Belén Lloveras (Hospital del Mar); Laia Alemany, F Xavier Bosch, Ignacio Bravo, Vanesa Camón, Xavier Castellsagué, Omar Clavero, Silvia de Sanjosé, Ion Espuña, Ana Esteban, Jose M Godínez, Yolanda Florencia, Joellen Klaustermeier, Nubia Muñoz, Nati Patón, Beatriz Quirós, Maëlle Saunier, Cristina Rajo, Sara Tous, Marleny Vergara (IDIBELL, Institut Català d’Oncologia – Catalan Institute of Oncology), August Vidal, Enric Condom (Hospital Universitari de Bellvitge), Jaume Ordi (Hospital Clínic), Julio Velasco, Cristina Pérez (Hospital San Agustín).
Taiwan: Chou Cheng-Yang (National Cheng Kung University Medical College, Taiwan Association of Gynecologic Oncologists); Tang-Yuan Chu (Buddhist Tzuchi Genral Hospital); Kuo-Feng Huang (Chi Mei Medical Center); Cheng Wen-Fang (National Taiwan University Hospital); Chih-Ming Ho (Gynecologic Cancer Center, Cathay General Hospital).
The Netherlands: Wim Quint, Anco C Molijn, Daan T Geraets, Nuria Guimera (DDL Diagnostic Laboratory); (Chris JLM Meijer (Vrije Universiteit Medical Center).
Turkey: Alp Usubutun (Hacettepe University).
United Kingdom: Henry Kitchener, Godfrey Wilson (School of Medicine, University of Manchester); Paul Cross (Queen Elizabeth Hospital, Sheriff Hill).
Uruguay: Adela Rosa Sica, Benedicta Caserta, Mabel Cedeira, Daniel Mazal, Guillermo Rodríguez (Laboratorio de Anatomía patológica del hospital de la Mujer, Montevideo).
USA: Wendy Cozen (Los Angeles, CA); Marc T Goodman and Brenda Y Hernández (Hawaii); Charles F Lynch, Daniel B Olson, Freda R Selk (Iowa).
Venezuela: Victoria García Barriola, Mirian Naranjo de Gómez, Adayza Figueredo, Janira Navarro (Universidad Central de Venezuela).
References
IARC Human papillomaviruses. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. A Review of Human Carcinogens—Part B: Biological Agents 2011.
Del Pino M, Rodriguez-Carunchio L, Ordi J . Pathways of vulvar intraepithelial neoplasia and squamous cell carcinoma. Histopathology 2013;62:161–175.
de Martel C, Ferlay J, Franceschi S et al, Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol 2012;13:607–615.
Thorstensen KA, Birenbaum DL . Recognition and management of vulvar dermatologic conditions: lichen sclerosus, lichen planus, and lichen simplex chronicus. J Midwifery Womens Health 2012;57:260–275.
Sideri M, Jones RW, Wilkinson EJ et al, Squamous vulvar intraepithelial neoplasia: 2004 modified terminology, ISSVD Vulvar Oncology Subcommittee. J Reprod Med 2005;50:807–810.
Committee on Gynecologic Practice of American College Obstetricians and Gynecologists. ACOG Committee Opinion No. 509: Management of vulvar intraepithelial neoplasia. Obstet Gynecol 2011;118:1192–1194.
Judson PL, Habermann EB, Baxter NN et al, Trends in the incidence of invasive and in situ vulvar carcinoma. Obstet Gynecol 2006;107:1018–1022.
Regauer S, Reich O . Etiology of vulvar cancer will impact on treatment options and therapy outcome: two major pathways of vulvar cancer. Gynecol Oncol 2013;131:246–247.
van Seters M, van Beurden M, ten Kate FJ et al, Treatment of vulvar intraepithelial neoplasia with topical imiquimod. N Engl J Med 2008;358:1465–1473.
Terlou A, van Seters M, Kleinjan A et al, Imiquimod-induced clearance of HPV is associated with normalization of immune cell counts in usual type vulvar intraepithelial neoplasia. Int J Cancer 2010;127:2831–2840.
Preti M, Scurry J, Marchitelli CE et al, Vulvar intraepithelial neoplasia. Best Pract Res Clin Obstet Gynaecol 2014;28:1051–1062.
De Vuyst H, Clifford GM, Nascimento MC et al, Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: a meta-analysis. Int J Cancer 2009;124:1626–1636.
Alemany L, Cubilla A, Halec G et al, Role of human papillomavirus in penile carcinomas worldwide. Eur Urol 2016;69:953–961.
Castellsague X, Alemany L, Quer M et al, HPV involvement in head and neck cancers: comprehensive assessment of biomarkers in 3680 patients. J Natl Cancer Inst 2016;108:djv403.
Lassen P . The role of human papillomavirus in head and neck cancer and the impact on radiotherapy outcome. Radiother Oncol 2010;95:371–380.
Halec G, Holzinger D, Schmitt M et al, Biological evidence for a causal role of HPV16 in a small fraction of laryngeal squamous cell carcinoma. Br J Cancer 2013;109:172–183.
de Sanjose S, Alemany L, Ordi J et al, Worldwide human papillomavirus genotype attribution in over 2000 cases of intraepithelial and invasive lesions of the vulva. Eur J Cancer 2013;49:3450–3461.
Halec G, Schmitt M, Dondog B et al, Biological activity of probable/possible high-risk human papillomavirus types in cervical cancer. Int J Cancer 2013;132:63–71.
de Sanjose S, Quint WG, Alemany L et al, Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol 2010;11:1048–1056.
von Knebel Doeberitz M . The causal role of human papillomavirus infections in non-anogenital cancers. It's time to ask for the functional evidence. Int J Cancer 2016;139:9–11.
Halec G, Alemany L, Lloveras B et al, Pathogenic role of the eight probably/possibly carcinogenic HPV types 26, 53, 66, 67, 68, 70, 73 and 82 in cervical cancer. J Pathol 2014.
Schmitt M, Pawlita M . The HPV transcriptome in HPV16 positive cell lines. Mol Cell Probes 2011;25:108–113.
Boscolo-Rizzo P, Pawlita M, Holzinger D . From HPV-positive towards HPV-driven oropharyngeal squamous cell carcinomas. Cancer Treat Rev 2015;42:24–29.
Jochmus I, Durst M, Reid R et al, Major histocompatibility complex and human papillomavirus type 16 E7 expression in high-grade vulvar lesions. Hum Pathol 1993;24:519–524.
Wentzensen N, Ridder R, Klaes R et al, Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene 2002;21:419–426.
Hillemanns P, Wang X . Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol 2006;100:276–282.
Riethdorf S, Neffen EF, Cviko A et al, p16INK4A expression as biomarker for HPV 16-related vulvar neoplasias. Hum Pathol 2004;35:1477–1483.
Crum CP, Nuovo G, Friedman D et al, Accumulation of RNA homologous to human papillomavirus type 16 open reading frames in genital precancers. J Virol 1988;62:84–90.
Park JS, Kurman RJ, Kessis TD et al, Comparison of peroxidase-labeled DNA probes with radioactive RNA probes for detection of human papillomaviruses by in situ hybridization in paraffin sections. Mod Pathol 1991;4:81–85.
Higgins GD, Uzelin DM, Phillips GE et al, Presence and distribution of human papillomavirus sense and antisense RNA transcripts in genital cancers. J Gen Virol 1991;72 (Pt 4):885–895.
Auvinen E, Kujari H, Arstila P et al, Expression of the L2 and E7 genes of the human papillomavirus type 16 in female genital dysplasias. Am J Pathol 1992;141:1217–1224.
Alonso I, Fuste V, del Pino M et al, Does human papillomavirus infection imply a different prognosis in vulvar squamous cell carcinoma? Gynecol Oncol 2011;122:509–514.
Guerrero D, Guarch R, Ojer A et al, Differential hypermethylation of genes in vulvar cancer and lichen sclerosus coexisting or not with vulvar cancer. Int J Cancer 2011;128:2853–2864.
Reyes MC, Cooper K . An update on vulvar intraepithelial neoplasia: terminology and a practical approach to diagnosis. J Clin Pathol 2014;67:290–294.
Stewart CJ, Crook ML . Fascin and cyclin D1 immunoreactivity in non-neoplastic vulvar squamous epithelium, vulvar intraepithelial neoplasia and invasive squamous carcinoma: correlation with Ki67 and p16 protein expression. J Clin Pathol 2014;67:319–325.
McLaughlin-Drubin ME, Park D, Munger K . Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc Natl Acad Sci USA 2013;110:16175–16180.
Munger K, Jones DL . Human papillomavirus carcinogenesis: an identity crisis in the retinoblastoma tumor suppressor pathway. J Virol 2015;89:4708–4711.
Hirama T, Miller CW, Wilczynski SP et al, p16 (CDKN2/cyclin-dependent kinase-4 inhibitor/multiple tumor suppressor-1) gene is not altered in uterine cervical carcinomas or cell lines. Mod Pathol 1996;9:26–31.
Soufir N, Queille S, Liboutet M et al, Inactivation of the CDKN2A and the p53 tumour suppressor genes in external genital carcinomas and their precursors. Br J Dermatol 2007;156:448–453.
Trietsch MD, Spaans VM, ter Haar NT et al, CDKN2A(p16) and HRAS are frequently mutated in vulvar squamous cell carcinoma. Gynecol Oncol 2014;135:149–155.
Lassen P, Primdahl H, Johansen J et al, Impact of HPV-associated p16-expression on radiotherapy outcome in advanced oropharynx and non-oropharynx cancer. Radiother Oncol 2014;113:310–316.
Koerber SA, Schoneweg C, Slynko A et al, Influence of human papillomavirus and p16(INK4a) on treatment outcome of patients with anal cancer. Radiother Oncol 2014;113:331–336.
Vinokurova S, Wentzensen N, von Knebel Doeberitz M . Analysis of p16INK4a and integrated HPV genomes as progression markers. Methods Mol Med 2005;119:73–83.
Holzinger D, Flechtenmacher C, Henfling N et al, Identification of oropharyngeal squamous cell carcinomas with active HPV16 involvement by immunohistochemical analysis of the retinoblastoma protein pathway. Int J Cancer 2013;133:1389–1399.
Terlou A, van Seters M, Ewing PC et al, Treatment of vulvar intraepithelial neoplasia with topical imiquimod: seven years median follow-up of a randomized clinical trial. Gynecol Oncol 2011;121:157–162.
Guimera N, Lloveras B, Lindeman J et al, The occasional role of low-risk human papillomaviruses 6, 11, 42, 44, and 70 in anogenital carcinoma defined by laser capture microdissection/PCR methodology: results from a global study. Am J Surg Pathol 2013;37:1299–1310.
Darragh TM, Colgan TJ, Cox JT et al, The Lower Anogenital Squamous Terminology Standardization Project for HPV-Associated Lesions: background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Arch Pathol Lab Med 2012;136:1266–1297.
Nuno T, Garcia F . The Lower Anogenital Squamous Terminology Project and its implications for clinical care. Obstet Gynecol Clin North Am 2013;40:225–233.
Holzinger D, Schmitt M, Dyckhoff G et al, Viral RNA patterns and high viral load reliably define oropharynx carcinomas with active HPV16 involvement. Cancer Res 2012;72:4993–5003.
Hay CM, Lachance JA, Lucas FL et al, Biomarkers p16, human papillomavirus and p53 predict recurrence and survival in early stage squamous cell carcinoma of the vulva. J Low Genit Tract Dis 2016;20:252–256.
Lee LJ, Howitt B, Catalano P et al, Prognostic importance of human papillomavirus (HPV) and p16 positivity in squamous cell carcinoma of the vulva treated with radiotherapy. Gynecol Oncol 2016;142:293–298.
Sznurkowski JJ, Zawrocki A, Biernat W . The overexpression of p16 is not a surrogate marker for high-risk human papilloma virus genotypes and predicts clinical outcomes for vulvar cancer. BMC Cancer 2016;16:465.
Acknowledgements
We thank Dr. Otoniel Martinez-Maza for his support during preparation of this manuscript and Mrs Crystal Freeman and Dr Geoff Smith for careful reading and their constructive comments. This work was supported by grant numbers from the following funders: Funder 1: Instituto de Salud Carlos III (Spanish Government): RCESP C03/09, RTICESP C03/10, RTIC RD06/0020/0095, RD12/0036/0056, and CIBERESP; Funder 2: Agència de Gestió d’AjutsUniversitarisi de Recerca (AGAUR): 2005SGR00695, 2009SGR126, and 2014SGR1077; Funder 3: Lilly Foundation. Premio de Investigación Biomédica Preclínica 2012; Funder 4: Asociación Española Contra el Cáncer, Programa de Formación Avanzada en Oncología 2012; and Funder 5: Sanofi Pasteur MSD and Merck: EP080140290702.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
MP has received research support through cooperation contracts of DKFZ with Roche and Qiagen in the field of development of HPV diagnostics. He is an inventor on patents owned by DKFZ in the field of HPV diagnostics. Authors LA and SdS have received occasional travel fund to attend scientific meetings from Merck and Sanofi Pasteur MSD. The other authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Halec, G., Alemany, L., Quiros, B. et al. Biological relevance of human papillomaviruses in vulvar cancer. Mod Pathol 30, 549–562 (2017). https://doi.org/10.1038/modpathol.2016.197
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.197
This article is cited by
-
Viral Venereal Diseases of the Skin
American Journal of Clinical Dermatology (2021)
-
Role of human papillomavirus infection in the etiology of vulvar cancer in Italian women
Infectious Agents and Cancer (2020)
-
Prognostic implications of genotyping and p16 immunostaining in HPV-positive tumors of the uterine cervix
Modern Pathology (2020)
