
Abstract
HMGA2, CDK4, and JUN genes have been described as frequently coamplified with MDM2 in atypical lipomatous tumor, well-differentiated liposarcoma, and dedifferentiated liposarcoma. We studied the frequency of amplification of these genes in a series of 48 dedifferentiated liposarcomas and 68 atypical lipomatous tumors/well-differentiated liposarcomas. We correlated their amplification status with clinicopathological features and outcomes. Histologically, both CDK4 (P=0.007) and JUN (P=0.005) amplifications were associated with dedifferentiated liposarcoma, whereas amplification of the proximal parts of HMGA2 (5′-untranslated region (UTR) and exons 1–3) was associated with atypical lipomatous tumor/well-differentiated liposarcoma (P=0.01). CDK4 amplification was associated with axial tumors. Amplification of 5′-UTR and exons 1–3 of HMGA2 was associated with primary status and grade 1. Shorter overall survival was correlated with: age >64 years (P=0.03), chemotherapy used in first intent (P<0.001), no surgery (P=0.003), grade 3 (P<0.001), distant metastasis (P<0.001), node involvement (P=0.006), and CDK4 amplification (P=0.07). In multivariate analysis, distant metastasis (HR=8.8) and grade 3 (HR=18.2) were associated with shorter overall survival. A shorter recurrence-free survival was associated with dedifferentiated liposarcoma (P<0.001), grade 3 (P<0.001), node involvement (P<0.001), distant metastasis (P=0.02), recurrent status (P=0.009), axial location (P=0.001), and with molecular features such as CDK4 (P=0.05) and JUN amplification (P=0.07). Amplification of 5′-UTR and exons 1–3 (P=0.08) and 3′-UTR (P=0.01) of HMGA2 were associated with longer recurrence-free survival. Distant metastasis was associated with shorter recurrence-free survival (HR=5.8) in multivariate analysis. Dedifferentiated liposarcoma type was associated with axial location, grade 3 and recurrent status. In conclusion, we showed that the amplification of HMGA2 was associated with the atypical lipomatous tumor/well-differentiated liposarcoma histological type and a good prognosis, whereas CDK4 and JUN amplifications were associated with dedifferentiated liposarcoma histology and a bad prognosis. In addition, we also provided the first description of the molecular evolution of a well-differentiated liposarcoma into four successive dedifferentiated liposarcoma relapses, which was consistent with our general observations.
Similar content being viewed by others
Main
Atypical lipomatous tumor, well-differentiated liposarcoma, and dedifferentiated liposarcoma are the most frequent types of liposarcomas. Atypical lipomatous tumor refers to surgically amenable tumors located in the limbs or the trunk, whereas well-differentiated liposarcomas are intra-abdominal or retroperitoneal lesions.1 Those tumors are locally aggressive but do not show the potential for metastasis unless they undergo dedifferentiation. ‘Dedifferentiation,’ defined as the transition from atypical lipomatous tumor/well-differentiated liposarcoma to a non-lipogenic and often high-grade liposarcoma, occurs in ~10% of atypical lipomatous tumors/well-differentiated liposarcomas. Cytogenetically, atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma are characterized by supernumerary ring or giant chromosomes made up of 12q14–15 amplification including the MDM2 gene. Other variable chromosomal regions are generally coamplified.2 Dedifferentiated liposarcoma may carry additional genetic abnormalities when compared with atypical lipomatous tumor/well-differentiated liposarcoma,3 suggesting that atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma represent an original model of sarcoma transformation.4
MDM2 is considered as the driver gene of the 12q amplicon as it is amplified and expressed in 100% of cases.5, 6 The detection of MDM2 amplification helps to distinguish atypical lipomatous tumor/well-differentiated liposarcoma from benign lipomatous tumors and dedifferentiated liposarcoma from other pleomorphic malignant proliferations, although MDM2 amplification can be observed in other malignant tumors.7 MDM2 amplification leads to the inactivation of TP53, which is rarely mutated in those tumors.8 The pathogenesis of atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma might also be linked to several molecular events in addition to the inactivation of TP53 such as the frequent amplification of HMGA2,5, 6 CDK4,6, 9 and FRS2,10 as well as other genes located nearby MDM2. These genes are probably not only passive passengers of the amplicons but may also have a crucial role in the genesis and progression of these tumors. HMGA2 is expressed during embryogenesis but not in normal adult tissues. It regulates transcription through DNA architectural modifications and interacts with other proteins of the enhanceosome. Its overexpression in benign and malignant tumors indicates oncogenic properties.11 CDK4 phosphorylates RB1 (retinoblastoma protein 1) and is implicated in the G1–S checkpoint of the cell cycle.12 CDK4 is frequently but not systematically coamplified with MDM2 in atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma.9
Among the mechanisms that have been reported to be involved in atypical lipomatous tumor/well-differentiated liposarcoma dedifferentiation, the amplification of JUN (1p32) or MAP3K5 (6q23.3) (that encodes a kinase acting upstream of JUN) has been suggested because of a correlation between JUN amplification and dedifferentiated liposarcoma histotype.13, 14 Remarkably, JUN can regulate the activity of transcription factors involved in adipogenesis. However, a systematic link between inhibition of adipocytic differentiation and JUN amplification was not confirmed.15, 16
Our aim was to assess the frequencies of HMGA2, CDK4, and JUN amplification, as well as the prognostic value of these amplifications in a large series of liposarcomas. In particular, we explored the quantitative status of intra- and extraexonic parts of the HMGA2 gene. In addition, we provided the detailed evolution of the amplification of these genes over a 15-year dedifferentiation process in the primary well-differentiated liposarcoma and recurrent dedifferentiated liposarcomas from the same patient.
Materials and methods
Patients and Samples
We have identified 116 MDM2-amplified cases among 210 formalin-fixed and paraffin-embedded samples of liposarcomas (Supplementary Table 1). These tumors had been submitted to our laboratory between July 2006 and January 2012 for molecular cytogenetic analyses because they were suspected to be atypical lipomatous tumors/well-differentiated liposarcomas/dedifferentiated liposarcomas. Pathological evaluation had been made by pathologists who are experts in sarcomas from 13 French centers. Differentiation of tumors and surgical margins were defined according to the French Sarcoma Group grading17 and the surgical staging system of Enneking et al,18 respectively. Clinicopathologic data were collected from medical records and from the European sarcoma database and tumor bank ‘SarcomaBCB Conticabase’ (https://conticabase.sarcomabcb.org/). Tumors located in the limbs were designed as peripheral, and the others as axial. This study was approved by the boards of the participating institutions. For simplification, atypical lipomatous tumors and well-differentiated liposarcomas were grouped under the term atypical lipomatous tumor/well-differentiated liposarcoma.
Molecular Cytogenetic Analysis
Bacterial artificial chromosomes were prepared as FISH probes for HMGA2 (Figure 1), CDK4, and JUN (Table 1) according to standard procedures.19 Genomic amplification was defined as the presence of at least 10 fluorescent signals (grouped in clusters) per cell in ≥1% of cells. Genomic gain was defined as the presence of an average of ~3–9 fluorescent signals evaluated in the same conditions. Microscopic analysis was performed using a DM6000B microscope (Leica Microsystems, Wetzlar, Germany) by two independent observers in a blinded manner. FISH images were processed using the ISIS software (Metasystems, Altlussheim, Germany).

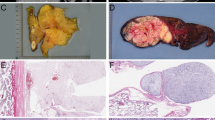
Schematic representation of the structure of the HMGA2 gene (12q14.3) and matching fluorescence in situ hybridization (FISH) probes: black solid rectangles represent the five exons. The upper part indicates the locations of BAC clones RP11-30I11 (upstream region of 5′-UTR of HMGA2), RP11-299L9 (5′-UTR and exons 1 and 2 of HMGA2), RP11-23C9 (5′-UTR and exons 1, 2, and 3 of HMGA2), CTC-782I9 (third intron and exon 4 of HMGA2), CTD-2240K5 (exons 4 and 5 and 3′-UTR of HMGA2) and RP11-118B13 (3′-UTR of HMGA2), according to http://genome.ucsc.edu/ (February 2009 release).
Comparative genomic hybridization on arrays (a-CGH) was performed in seven cases (07T082, 09T033, 09T098, 10T542, 10T543, 11T1030, 11T1024) to complete the FISH data and in the primary and four successive recurrences of case 74. Male and female human genomic DNA (Promega Corp., Madison, WI, USA) was used as the control DNA. Following hybridization, washing, and drying steps, the microarray slides were scanned at 3 μm resolution, using the Agilent SureScan (Agilent Technologies). Features were extracted from the scanned images and analyzed using Cytogenomics software (version 2.5.8.11; Agilent Technologies) using the aberration detection method-2 algorithm. Thresholds were set at a minimum of 3 probes and 0.25 average log ratios. Raw data have been submitted to Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) with the reference GSE64597. Gene amplification was defined by a log ratio >1.1 and genomic gain was defined by a log ratio between 0.2 and 1.1.
Immunohistochemistry
Immunohistochemical analysis was carried out in 44 cases for CDK4 (clone DCS 31; Biosource International, Camarillo, CA, USA), 57 cases for MDM2 (clone IF2; Zymed Laboratories, South San Francisco, CA, USA), and 30 cases for HMGA2 (clone HMGA2-P1; Biocheck, Foster City, CA, USA) according to standard procedures.19, 20 The staining was considered as positive when at least one cell nucleus per high-power field was strongly stained.
Statistical Analysis
Date of histological diagnosis was used for date of diagnosis. Statistical comparisons were performed by using χ2 or Fisher’s exact tests for categorical data. The Kaplan–Meier method was used to estimate the time to survival rates, which were compared by the log-rank test. P-value ≤0.05 was significant. For analysis of factors influencing survival, the variables tested were gender, age at diagnosis, history of cancer, histological subtype, the French Sarcoma Group grading, complete remission after first treatment, node involvement, metastatic status, CDK4 amplification, HMGA2 amplification, and JUN amplification. If variables were significantly associated with survival in the univariate analysis, a multivariate analysis using Cox proportional hazards regression was performed. The R-3.1.1 statistical software for Windows was used.
Results
Clinical and Histological Features
The 116 MDM2-amplified samples included 48 dedifferentiated liposarcomas and 68 atypical lipomatous tumors/well-differentiated liposarcomas (Table 2). Median age was 64 years (38–88 years) for atypical lipomatous tumors/well-differentiated liposarcomas and 69 years (42–90 years) for dedifferentiated liposarcomas. There were 72 men and 44 women. Median tumor sizes and ranges were 160 mm (35–400 mm) for atypical lipomatous tumors/well-differentiated liposarcomas and 150 mm (27–410 mm) for dedifferentiated liposarcomas. When compared with atypical lipomatous tumors/well-differentiated liposarcomas, dedifferentiated liposarcomas were associated with: (i) axial location (P<0.001), (ii) grade 3 (P<0.001), (iii) prescription of chemotherapy (P=0.02), (iv) recurrent status (P=0.07), and (v) lack of complete remission after the first treatment (P=0.02).
Expression and Amplification of MDM2, CDK4, JUN, and HMGA2 in Atypical Lipomatous Tumors/Well-Differentiated Liposarcomas and Dedifferentiated Liposarcomas
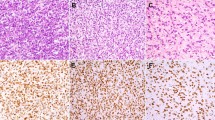
A positive MDM2 staining using immunohistochemistry was observed in 49 of the 57 analyzed cases (86%): the eight negative cases were atypical lipomatous tumors/well-differentiated liposarcomas; all the dedifferentiated liposarcomas showed a positive expression for MDM2 (P=0.02). CDK4 amplification was significantly associated with dedifferentiated liposarcoma histotype (88.2% dedifferentiated liposarcomas vs 58.9% atypical lipomatous tumors/well-differentiated liposarcomas, P=0.007) (Figure 2b). A positive CDK4 staining using immunohistochemistry was observed in 15 (60%) atypical lipomatous tumors/well-differentiated liposarcomas and 16 (84%) dedifferentiated liposarcomas. The amplification of the 5′-untranslated region (UTR) and exons 1–3 of HMGA2 was associated with atypical lipomatous tumor/well-differentiated liposarcoma histotype: 95.3% of atypical lipomatous tumors/well-differentiated liposarcomas vs 75% of dedifferentiated liposarcomas (P=0.01) (Figure 2a and Table 3) and HMGA2 was expressed in 12 (93%) atypical lipomatous tumors/well-differentiated liposarcomas and 15 (88%) dedifferentiated liposarcomas. JUN amplification was also significantly associated with dedifferentiated liposarcoma (60.5% dedifferentiated liposarcomas vs 16.7% atypical lipomatous tumors/well-differentiated liposarcomas, P=0.005) (Figure 2b).

Histograms representing (a) the proportion of tumors with amplification of HMGA2 domains according to histological subtype and (b) the proportion of tumors with amplification of HMGA2 (5′-UTR and exons 1–3), CDK4, and JUN according to the histological subtype. *Statistical significance (P≤0.05).
Molecular Markers and Clinical Correlations
CDK4 amplification was associated with an axial tumor location (66% vs 39%, P=0.05). Amplification of 5′-UTR and exons 1–3 of HMGA2 was associated with a tumor size >150 mm (57% vs 17%, P=0.05), primary status (82% vs 33%, P=0.02), and grade 1 (68% vs 0%, P=0.004). JUN amplification was associated with grade 3 (35% vs 12%, P=0.07) and use of radiotherapy (44% vs 12%, P=0.06).
Survival and Prognosis Data
Median follow-up duration was 40.7 months (95% CI: 32.0–45.5). Median overall survival was not reached at the time of analysis for the atypical lipomatous tumor/well-differentiated liposarcoma group and was equal to 27.7 months for dedifferentiated liposarcomas. The 2-year (2y) overall survival rate was 98% for atypical lipomatous tumors/well-differentiated liposarcomas and 55.3% for dedifferentiated liposarcomas (P<0.001) (Figure 3a). Patients for whom a complete remission was initially obtained had a better prognosis compared with those with residual tumors (2y overall survival: 95.7% vs 45%, P<0.001). Shorter overall survival was observed in patients aged >64 years (2y overall survival: 74.5% vs 87.3%, P=0.03), patients treated by chemotherapy in first intent (2y overall survival: 42.4% vs 93.8%, P<0.001) and absence of surgery (2y overall survival: 33.3% vs 86.2%, P=0.003), both because of initial unresectability of the tumor. Shorter overall survival was correlated with grade 3 (2y overall survival: 41% vs 100% for grade 1, P<0.001) (Figure 3b), distant metastasis (2y overall survival: 40% vs 87.3%, P<0.001) (Figure 3c), and node involvement (2y overall survival: 50% vs 86%, P=0.006). Shorter overall survival was associated with CDK4 amplification (2y overall survival: 79.8% vs 94.7%, P=0.07) (Figure 3d) and CDK4-positive immunohistochemistry staining (2y overall survival: 79.7% vs 100%, P=0.03) but neither with amplification of JUN (P=0.2) nor 5′-UTR and exons 1–3 of amplification of HMGA2 (P=0.3). Multivariate analysis confirmed the prognostic role of distant metastasis (HR=8.8; 95% CI: 1.6–49–2) and grade 3 (HR=18.2; 95% CI: 1.8–182.7).

Overall survival (OS) according to (a) histological subtype, (b) differentiation, (c) distant metastasis, and (d) CDK4 amplification. ALT/WDLPS, atypical lipomatous tumor/well-differentiated liposarcoma; DDLPS, dedifferentiated liposarcoma.
A shorter recurrence-free survival was associated with histology of dedifferentiated liposarcoma (2y recurrence-free survival: 38.5% vs 92%, P<0.0001) (Figure 4a), grade 3 (2y recurrence-free survival: 44% vs 97%, P<0.0001) (Figure 4b), node involvement (2y recurrence-free survival: 0 vs 74%, P<0.0001), distant metastasis (2y recurrence-free survival: 0 vs 74%, P=0.02), recurrent status (P=0.009), axial location (P=0.001), and absence of complete remission after first treatment (2y recurrence-free survival: 25% vs 73%, P=0.0002). CDK4 amplification (2y recurrence-free survival: 67% vs 93%, P=0.05) (Figure 4c) and JUN amplification (2y recurrence-free survival: 38% vs 64.2%, P=0.07) (Figure 4d) were associated with shorter recurrence-free survival. Amplification of 5′-UTR and exons 1–3 (2y recurrence-free survival: 69% vs 40%, P=0.08) (Figure 4e) and 3′-UTR (2y recurrence-free survival: 84% vs 52%, P=0.01) (Figure 4f) of HMGA2 was associated with longer recurrence-free survival. Multivariate analysis confirmed a shorter recurrence-free survival for metastatic patients (HR=5.8; 95% CI: 1.1–30).

Recurrence-free survival (RFS) according to (a) histological subtype, (b) differentiation, (c) CDK4 amplification, (d) JUN amplification, (e) 5′-UTR and exons 1–3 amplification of HMGA2, and (f) 3′-UTR amplification of HMGA2. ALT/WDLPS, atypical lipomatous tumor/well-differentiated liposarcoma; DDLPS, dedifferentiated liposarcoma; UTR, untranslated region.
Detailed Medical History and Molecular Data of Case 74
The tumor sample of case 74 was a dedifferentiated liposarcoma analyzed in 2009. Because we had studied several preceding and subsequent samples from this patient (from 1999 to 2014), we compared the histological and genomic features of these successive samples. The patient was 53 years old when she was diagnosed with a retroperitoneal grade 1 well-differentiated liposarcoma in 1999. Molecular cytogenetic analysis of the tumor after complete resection showed ring chromosomes containing amplified sequences from the chromosomes 12 and 1. In particular, MDM2 and CDK4 were amplified (Table 4). Only a moderate gain of HMGA2 was detected. JUN and MAP3K5 were not amplified. A second enlarged surgery of intra-abdominal and retroperitoneal relapse was performed in 2008 and followed by hyperthermic intraperitoneal chemotherapy. The recurrent tumor was an undifferentiated grade 3 sarcoma. The presence of amplification of MDM2 and CDK4 and absence of HMGA2 amplification detected by FISH was in favor of a dedifferentiation of the previous well-differentiated liposarcoma. CGH analysis showed more complex anomalies than for the initial well-differentiated liposarcoma (Supplementary Table 2), including the occurrence of JUN amplification (Table 4). In July 2009, a lytic lesion of the 11th right rib and a mass of the right flank were both completely resected. Histological analyses indicated a relapse of the dedifferentiated liposarcoma. FISH and CGH analyses showed amplification of MDM2, CDK4, JUN, and gain of HMGA2. A macroscopically complete resection was performed and a regular follow-up was resumed. In 2011, a mass of the right thoracic wall was resected. Margins were microscopically involved; histological and molecular analyses confirmed a third relapse with amplification of MDM2 and JUN, a gain of CDK4, and absence of amplification of HMGA2. External radiotherapy was delivered in January and February 2012. In 2014, a fourth relapse was resected with microscopic clear margins. The CGH and FISH analyses showed an amplification of the regions including CDK4, WIF1, HMGA2, MDM2, and JUN, and the occurrence of MAP3K5 amplification.
Discussion
The role of HMGA2 in adipose tumors is intriguing and not yet elucidated. When we started this study, information about the genomic status of HMGA2 in liposarcoma was scarce. A high frequency of the amplification of HMGA2 in well-differentiated liposarcoma had indeed been reported by our group5, 19 and by others.6, 10, 21 Altogether, the available data had been obtained by heterogeneous methods including FISH,5, 19, 22 qRT-PCR,19 Southern blotting,21 or CGH,6 and the total number of patients studied was small. Here, we used FISH in a large series of 116 atypical lipomatous tumors/well-differentiated liposarcomas and dedifferentiated liposarcomas and we demonstrated that amplification of HMGA2 was significantly associated with atypical lipomatous tumor/well-differentiated liposarcoma, whereas the absence of amplification of HMGA2 was associated with dedifferentiated liposarcoma. We chose FISH analysis because we believed that it was a more appropriate and a more sensitive method than array-CGH to detect amplifications either concerning only a segment of a gene (in particular, various segments of HMGA2) or only a fraction of the cell population. Our conclusions are in agreement with those of Dreux et al23 who observed a positive HMGA2 protein expression using immunohistochemistry in 86% of 43 atypical lipomatous tumors/well-differentiated liposarcomas and 67% of 69 dedifferentiated liposarcomas (P=0.02) and with those of Tap et al6 who found HMGA2 amplified in 100% of 21 atypical lipomatous tumor/well-differentiated liposarcomas, whereas it was amplified only in 15 of 17 dedifferentiated liposarcomas (88%). HMGA2 amplification was associated with favorable features such as absence of node involvement, primary status, and grade 1, as well as longer recurrence-free survival in both atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma patients. This better prognosis seems specific to adipose tumors as alterations of HMGA2 have been associated with bad prognosis in several other tumor types, mainly carcinomas.24, 25, 26 Our observations are reminiscent of the positive role of HMGA2 in adipocytic differentiation. Indeed, hmga2−/− mice show a pygmy phenotype characterized by a small size and a deficiency in fat tissue,27 whereas overexpression of a truncated hmga2 gene induces gigantism and lipomatosis in murine models.28 Moreover, HMGA2 is involved in cell differentiation.29 For instance, HMGA2 is expressed in yolk sac testicular germ cell tumors and in embryonal carcinomas but not in intratubular germ cell tumor and seminomas;30 it is expressed in low-grade gliomas but not in glioblastomas.31 Altogether, these observations suggest that HMGA2 also interferes with cell proliferation by promoting cell differentiation.
We also confirmed that the exons 1–3 of HMGA2—which encode the three DNA AT-hook-binding domains—were more frequently amplified compared with exons 4–5 (88% vs 76%).5, 19 Importantly, it has been previously shown that the expression of a truncated form of HMGA2 that retained only the three DNA-binding domains was sufficient to induce malignant transformation of fibroblasts.32 More recently, Taylor et al33 studied the genome, exome, transcriptome, and methylome of primary and recurrent cases of dedifferentiated liposarcomas and showed that HMGA2 lacked its 3′-UTR in both tumors. Altogether, those results support the notion that HMGA2 oncogenicity is mediated by its first three exons and the loss of its regulatory negative 3′-UTR.
HMGA2 is rearranged and overexpressed in a majority of lipomas,19 amplified, rearranged, and overexpressed in most atypical lipomatous tumors/well-differentiated liposarcomas5 and in a significant proportion of dedifferentiated liposarcomas. This suggests that HMGA2 may be the cornerstone of a genetic continuum among the lineage of differentiated adipocytic tumors. The expression of HMGA2 may maintain a degree of adipocytic differentiation.28, 34 Conversely, lack of HMGA2 expression when MDM2 is amplified may favor dedifferentiation.27 The role of HMGA2 in tumorigenesis might therefore be alternately pro- or antioncogenic35 according to the molecular context. Indeed, HMGA2 accumulates on the chromatin of senescent fibroblasts and cooperates with p16INK4a to stop proliferation and contribute to oncogene repression.35 Interestingly, this antiproliferative action was bypassed by an overexpression of MDM2 or CDK4,35 which is the particular cellular context of atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma. This may explain how HMGA2 might be the lever arm of the proliferation/dedifferentiation in MDM2/CDK4-amplified adipocytic tumors.
In the present series, CDK4 was not amplified in 24% of atypical lipomatous tumors/well-differentiated liposarcomas and 14% of dedifferentiated liposarcomas. This confirms that the coamplification of CDK4 is not necessary in MDM2-amplified atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma.6, 9 CDK4 amplification was significantly associated with axial location, dedifferentiated liposarcoma phenotype, shorter recurrence-free survival, and overall survival. Altogether, the amplification of CDK4 appears as a negative event in liposarcoma.9 Moreover, some of the genes located within the sequence targeted by the BAC clone RP11-571M6 containing CDK4 may also have a role. In this setting, Tap et al6 studied the amplification status of genes in the vicinity of CDK4 in well-differentiated liposarcomas/dedifferentiated liposarcomas. They found a frequent amplification of the genes involved in cell growth, motility, and proliferation.
JUN amplification was significantly associated with dedifferentiated liposarcoma subtype, as suggested in smaller series.6, 15, 33 This correlation might suggest a role of JUN in dedifferentiation.15 However, JUN amplification is also observed in some atypical lipomatous tumors/well-differentiated liposarcomas without any sign of dedifferentiation. In previous reports, a majority of dedifferentiated liposarcomas did not show any amplification of JUN.6, 15, 36, 37 Moreover, Chibon et al13 showed that neither JUN nor MAP3K5 (ASK1) were overexpressed or amplified in one-third of dedifferentiated liposarcoma cases. In vitro and in vivo analyses showed that overexpression and amplification of JUN did not influence adipocytic differentiation but had oncogenic properties in dedifferentiated liposarcoma.15 This oncogenicity might better explain the worse recurrence-free survival associated with JUN amplification in our series. Recently, plitidepsin (Aplidin®), a drug that requires an activation of the JUN pathway to induce apoptosis, was evaluated in a phase II trial in advanced dedifferentiated liposarcomas and failed to demonstrate any benefit.38
To the best of our knowledge, we describe for the first time the comprehensive genomic analysis of several tumors from a same patient, including a primary well-differentiated liposarcoma and the four successive dedifferentiated liposarcoma recurrences. This analysis gives some clues on well-differentiated liposarcoma-dedifferentiated liposarcoma genesis. Besides the early and constant amplification of MDM2, several additional features are of interest: (i) CDK4 was amplified in all tumors (except the third recurrence where a gain was noted, which could be explained by a technical artifact); (ii) JUN amplification was not initially detectable but occurred concomitantly with the dedifferentiation of the tumor and persisted in all successive relapses; (iii) MAP3K5 amplification was detected only in the fourth recurrence, demonstrating that MAP3K5 and JUN amplifications are not mutually exclusive events; (iv) HMGA2 showed only a moderate gain in the primary tumor and first recurrences. This absence of amplification may have favored the dedifferentiation process. Remarkably, HMGA2 was amplified in the fourth relapse (2014). This late amplification is perhaps related to a functional determinism to counteract the synergistic effect of the coamplification of JUN and MAP3K5 in this last relapse; (v) we observed both variability and an increasing complexity of the additional genomic abnormalities throughout the successive recurrences. The genomic profile of the primary well-differentiated liposarcoma was quite simple, which underlines the predominance of the 12q13–15 region amplification. In this setting, Taylor et al33 confirmed that the 12q amplicon structure was organized around the MDM2 amplification. Those specific amplicons follow progressive rearrangements and amplifications that lead to distinct clustering rearrangements between primary and recurrent dedifferentiated liposarcoma. On the other hand, one cannot exclude the existence of a potential minority dedifferentiated clone pre-existing in the initial well-differentiated liposarcoma. Indeed, the occurrence of JUN amplification was correlated with a dedifferentiated phenotype. Although no dedifferentiated liposarcoma cells were detected in the multiple tumor blocks of the primary tumor, the early presence of overlooked dedifferentiated foci cannot be definitely excluded. Both the hypothesis of a clonal heterogeneity and of a progressive onset of JUN or MAP3K5 amplification have to be considered. Nord et al16 showed that HMGA2, JUN, CDK4, or MAP3K5 are among other oncogenes amplified in sarcomas with MDM2-positive rings. In contrast with the genetic complexity of the ring chromosome, the karyotype of the MDM2-positive ring chromosomes was quite simple, suggesting that their formation was an early genetic event and that the content of these rings was sufficient for tumor development.16
In summary, we have shown that, among atypical lipomatous tumors/well-differentiated liposarcomas and dedifferentiated liposarcomas, the amplification of HMGA2 was associated with the atypical lipomatous tumor/well-differentiated liposarcoma subtype and a good prognosis, whereas CDK4 and JUN amplification were associated with dedifferentiated liposarcoma histology and a poor outcome. Although HMGA2 and MDM2 might be implicated in the early process of the atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma genesis, CDK4 and JUN may have a secondary role in the genesis of those tumors but a negative oncogenic effect.
References
Dei Tos AP, Pedeutour F . Well-differentiated liposarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW et al (eds). WHO Classification of Tumors of Soft Tissue and Bone, 4th edn. International Agency for Research on Cancer (IARC): Lyon, France, 2013, pp 33–36.
Pedeutour F, Forus A, Coindre JM et al. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer 1999;24:30–41.
Crago AM, Socci ND, DeCarolis P et al. Copy number losses define subgroups of dedifferentiated liposarcoma with poor prognosis and genomic instability. Clin Cancer Res 2012;18:1334–1340.
Crago AM, Singer S . Clinical and molecular approaches to well differentiated and dedifferentiated liposarcoma. Curr Opin Oncol 2011;23:373–378.
Italiano A, Bianchini L, Keslair F et al. HMGA2 is the partner of MDM2 in well-differentiated and dedifferentiated liposarcomas whereas CDK4 belongs to a distinct inconsistent amplicon. Int J Cancer 2008;122:2233–2241.
Tap WD, Eilber FC, Ginther C et al. Evaluation of well-differentiated/de-differentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridization. Genes Chromosomes Cancer 2011;50:95–112.
Ragazzini P, Gamberi G, Pazzaglia L et al. Amplification of CDK4, MDM2, SAS and GLI genes in leiomyosarcoma, alveolar and embryonal rhabdomyosarcoma. Histol Histopathol 2004;19:401–411.
Ray-Coquard I, Blay JY, Italiano A et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 2012;13:1133–1140.
Italiano A, Bianchini L, Gjernes E et al. Clinical and biological significance of CDK4 amplification in well-differentiated and dedifferentiated liposarcomas. Clin Cancer Res 2009;15:5696–5703.
Wang X, Asmann YW, Erickson-Johnson MR et al. High-resolution genomic mapping reveals consistent amplification of the fibroblast growth factor receptor substrate 2 gene in welldifferentiated and dedifferentiated liposarcoma. Genes Chromosomes Cancer 2011;50:849–858.
Cleynen I, Van de Ven WJ . The HMGA proteins: a myriad of functions (Review). Int J Oncol 2008;32:289–305.
Brown VD, Phillips RA, Gallie BL . Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol Cell Biol 1999;19:3246–3256.
Chibon F, Mariani O, Derre J et al. ASK1 (MAP3K5) as a potential therapeutic target in malignant fibrous histiocytomas with 12q14-q15 and 6q23 amplifications. Genes Chromosomes Cancer 2004;40:32–37.
Mariani O, Brennetot C, Coindre JM et al. JUN oncogene amplification and overexpression block adipocytic differentiation in. Cancer Cell 2007;11:361–374.
Snyder EL, Sandstrom DJ, Law K et al. c-Jun amplification and overexpression are oncogenic in liposarcoma but not always sufficient to inhibit the adipocytic differentiation programme. J Pathol 2009;218:292–300.
Nord KH, Macchia G, Tayebwa J et al. Integrative genome and transcriptome analyses reveal two distinct types of ring chromosome in soft tissue sarcomas. Hum Mol Genet 2014;23:878–888.
Coindre JM . Histologic grading of adult soft tissue sarcomas. Verh Dtsch Ges Pathol 1998;82:59–63.
Enneking WF, Spanier SS, Goodman MA . A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res 1980;153:106–120.
Bianchini L, Saada E, Gjernes E et al. Let-7 microRNA and HMGA2 levels of expression are not inversely linked in adipocytic tumors: analysis of 56 lipomas and liposarcomas with molecular cytogenetic data. Genes Chromosomes Cancer 2011;50:442–455.
Binh MB, Sastre-Garau X, Guillou L et al. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol 2005;29:1340–1347.
Dei Tos AP, Doglioni C, Piccinin S et al. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumors. J Pathol 2000;190:531–536.
Marino-Enriquez A, Fletcher CD, Dal Cin P et al. Dedifferentiated liposarcoma with 'homologous' lipoblastic (pleomorphic liposarcoma-like) differentiation: clinicopathologic and molecular analysis of a series suggesting revised diagnostic criteria. Am J Surg Pathol 2010;34:1122–1131.
Dreux N, Marty M, Chibon F et al. Value and limitation of immunohistochemical expression of HMGA2 in mesenchymal tumors: about a series of 1052 cases. Mod Pathol 2010;23:1657–1666.
Meyer B, Loeschke S, Schultze A et al. HMGA2 overexpression in non-small cell lung cancer. Mol Carcinogen 2007;46:503–511.
Miyazawa J, Mitoro A, Kawashiri S et al. Expression of mesenchyme-specific gene HMGA2 in squamous cell carcinomas of the oral cavity. Cancer Res 2004;64:2024–2029.
Califano D, Pignata S, Losito NS et al. High HMGA2 expression and high body mass index negatively affect the prognosis of patients with ovarian cancer. J Cell Physiol 2014;229:53–59.
Zhou X, Benson KF, Ashar HR et al. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 1995;376:771–774.
Battista S, Fidanza V, Fedele M et al. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res 1999;59:4793–4797.
Hirning-Folz U, Wilda M, Rippe V et al. The expression pattern of the Hmgic gene during development. Genes Chromosomes Cancer 1998;23:350–357.
Franco R, Esposito F, Fedele M et al. Detection of high-mobility group proteins A1 and A2 represents a valid diagnostic marker in post-pubertal testicular germ cell tumors. J Pathol 2008;214:58–64.
Akai T, Ueda Y, Sasagawa Y et al. High mobility group I-C protein in astrocytoma and glioblastoma. Pathol Res Pract 2004;200:619–624.
Fedele M, Berlingieri MT, Scala S et al. Truncated and chimeric HMGI-C genes induce neoplastic transformation of NIH3T3 murine fibroblasts. Oncogene 1998;17:413–418.
Taylor BS, DeCarolis PL, Angeles CV et al. Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov 2011;1:587–597.
Sun T, Fu M, Bookout AL et al. MicroRNA let-7 regulates 3T3-L1 adipogenesis. Mol Endocrinol 2009;23:925–931.
Narita M, Krizhanovsky V, Nunez S et al. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006;126:503–514.
Horvai AE, DeVries S, Roy R et al. Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma. Mod Pathol 2009;22:1477–1488.
Chibon F, Mariani O, Derre J et al. A subgroup of malignant fibrous histiocytomas is associated with genetic changes similar to those of well-differentiated liposarcomas. Cancer Genet Cytogenet 2002;139:24–29.
Toulmonde M, Le Cesne A, Piperno-Neumann S et al. Aplidin in patients with advanced dedifferentiated liposarcomas: a French Sarcoma Group Single-Arm Phase II study. Ann Oncol 2015;26:1465–1470.
Acknowledgements
We thank Dr Agnès Leroux (Centre Alexis Vautrin, Nancy, France), Dr Françoise Collin (Centre Georges Francois Leclerc, Dijon, France), Dr Florence Dupré (Princesse Grace Hospital Center, Monaco, France), Dr Hélène Perrochia (University Hospital of Montpellier, Montpellier, France), Dr Philippe Rochaix (Institut Claudius Régaud, Toulouse, France), Dr Nicolas Weinbreck (Medipath, Fréjus, France), and Dr Philippe Terrier (Institut Gustave Roussy, Villejuif, France) for providing some of the tumor samples, and Dr Olivier Collard (Institut de Cancérologie de la Loire, Saint-Priest-en-Jarez, France), Pr François Bertucci (Institut Paoli-Calmettes, Marseille, France), Pr Antoine Thyss (Centre Antoine-Lacassagne, Nice, France), and Pr Jean-Yves Blay (Centre Léon-Bérard, Lyon, France) for providing clinical data. We also thank Frédérique Keslair and Roger Grattery for helpful assistance.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Saâda-Bouzid, E., Burel-Vandenbos, F., Ranchère-Vince, D. et al. Prognostic value of HMGA2, CDK4, and JUN amplification in well-differentiated and dedifferentiated liposarcomas. Mod Pathol 28, 1404–1414 (2015). https://doi.org/10.1038/modpathol.2015.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2015.96
This article is cited by
-
Molecular and clinicopathological analysis revealed an immuno-checkpoint inhibitor as a potential therapeutic target in a subset of high-grade myxofibrosarcoma
Virchows Archiv (2022)
-
The clinical significance of perioperative inflammatory index as a prognostic factor for patients with retroperitoneal soft tissue sarcoma
International Journal of Clinical Oncology (2022)
-
Update on genomic and molecular landscapes of well-differentiated liposarcoma and dedifferentiated liposarcoma
Molecular Biology Reports (2021)
-
Primary hepatic carcinoma with inhibin positivity in a young male patient: a rare tumor previously only reported in females—case report and review of literature
Virchows Archiv (2021)
-
Establishment and characterization of a novel cell line, NCC-DDLPS2-C1, derived from a patient with dedifferentiated liposarcoma
Human Cell (2021)
