
Abstract
Lipoblastomas are benign neoplasms of embryonal white fat that typically present in the first 3 years of life and show a lobular arrangement of maturing adipocytes with variable degrees of myxoid change. We systematically studied the clinicopathologic and genetic features of lipoblastomas arising in older children and adults. Cases with a diagnosis of lipoblastoma or maturing lipoblastoma in patients >3 years of age were retrieved from our archives. Immunostaining for CD34 and desmin and molecular studies (FISH, RNA sequencing) were performed. Twenty-two cases (8F; 14M) were identified in patients ranging from 4 to 44 years of age (median 10 years). Sites included extremity (n = 15), head and neck (n = 4), and trunk (n = 3) with tumor sizes varying from 1.6 to 17.5 cm (median 5). Only three tumors had histologic features of “conventional” lipoblastoma. The majority of tumors (n = 14) were composed of variably sized lobules of mature adipose tissue partitioned by thin fibrous septa (“maturing”). The remaining five cases consisted predominantly of bland spindled to plump ovoid cells embedded in a fibrous stroma, with a vaguely plexiform arrangement of small myxoid and adipocytic nodules (“fibroblastic”). CD34 was diffusely positive in all cases tested (21/21), while desmin immunoreactivity was identified in 12 of 21 cases (diffuse = 7, focal = 5). PLAG1 rearrangements were identified in 13 tumors in the entire cohort (59%), including all 5 fibroblastic tumors. RNA sequencing detected eight PLAG1 fusion partners, of which two were known (CHCHD7 and COL3A1) and six were novel (SRSF3, HNRNPC, PCMTD1, YWHAZ, CTDSP2, and PPP2R2A). Twelve cases had follow-up (1–107 months; median 21 months), and no recurrences were reported. Lipoblastomas may occur in older children and adults and may be difficult to recognize due to their predominantly adipocytic or fibrous appearance. Awareness that lipoblastomas may occur in older patients, careful evaluation for foci showing more typical morphologic features, ancillary immunohistochemistry for CD34 and desmin, and molecular genetic studies to identify PLAG1 rearrangements are the keys to recognizing these tumors.
Similar content being viewed by others
Introduction
Lipoblastomas, benign neoplasms of embryonal white fat characterized by PLAG1 rearrangements, overwhelming affect children 3 years of age or younger [1,2,3,4,5,6,7]. When these tumors present in this characteristic age group they are readily recognized morphologically, consisting of a distinctly lobular, variably myxoid, highly vascular proliferation of bland, round to spindled cells and maturing adipocytes, partitioned by fibrous septa. However, as the patient ages, the adipocytic population is thought to “mature”, resulting in a well-differentiated fibro-fatty proliferation devoid of myxoid stroma, resembling an unusually septated lipoma. Consequently, lipoblastomas in older children and adults are often difficult to diagnose. Immunohistochemically, lipoblastomas are typically CD34-positive and often show desmin expression in spindled cells, a useful diagnostic clue [8, 9]. Prompted by a recent case of lipoblastoma arising in the back of a 10-year-old male, which was composed predominantly of fibrous stroma and harbored a CTDSP2-PLAG1 fusion, we sought to explore the pathologic and genetic spectrum of these tumors in older children and adults.
Materials and methods
The Institutional Review Boards of all participating institutions approved this study. The consultation and institutional anatomic pathology archives of Mayo Clinic were searched for cases of lipoblastoma arising in patients older than 3 years of age. All available slides were reviewed, and the cases were classified into three morphologic subtypes: conventional, maturing and fibroblastic. One additional case with fibroblastic morphology from St. Jude Children’s Research Hospital was included. Clinical and follow-up data were collected from institutional medical records and the medical records of the submitting pathologists.
Immunohistochemistry
Immunohistochemistry was performed on deparaffinized, rehydrated sections obtained from a representative formalin-fixed, paraffin-embedded (FFPE) block from each case using antibody-specific epitope retrieval techniques with the Dako Envision (Dako, Carpinteria, CA, USA) automated system for detection of the following primary antigens: CD34 (Leica, QBEnd/10, 1:100) and desmin (Leica, DE-R-11, 1:50–1:100).
Fluorescence in situ hybridization
Interphase fluorescence in situ hybridization (FISH) for PLAG1 rearrangement was performed on FFPE tumor tissues of a subset of cases using a dual-color break-apart probe set derived from bacterial artificial chromosome (BAC) clones RP11-22E14 and RP11-1130K23 as previously described [10]. In brief, RP11-22E14 mapped to the downstream (centromeric) flanking region of PLAG1 is labeled with AF 488 FITC (green), and RP11-1130K23 targeted 5′ end and the upstream sequence (telomeric) of PLAG1 is labeled with AF555 Rhodamine (red). Prior to testing the samples accrued in this study, the FISH assay was validated in CLIA-certified laboratory at St. Jude Children’s Research Hospital, and the cutoff to define PLAG1 rearrangement was settled at 17% empirically. For each sample, 200 nuclei were analyzed, and FISH signals were scored by two experienced cytogenetic technologists independently.
RNA extraction and next-generation sequencing
FFPE tumor tissues were subjected to RNA extraction using Zymo quick-RNA FFPE kit (Zymo Research, CA). Random priming was used to synthesize cDNAs from the RNA samples extracted. For all cases except #18 and #19, an anchored multiplex PCR assay based on the Rapid Amplification of cDNA Ends (RACE) strategy followed by next-generation sequencing (Archer FusionPlex, ArcherDX, USA) was employed to identify gene fusions. We used Archer FusionPlex Sarcoma panel, which is designed to simultaneously screen for fusions of 26 genes associated with soft tissue tumors. The library of multiplex RACE products was sequenced on MiSeq (Illumina) and sequencing data were analyzed by Archer software (version 6.2). In addition to fusion detection, data from these 20 cases went through the RNA expression pipeline available with Archer v6.2 software, and PLAG1 RNA expression was analyzed. In brief, median-normalized RNA-seq result of each case was exported in TSV format from the Archer Analysis software platform. Data for each sample were next filtered for only those amplicon locations corresponding to PLAG1. The median of the normalized expression values for PLAG1 amplicons of each sample was calculated. The RNA-seq sample extracted from #18 and #19 were subjected to whole transcriptome sequencing (RNA-seq) analysis following the procedure and pipeline that have been described previously [11].
RT-PCR and Sanger Sequencing
RNA samples extracted were subjected to reverse transcription using SuperScript™ II Reverse Transcriptase (ThermoFisher Scientific, USA). Primer sequences used for PCR to verify fusions in selected cases are as follows: CHCHD7 forward primer 5′-AAGTTGGGATGCGCGCTAC-3′; COL3A1 forward primer 5′-CTCATGTCTGATATTTAGACATGATGAGC-3′; HNRNPC forward primer 5′-CAGCAGCAGTCGGCTTCTCTA-3′; SRSF3 forward primer 5′-GTGAGAGAGTTGGTTGGTGTTGG-3′; and two reverse primers targeting PLAG1 exons 2 and 3, respectively. PLAG1 exon 2 reverse primer 5′-CCAAATACGGCCAAGGCAG-3′ and PLAG1 exon 3 reverse primer 5′-GAAGAGAGTGGAATCCAATCCTTC-3′. RT-PCR products were evaluated by running agarose gel along with a 100-bp ladder DNA marker, and for SRSF3-PLAG1 and HNRNPC-PLAG1 fusions, bands with expected size were purified from the gel and sequenced (Sanger Sequencing).
Results
Twenty-four cases of lipoblastomas from 24 patients arising in patients >3 years of age were identified in our institutional and consultation archives out of a total of 74 total lipoblastoma cases (32%). Two cases were excluded due to insufficient quality and quantity of the RNA extracted leaving 22 cases (8 females and 14 males) occurring in patients ranging in age from 4 to 44 years (median 10 years) (Table 1). The median ages of those with maturing, fibroblastic, and conventional morphologies were 10.5 years (range 4–44 years), 10 years (range 4–16) and 13 years (range 7–41), respectively. Sites included extremity (n = 15; 68%), head and neck (n = 4; 18%), and trunk (n = 3; 14%), and tumor sizes varied from 1.6 to 11.9 cm (median 5 cm).
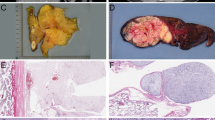
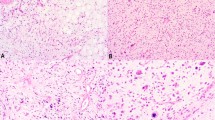
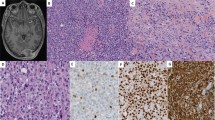
The number of sections available per case ranged from 1 to 15 (median 3.5 sections). The majority of tumors (n = 14; 64%) were composed of solely of variably sized lobules of mature adipose tissue partitioned by thin fibrous septa without myxoid stroma (Fig. 1). These tumors were classified as “maturing” lipoblastoma. A smaller subset (n = 3; 14%) showed features of conventional lipoblastoma, with the characteristic lobules of adipocytes in various stages of maturation in a myxoid stroma with a fine capillary vascular network (Fig. 2). The remaining five cases (23%) consisted predominantly of bland spindled to plump ovoid cells embedded in a fibrous stroma with a vaguely plexiform arrangement of small myxoid and adipocytic nodules. These tumors were classified as “fibroblastic” lipoblastoma. (Fig. 3). Three fibroblastic lipoblastomas showed a predominance of fibrous stroma, largely obscuring the adipocytic nature of the tumor (Fig. 4). The cells within the myxoid and fibroblastic areas displayed thin tapered nuclei without cytologic atypia or significant mitotic activity (<1 mitotic figure/10 high power fields). CD34 was diffusely positive in this population in all cases tested (21/21), while desmin immunoreactivity was identified in 12 of 21 cases (diffuse = 7, focal = 5) (Fig. 5).

Lipoblastomas with maturing morphology were composed predominantly of mature adipose tissue with intervening fibrous setpa (A) composed of bland spindled cells (B).

Tumors with conventional morphology harbored myxoid nodules with adipocytes in varying stages of maturation and a prominent thin-walled capillary network.

Fibroblastic lipoblastomas (A) harbored a plexiform arrangement of myxoid (B) and adipocytic (C) nodules set in a densely hyalinized background.

Case 15 with fibroblastic morphology exhibited large amounts of fibrous stroma without myxoid or adipocytic foci (A). The spindle cell population was bland without significant atypia or mitotic activity (B, C).

Lipoblastomas showed expression of CD34 (A) and desmin (B).
PLAG1 (8q12.1) rearrangements were detected in 13 of 22 cases (59%) by RNA sequencing and/or FISH. RNA sequencing identified eight fusion partner genes across the cohort, of which two were known (COL3A1 (2q32.2) and CHCHD7 (8q12.1)) and six were novel. The novel partner genes, PCMTD1 (8q11.23), YWHAZ (8q22.3), CTDSP2 (12q14.1), and PPP2R2A (8q21.2), were found in fibroblastic tumors, whereas SRSF3 (6p21.3-p21.1) and HNRNPC (14q11.2) were identified in maturing tumors. The most common fusion seen in seven lipoblastomas was CHCHD7-PLAG1, resulting from a cryptic intrachromosomal rearrangement at 8q12. In five lipoblastomas (four maturing and one conventional), CHCHD7-PLAG1 was the sole fusion, whereas in two lipoblastomas (maturing), it was identified as the second PLAG1 fusion (Table 1). All PLAG1 fusions (except PPP2R2A-PLAG1) identified in this study involved the 5′ untranslated regions of both PLAG1 and a fusion partner gene. In PPP2R2A-PLAG1, PPP2R2A (NM_002717) exon 2 fused to PLAG1 (NM_002655) exon 3. Consequently, all fusions resulted in having the entire PLAG1 coding sequence, which begins in exon 4, placed under the transcriptional control of promoter regions of fusion partner genes (promoter swap).
A comparison of PLAG1 RNA expression between fusion-positive and fusion-negative groups defined by the targeted RNA-seq analysis is presented in Fig. 6, and increased expression of PLAG1 due to promoter swap was observed in all cases except #3, #7, (borderline) and #14. The fusions transcripts revealed by targeted RNA-seq analysis in these three cases were successfully verified by RT-PCR (Supplementary Fig. 1). The relatively low expression of PLAG1 in the three cases may be due to the low tumor content in the sample studied as these represented the maturing variant. Considering that the SRSF3 is a novel partner to PLAG1 fusions, the RT-PCR product of case #14 was sequenced by Sanger Sequencing, and the same fusion transcript was detected (Fig. 7). In case 5, two PLAG1 fusions were identified, one is a known fusion (CHCHD7-PLAG1) and the other one showed a novel 5′ partner (HNRNPC-PLAG1). Although high expression of PLAG1 was observed in this case, we performed RT-PCR followed by Sanger Sequencing to verify the validity of the novel HNRNPC-PLAG1. Fig. 7 shows the fusion transcript detected by RT-PCR and Sanger Sequencing. Case #18 was analyzed by Whole Transcriptome Sequencing and a PPP2R2A-PLAG1 fusion was detected; high expression of PLAG1 was observed as well.

Comparison of PLAG1 RNA expression between fusion-positive and fusion-negative groups defined by the targeted RNA-seq analysis. The median of the normalized expression values for PLAG1 amplicons of each sample is presented.

RT-PCR and Sanger Sequencing confirmation of SRSF3-PLAG1 fusion in case #14 and HNRNPC-PLAG1 fusion in case #5. Upper panel: SRSF3-PLAG1 fusion transcript was verified by RT-PCR using SRSF3 forward and PLAG1 exon 2 reverse primers. Sequencing of the band at 128 bp confirmed the fusion. Lower panel: HNRNPC-PLAG1 fusion transcript was verified by RT-PCR using HNRNPC forward and PLAG1 exon 3 reverse primers. Sequencing of the band at 115 bp confirmed the fusion.
Clinical follow-up was available for 12 cases (1–107 months; median 21 months); no recurrences were reported.
Discussion
Lipoblastomas generally occur in children 3 years of age or younger. Approximately one-third of patients with lipoblastomas in our archives presented over the age of three; this relatively high percentage likely reflects referral bias. Although the literature contains reports of lipoblastomas arising in older children and adults, systematic reviews of the histology and genetics of these tumors beyond childhood are lacking [1,2,3,4,5, 7, 8, 12, 13]. Furthermore, lipoblastomas are thought to mature over time, eventually resembling ordinary lipomas or “fibrolipomas,” although evidence for this is quite scant [4, 8, 14, 15].
The results of our study suggest that lipoblastomas in older children and adults tend to follow the same anatomic site distribution as their younger counterparts, most often occurring in the extremities with a median size of 5 cm and a male predominance [1, 2, 4, 7, 16]. Although the recurrence rates in young children reach 46% in some studies, no cases in our series recurred [7]. This discrepancy may be secondary to limited follow-up; alternatively, complete excision may be more feasible in older children and adults resulting in lower recurrence rates, or these lesions might simply have a lesser potential for continued growth in older children. One other explanation might be that none of our cases had a diffuse growth pattern characteristic of lipoblastomatosis, which has a greater recurrence risk than lipoblastoma [17].
Nearly two-thirds of lipoblastomas in our cohort were composed chiefly of a lobular arrangement of mature adipose tissue with thin intervening fibrous septa, possibly lending support to prior work suggesting that these tumors mature over time [4, 14, 18, 19]. A smaller percentage of cases harbored features of conventional lipoblastoma including myxoid lobules of adipocytes and more primitive-appearing mesenchymal cells admixed with a delicate capillary network. While we initially suspected that this conventional morphology might correlate with younger age, the median age of these patients was in fact older than in those whose tumors showed maturing histology (13 years vs. 10.5 years). The remaining five cases in our series were predominantly fibroblastic, composed of small adipocytic and mxyoid nodules embedded in a dense collagenous background, with a vaguely plexiform low-power appearance. While there are occasional reports of this unusual variant, lipoblastomas with this morphology are likely rare, under recognized or both [20, 21]. The spindle cell component of the fibroblastic variant, similar to those with conventional and maturing histologies, co-expressed CD34 and desmin [8, 9, 22].
The genetic hallmark of lipoblastoma is chromosomal translocations resulting in PLAG1 rearrangement and its subsequent upregulation as a result of promoter swapping [23,24,25,26,27,28,29]. To date, various fusion partners of PLAG1 have been reported including COL1A2 (7q21.3), HAS2 (8q24.13), RAD51B (14q24.1), COL3A1 (2q32.2), RAB2A (8q12.1-q12.2), and BOC (3q13.2) [28, 30,31,32,33,34,35]. Approximately 60% of cases in our group showed PLAG1 rearrangement, similar to the rates reported in the literature [36, 37]. Although the PLAG1 negative cases tended to occur in older patients for each subtype, these cases were reviewed by two soft tissue pathologists and felt to represent lipoblastomas. CHCHD7 represented the most common fusion partner in our series. CHCHD7-PLAG1 fusions, also resulting in promotor swap and activation of PLAG1, have been reported as recurrent events in pleomorphic adenomas of the salivary gland, but not described previously in lipoblastoma [38,39,40,41].
We identified six novel fusions including HNRNPC-PLAG1, SRSF3-PLAG1, PCMTD1-PLAG1, YWHAZ-PLAG1, CTDSP2-PLAG1, and PPP2R2A-PLAG1. These novel fusion partners revealed in this study may help to elucidate molecular genetic alterations of some previously reported cytogenetic abnormalities in lipoblastomas. For instance, McVay et al. reported a case with an inv (p21.3q11.23) [42]. As PPP2R2A is located at 8p21.3, the inv [8] in this case may produce a PPP2R2A-PLAG1 fusion. Another example is a case with t(6;8)(p21;q12) reported by Coffin et al. [7]. This chromosome translocation could result in a SRSF3-PLAG1 fusion as SRSF3 is located at 6p21.31-p21.2. The novel fusions all were identified in the non-conventional lipoblastomas in our cohort (four fibroblastic and two maturing).
In three cases, PLAG1 rearrangements identified by RNA sequencing were undetected by FISH studies. CHCHD7 and PLAG1 are neighbor genes only a few hundred base pairs apart at 8q12.1 and are transcribed in opposite directions. The FISH probes derived from BAC clones are not capable to detect this extremely cryptic fusion. Interestingly, the FISH assay failed to detect the other two fusions, CTDSP2-PLAG1 and PPP2R2A-PLAG1. CTDSP2 and PPP2R2A are located at 12q13 and 8p21, respectively. The discrepancy between RNA sequencing analysis and the current FISH study may suggest that cryptic chromosomal rearrangements are common in PLAG1 fusions and these fusions may be overlooked if high-resolution sequencing is not performed.
The differential diagnosis of lipoblastomas in older children and adults depends on the morphologic subtype. “Maturing” lipoblastomas are most likely to mimic atypical lipomatous tumor/well-differentiated liposarcomas. Careful examination of the fibrous septa for the atypical hyperchromatic stromal cells present in the latter in conjunction with FISH studies for MDM2 amplification should aide in appropriate classification. Outside of infancy, the conventional variant of lipoblastoma must be differentiated from myxoid liposarcoma, the most common form of pediatric liposarcoma [43]. Although lipoblastomas typically show more prominent lobulation, display maturation toward the periphery of lobules, and lack “myxoid pools”, molecular genetic studies for DDIT3 and/or PLAG1 may be necessary for definitive classification. The fibroblastic variant may simulate other entities with a plexiform growth pattern such as cellular neurothekeoma and plexiform fibrohistiocytic tumor. The former may show myxoid change but lacks the adipocytic component present in lipoblastoma, while plexiform fibrohistiocytic tumors show giant cell and histiocyte-rich nodules without myxoid stroma. Tumors with a predominance of fibroblastic stroma may also raise the possibility of low-grade fibromyxoid sarcoma, a tumor which can be excluded by MUC4 immunohistochemistry. Finally, fibrous hamartoma of infancy usually occurs within the first 2 years of life and shows a distinct triphasic appearance of bland fibroblasts/myofibroblasts, primitive-appearing spindle cells, and mature fat.
In summary, we have reported the largest series of lipoblastomas arising in patients older than 3 years of age, with characterization of their immunohistochemical and genetic features, including seven novel fusions not previously identified in lipoblastomas. As lipoblastomas in this population may harbor fibroblastic areas, careful examination for adipocytic or myxoid stroma in conjunction with CD34/desmin immunostains and PLAG1 molecular studies should aid in appropriate diagnosis.
References
Chung EB, Enzinger FM. Benign lipoblastomatosis. An analysis of 35 cases. Cancer. 1973;32:482–92.
Mentzel T, Calonje E, Fletcher CD. Lipoblastoma and lipoblastomatosis: a clinicopathological study of 14 cases. Histopathology. 1993;23:527–33.
Coffin CM, Williams RA. Congenital lipoblastoma of the hand. Pediatr Pathol. 1992;12:857–64.
Collins MH, Chatten J. Lipoblastoma/lipoblastomatosis: a clinicopathologic study of 25 tumors. Am J Surg Pathol. 1997;21:1131–7.
Hicks J, Dilley A, Patel D, Barrish J, Zhu SH, Brandt M. Lipoblastoma and lipoblastomatosis in infancy and childhood: histopathologic, ultrastructural, and cytogenetic features. Ultrastruct Pathol. 2001;25:321–33.
Jung SM, Chang PY, Luo CC, Huang CS, Lai JY, Hsueh C. Lipoblastoma/lipoblastomatosis: a clinicopathologic study of 16 cases in Taiwan. Pediatr Surg Int. 2005;21:809–12.
Coffin CM, Lowichik A, Putnam A. Lipoblastoma (LPB): a clinicopathologic and immunohistochemical analysis of 59 cases. Am J Surg Pathol. 2009;33:1705–12.
Abdul-Ghafar J, Ahmad Z, Tariq MU, Kayani N, Uddin N. Lipoblastoma: a clinicopathologic review of 23 cases from a major tertiary care center plus detailed review of literature. BMC Res Notes. 2018;11:42.
Kubota F, Matsuyama A, Shibuya R, Nakamoto M, Hisaoka M. Desmin-positivity in spindle cells: under-recognized immunophenotype of lipoblastoma. Pathol Int. 2013;63:353–7.
Bahrami A, Dalton JD, Krane JF, Fletcher CD. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer. 2012;51:140–8.
Rusch M, Nakitandwe J, Shurtleff S, Newman S, Zhang Z, Edmonson MN, et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat Commun. 2018;9:3962.
Sciot R, De Wever I, Debiec-Rychter M. Lipoblastoma in a 23-year-old male: distinction from atypical lipomatous tumor using cytogenetic and fluorescence in-situ hybridization analysis. Virchows Arch. 2003;442:468–71.
de Saint Aubain Somerhausen N, Coindre JM, Debiec-Rychter M, Delplace J, Sciot R. Lipoblastoma in adolescents and young adults: report of six cases with FISH analysis. Histopathology. 2008;52:294–8.
Duhaime AC, Chatten J, Schut L, Rorke L. Cervical lipoblastomatosis with intraspinal extension and transformation to mature fat in a child. Childs Nerv Syst. 1987;3:304–6.
Willen H, Akerman M, Dal Cin P, De Wever I, Fletcher CD, Mandahl N, et al. Comparison of chromosomal patterns with clinical features in 165 lipomas: a report of the CHAMP study group. Cancer Genet Cytogenet. 1998;102:46–9.
Han JW, Kim H, Youn JK, Oh C, Jung SE, Park KW, et al. Analysis of clinical features of lipoblastoma in children. Pediatr Hematol Oncol. 2017;34:212–20.
Dao D, Najor AJ, Sun PY, Farrokhyar F, Moir CR, Ishitani MB. Follow-up outcomes of pediatric patients who underwent surgical resection for lipoblastomas or lipoblastomatosis: a single-institution experience with a systematic review and meta-analysis. Pediatr Surg Int. 2020;36:341–55.
Van Meurs DP. The transformation of an embryonic lipoma to a common lipoma. Br J Surg. 1947;34:282–4.
Miller GG, Yanchar NL, Magee JF, Blair GK. Tumor karyotype differentiates lipoblastoma from liposarcoma. J Pediatr Surg. 1997;32:1771–2.
Craver RD, Henrich S, Kao YS. Fibrous lipoblastoma with 8q11.2 abnormality. Cancer Genet Cytogenet. 2006;171:112–4.
Stringel G, Shandling B, Mancer K, Ein SH. Lipoblastoma in infants and children. J Pediatr Surg. 1982;17:277–80.
Miyano G, Hayashi T, Arakawa A, Goto S, Lane GJ, Okazaki T, et al. Giant omental lipoblastoma and CD56 expression. Afr J Paediatr Surg. 2013;10:32–4.
Sandberg AA, Gibas Z, Saren E, Li FP, Limon J, Tebbi CK. Chromosome abnormalities in two benign adipose tumors. Cancer Genet Cytogenet. 1986;22:55–61.
Ohjimi Y, Iwasaki H, Kaneko Y, Ishiguro M, Ohgami A, Kikuchi M. A case of lipoblastoma with t(3;8)(q12;q11.2). Cancer Genet Cytogenet. 1992;62:103–5.
Dal Cin P, Sciot R, De Wever I, Van Damme B, Van den Berghe H. New discriminative chromosomal marker in adipose tissue tumors. The chromosome 8q11-q13 region in lipoblastoma. Cancer Genet Cytogenet. 1994;78:232–5.
Fletcher JA, Kozakewich HP, Schoenberg ML, Morton CC. Cytogenetic findings in pediatric adipose tumors: consistent rearrangement of chromosome 8 in lipoblastoma. Genes Chromosomes Cancer. 1993;6:24–9.
Astrom A, D’Amore ES, Sainati L, Panarello C, Morerio C, Mark J, et al. Evidence of involvement of the PLAG1 gene in lipoblastomas. Int J Oncol. 2000;16:1107–10.
Hibbard MK, Kozakewich HP, Dal Cin P, Sciot R, Tan X, Xiao S, et al. PLAG1 fusion oncogenes in lipoblastoma. Cancer Res. 2000;60:4869–72.
Gisselsson D, Hibbard MK, Dal Cin P, Sciot R, Hsi BL, Kozakewich HP, et al. PLAG1 alterations in lipoblastoma: involvement in varied mesenchymal cell types and evidence for alternative oncogenic mechanisms. Am J Pathol. 2001;159:955–62.
Morerio C, Rapella A, Rosanda C, Tassano E, Gambini C, Romagnoli G, et al. PLAG1-HAS2 fusion in lipoblastoma with masked 8q intrachromosomal rearrangement. Cancer Genet Cytogenet. 2005;156:183–4.
Brinkman AS, Maxfield B, Gill K, Patel NJ, Gosain A. A novel t(3;8)(p13;q21.1) translocation in a case of lipoblastoma. Pediatr Surg Int. 2012;28:737–40.
Deen M, Ebrahim S, Schloff D, Mohamed AN. A novel PLAG1-RAD51L1 gene fusion resulting from a t(8;14)(q12;q24) in a case of lipoblastoma. Cancer Genet. 2013;206:233–7.
Yoshida H, Miyachi M, Ouchi K, Kuwahara Y, Tsuchiya K, Iehara T, et al. Identification of COL3A1 and RAB2A as novel translocation partner genes of PLAG1 in lipoblastoma. Genes Chromosomes Cancer. 2014;53:606–11.
Warren M, Turpin BK, Mark M, Smolarek TA, Li X. Undifferentiated myxoid lipoblastoma with PLAG1-HAS2 fusion in an infant; morphologically mimicking primitive myxoid mesenchymal tumor of infancy (PMMTI)-diagnostic importance of cytogenetic and molecular testing and literature review. Cancer Genet. 2016;209:21–9.
Nitta Y, Miyachi M, Tomida A, Sugimoto Y, Nakagawa N, Yoshida H, et al. Identification of a novel BOC-PLAG1 fusion gene in a case of lipoblastoma. Biochem Biophys Res Commun. 2019;512:49–52.
Dadone B, Refae S, Lemarie-Delaunay C, Bianchini L, Pedeutour F. Molecular cytogenetics of pediatric adipocytic tumors. Cancer Genet. 2015;208:469–81.
Fallon SC, Brandt ML, Rodriguez JR, Vasudevan SA, Lopez ME, Hicks MJ, et al. Cytogenetic analysis in the diagnosis and management of lipoblastomas: results from a single institution. J Surg Res. 2013;184:341–6.
Asp J, Persson F, Kost-Alimova M, Stenman G. CHCHD7-PLAG1 and TCEA1-PLAG1 gene fusions resulting from cryptic, intrachromosomal 8q rearrangements in pleomorphic salivary gland adenomas. Genes Chromosomes Cancer. 2006;45:820–8.
Matsuyama A, Hisaoka M, Hashimoto H. PLAG1 expression in cutaneous mixed tumors: an immunohistochemical and molecular genetic study. Virchows Arch. 2011;459:539–45.
Matsuyama A, Hisaoka M, Nagao Y, Hashimoto H. Aberrant PLAG1 expression in pleomorphic adenomas of the salivary gland: a molecular genetic and immunohistochemical study. Virchows Arch. 2011;458:583–92.
Asahina M, Saito T, Hayashi T, Fukumura Y, Mitani K, Yao T. Clinicopathological effect of PLAG1 fusion genes in pleomorphic adenoma and carcinoma ex pleomorphic adenoma with special emphasis on histological features. Histopathology. 2019;74:514–25.
McVay MR, Keller JE, Wagner CW, Jackson RJ, Smith SD. Surgical management of lipoblastoma. J Pediatr Surg. 2006;41:1067–71.
Alaggio R, Coffin CM, Weiss SW, Bridge JA, Issakov J, Oliveira AM, et al. Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol. 2009;33:645–58.
Acknowledgements
This work was supported in part by ALSAC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Fritchie, K., Wang, L., Yin, Z. et al. Lipoblastomas presenting in older children and adults: analysis of 22 cases with identification of novel PLAG1 fusion partners. Mod Pathol 34, 584–591 (2021). https://doi.org/10.1038/s41379-020-00696-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00696-4
This article is cited by
-
Pediatric fibromyxoid brachial plexus tumor with YWHAZ::PLAG1 gene fusion: a case report
Child's Nervous System (2024)
-
Chromothripsis in lipoblastoma: second reported case with complex PLAG1 rearrangement
Molecular Cytogenetics (2023)
-
Lipoblastoma Arising in the Head and Neck: A Clinicopathologic Analysis of 20 Cases
Head and Neck Pathology (2023)
-
Expanding the spectrum of PLAG1-rearranged lipoblastomas arising in patients over 45, with identification of novel fusion partners
Modern Pathology (2022)
