
Abstract
Primary myelofibrosis is a type of chronic myeloproliferative neoplasm characterized by progressive bone marrow failure with worsening cytopenia and in a subset of patients, progression to acute leukemia. Published data in patients with myelodysplastic syndromes have shown that the development of monocytosis in the course of myelodysplastic syndromes is associated with a poor prognosis. A similar occurrence has been only sporadically reported in patients with primary myelofibrosis. Over a period of four years we identified 10 out of 237 cases of primary myelofibrosis who developed persistent absolute monocytosis (>1 × 109/l) during the course of disease (5 men and 5 women; median age/range: 68 years/52–82). Monocytosis developed at a median interval of 42 months from diagnosis (range: 1–180) and persisted for a median period of 23 months (range: 2–57). Five patients died after developing monocytosis (range: 20–188 months) and two experienced worsening disease and became transfusion dependent. Monocytosis was associated with increased white blood cells, decreased hemoglobin, decreased platelet count, and the presence of circulating blasts. In three cases, bone marrow biopsies after the onset of monocytosis showed marked myelomonocytic proliferation with morphological shifting from a typical primary myelofibrosis marrow appearance to aspects compatible with an overt ‘secondary’ chronic myelomonocytic leukemia. Before the development of monocytosis, 5 of 10 patients carried the JAK2V617F mutation; five patients showed karyotypic alterations. No change in JAK2 mutational status or cytogenetic evolution were associated with the development of monocytosis. Four of nine patients analyzed showed KRAS mutation in codon 12 or 13 with low allele burden. This is the first study correlating monocytosis developing in primary myelofibrosis patients with bone marrow morphology, laboratory data, molecular analysis and clinical follow-up. Development of monocytosis in patients with established primary myelofibrosis is associated with rapid disease progression and these patients should be considered as a high-risk group associated with short survival.
Similar content being viewed by others
Main
Primary myelofibrosis, one of the classic Phidalelphia chromosome/BCR-ABL1-negative myeloproliferative neoplasms, is characterized in its overt stage by typical marrow changes such as cytoarchitectural disarray and severe marrow fibrosis in association with splenomegaly due to extramedullary hematopoiesis (also known as myeloid metaplasia).1 Although its prognosis is highly variable, primary myelofibrosis has the worst outcome among the Ph-myeloproliferative neoplasms with a median survival of 5 years (range: 1–10 years). The disease has a strong tendency to progress towards marrow failure and complications related to cytopenia are the most common cause of death. In addition, a significant subset of primary myelofibrosis patients (5–30%) tranform to acute myeloid leukemia. In a proportion of the patients transformation to acute myeloid leukemia is heralded by a usually brief myelodysplastic syndrome-like progression termed accelerated phase, characterized by the presence of an increased number of blasts and often multilineage dysplasia including numerous micromegakaryocytes. In these patients, the bone marrow, at least in part, resembles a high-grade myelodysplastic syndrome (eg, a refractory anemia with excess of blasts).1 In others, transformation to acute leukemia is more abrupt and is not preceded by a myelodysplastic syndrome-like phase.
The prognostic heterogeneity of primary myelofibrosis causes clinical challenges in, eg, deciding the type of therapy and when to start to treat a patient. Over time, multiple parameters have been evaluated to try to predict the speed of progression toward marrow failure or the risk of developing acute leukemia. Clinical parameters that have been associated with worse outcome include older age, anemia, red blood cell transfusion dependency, presence of leukopenia, leukocytosis or thrombocytopenia, increased peripheral blast count, manifestation of systemic symptoms, detection of cytogenetic abnormalities, and low JAK2V617F allele burden.2 However, better prognostic parameters still need to be identified for these patients.
Chronic myelomonocytic leukemia is a de novo myelodysplastic/myelopoliferative neoplasm of variable but usually unfavorable prognosis which is characterized by the present of absolute monocytosis of >1 × 109/l and usually >10% of the white blood count (WBC), sustained for >3 months.3, 4, 5 Although more often a ‘primary neoplasm’, rare cases of secondary chronic myelomonocytic leukemia have been sporadically reported. In a recent study,6 it was noted that a subset of patients with myelodysplastic syndrome developed monocytosis in the course of their disease. In these patients, the peripheral blood monocyte counts reached levels similar to those seen in patients with de novo chronic myelomonocytic leukemia and that this event portended a poor prognosis.6 In addition, monocytosis has also been reported to indicate a worse prognosis in patients with adult T-cell leukemia/lymphoma7 and head and neck cancers.8 In patients with primary myelofibrosis, there is lack of information in relation to the development of monocytosis during the course of the disease. One study, however, has reported that the presence of absolute monocytosis at disease outset had a statistically significant, independent predictive value of inferior survival in younger patients with primary myelofibrosis.9
In this study, we describe a rare subset of primary myelofibrosis patients who developed monocytosis and/or a complete chronic myelomonocytic leukemia-type mophologic shift during the course of their disease. Our study is based on a comprehensive hematopathological evalution which includes clinical and laboratory data, molecular analysis, cytogenetics, and bone marrow morphology results in an attempt to facilitate the identification of a previously unrecognized type of disease progression in primary myelofibrosis associated with adverse prognosis.
Materials and methods
Case Selection
We retrospectively identified in the files of the Department of Pathology and Laboratory Medicine of Weill Cornell Medical College/New York-Presbyterian Hospital in New York City 237 consecutive patients diagnosed with primary myelofibrosis over a period spanning from 2008 to 2011. These bone marrow biopsies were taken during the normal clinical follow-up of primary myelofibrosis patients in our institution: peripheral blood counts were reviewed to identify primary myelofibrosis patients that at some point during their clinical history developed absolute monocytosis (>1 × 109 /l) persistent for >3 months. We then investigated the clinical files of patients with monocytosis and excluded those with any clinical condition known to be possibly associated with reactive monocytosis. Ten patients out of 237 with a previous diagnosis of primary myelofibrosis and who had developed persistent monocytosis were selected. For these patients we looked for the first bone marrow biopsy diagnostic for primary myelofibrosis: this was available in 5 cases. When it was not available, for example, when a case was an outside consult, we carefully studied the reports and their clinical history to make sure the clinal diagnosis was also confirmed histologically both at the time of disease presentation and during the ensuing follow-up, following the criteria published by the WHO in its 2008 monograph.1
Thirteen additional bone marrow biopsies pathologically and clinically classified as primary myelofibrosis in fibrotic phase were also selected from the files of the Department of Pathology of the University of Tubingen, Germany, to be used as a control group in the comparison of the molecular results obtained from the primary myelofibrosis cases that developed monocytosis.
The design of this study was approved by the Institutional Review Board of Weill Cornell Medical College and of University of Tubingen.
Morphological Analysis
All bone marrow biopsies were fixed in Bouin’s solution and acid decalcified. Sections were stained with hematoxylin and eosin for morphological examination; Gomori silver impregnation and trichrome staining were used to evaluate fibrosis.10 Reticulin and trichrome histochemical staining was performed using an automated stainer (Leica Microsystems, Bannockburn, IL, USA) and manufacturer’s specific kits and protocols with only minor modifications. Wright–Giemsa stained bone marrow aspirates and peripheral blood smears corresponding to the bone marrow biopsies were also reviewed. The degree of bone marrow fibrosis was assessed by using the European Consensus Grading System.11 Trichrome stain was used to confirm the presence of collagen fibrosis.
Immunohistochemistry
Bone marrow biopsies, preceding and following the development of monocytosis, were stained for CD14 (Clone 7, Novocastra, Newcastle Upon Tyne, UK) on a Bond–Max autostainer (Vision BioSystems, Hingham, MA, USA) using a polymer-based peroxidase detection system after antigen retrieval in pH 8 EDTA solution at 100 °C for 20 min.
Cytogenetic Analysis
Bone marrow and/or peripheral blood specimens were cultured unstimulated for 24 h in MarrowMax medium (GIBCO; Invitrogen, Carlsbad, CA, USA) and harvested according to standard protocols. G-banded metaphases were analyzed and described according to the International System for Human Cytogenetic Nomenclature (ISCN 2009).12
Fluorescence in situ hybridization was performed on peripheral blood or bone marrow aspirates using the LSI BCR-ABL1 dual color dual fusion probe (Vysis/Abbott Molecular Inc., Des Plaines, IL, USA). Hybridizations were performed following the manufacturer’s instructions with minor modifications. Two hundred interphase nuclei were screened.
Molecular Studies
Molecular analysis for the determination of the JAK2 phenotypes was performed on peripheral blood samples obtained close to the time of bone marrow examination. Cells were lysed and DNA extracted. A multiplex PCR assay based on amplification refractory mutation system technology was performed to amplify the mutant and/or normal alleles plus a DNA quality control with two pairs of allele-specific primers. PCR products were analyzed by agarose gel electrophoresis.13
Detection of activating point mutations of KRAS (codons 12 and 13) and NRAS (codons 12, 13 and 61) was performed on DNA extracted from sections of trephine biopsies and bone marrow and peripheral blood smears, using proteinase K digestion and standard phenol/chloroform extraction. After PCR amplification, purified PCR products were sequenced in both directions using the GenomeLab DTCS-Quick Start Kit (Beckman Coulter, Brea, CA, USA) and analyzed in a GenomeLab GeXP Genetic Analysis System (Beckman Coulter).
In addition, KRAS codons 12 and 13 were investigated for mutations using a clamped probe assay with a wild-type-specific peptide locked nucleic acid (0.02 and 0.04 nM) and melting point analysis of amplification products with the Light Cycler System (Roche Applied Science, Mannheim, Germany) as previously described, with modifications.14 This approach significantly increases the sensitivity of mutation detection. PCR products were re-amplified with nested primers and sequenced for confirmation.
Results
Cases
Two hundred and thirty-seven cases of primary myelofibrosis were identified between 2008 and 2010. After review of the clinical files and having excluded those cases with clinical conditions associated with reactive monocytosis, 10 primary myelofibrosis patients (<5% of all primary myelofibrosis cases) who had developed persistent absolute monocytosis (>1 × 109/l) following their primary myelofibrosis diagnosis were identified. The cohort was composed of five men and five women with a median age of 68 years (mean: 69; range: 52–82). In these ten patients, absolute monocytosis developed after the diagnosis of primary myelofibrosis at a median interval of 42 months (mean: 60.3; range: 1–180). In all the ten cases monocytosis was present at the moment of the follow-up biopsy: its duration was calculated by reviewing all available peripheral blood counts preceding and following the follow-up bone marrow biopsy, to establish the time period during which monocytosis remained sustained with no interruption. The median duration of monocytosis was 23 months (mean: 26.1, range: 3–56) (see Table 1). Absolute monocytosis (Table 2) was associated with increased WBC (n=6), decreased hemoglobin (n=3), decreased platelet count (n=3), and circulating blasts (n=4). Monocyte count ranged between 1.1–28.0 × 109/l. Data about treatment were available for 9 of the 10 patients. Seven of ten patients had received treatment before the development of monocytosis, including hydroxyurea, thalidomide, arsenic, cytarabine, danazol, darbopetin alfa, anagrelide, and interferon; eight patients were treated following the development of monocytosis with different combinations of hydroxyurea, imatinib, anagrelide, decitabine, panibinostat, incite, and interferon; one patient received allogeneic stem cell transplant (Table 3). In some cases treatment was initiated after the development of monocytosis to control peripheral blood counts; in some case monocytosis resolved. The median follow-up time after development of monocytosis was 28 months (mean: 32.6; range: 2–72) (see Table 1). Five (50%) patients died following the onset of monocytosis, with a median survival interval of 45 months (mean: 68.6; range: 20–188 months): one of these (patient 4) developed an acute myeloid leukemia. The three patients whose bone marrow had CMML-like morphology died after a median survival of 20 months. Two of the alive patients had worsening blood counts and became transfusion dependent (cases 2 and 9), two patients have had stable disease (cases 7 and 10), and no clinical status information was available for another patient (case 3) (Table 3).
Morphological and Immunohistochemical Analysis
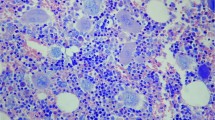
All patients had an initial diagnosis of primary myelofibrosis in fibrotic stage. The diagnosis was reviewed and confirmed at our institution. In the five cases in which a pre-monocytosis bone marrow biopsy was available for review at the time of this study, the morphological features were typical for primary myelofibrosis, including the presence of myelofibrotic marrow (MF-2 or MF-3) with increased cellularity and increased granulopoiesis and megakaryopoiesis. Megakaryocytes were predominantly large and pleomorphic with evidence of defective maturation (ie, showing deviations from the normal nuclear:cytoplasmic ratio) and abnormal chromatin clumping. They were often arranged in dense clusters of several cells; bare megakaryocytic nuclei were also frequently seen (Figure 1a, c).


Morphological features from bone marrow biopsies before and after the development of monocytosis in one case of primary myelofibrosis (patient 1). Sections were stained with hematoxylin and eosin, reticulin and trichrome stains as indicated. The first biopsy shows diagnostic features typical for primary myelofibrosis, such as increased cellularity, increased granulopoiesis and an increased number of predominantly large and pleomorphic megakaryocytes with defective maturation and abnormal chromatin clumping, often arranged in dense clusters of several cells (a, 10 × magnification; c 20 × magnification). Following the development of monocytosis, bone marrow shows strikingly different features, closely resembling those seen in de novo chronic myelomonocytic leukemia: a marked myelomonocytic proliferation with an increased number of neutrophils and monocytes and small and hypolobated megakaryocytes (b, 10 × magnification; d, 20 × magnification). Fibrosis, in this case, decreases from MF-3 to MF-2, as both reticulin (e and f, 20 × magnification) and trichrome staining (g and h, 20 × magnification) show.
In three of the primary myelofibrosis cases, the post-monocytosis bone marrows samples were characterized by the presence of a marked myelomonocytic proliferation with a high number of neutrophils and monocytes associated with a variable proportion of dwarf hypolobated megakaryocytes (Table 2). In two of these three cases, bone marrow biopsy samples obtained before the development of monocytosis were available for comparison. In both patients the findings in the post-monocytosis biopsy (Figure 1b, d) were strikingly different from those present in the initial (pre-monocytosis) biopsy, which showed the typical findings associated with primary myelofibrosis (Figure 1a, c). The post-monocytosis biopsy findings closely resembled those seen in cases of chronic myelomonocytic leukemia. In addition, bone marrow fibrosis in these two patients decreased from MF-3 to MF-2 (Figure 1e–h). In the third patient with a chronic myelomonocytic leukemia-like bone marrow morphology post onset of monocytosis (no pre-monocytosis was available for comparison), the marrow fibrosis in the post-monocytosis sample was graded as MF-3 (Table 2). In two other patients in which pre- and post-monocytosis biopsies were compared, there were no significant changes in marrow morphology or fibrosis grade. In a single patient (case 7), there was an increase in the degree of marrow fibrosis from MF-2 to MF-3 but not change in the remaining marrow morphology. Among the five cases in which only the biopsy following the onset of monocytosis was available, one revealed a variably prominent myelomonocytic proliferation within a cellular background typical for of primary myelofibrosis, whereas the remaining four showed only a typical primary myelofibrosis morphology (Table 2).
After the onset of peripheral blood monocytosis, an increase in the percentage of CD14-positive bone marrow cells ranging from 10 to 15%, was noted in 5 of 10 patients, including two of the three patients with chronic myelomonocytic leukemia-like morphological changes in the marrow (Table 2).
In most of the cases the bone marrow aspirate yielded a dry tap, precluding cytological assessment for morphological dysplasia. However, the cases showing chronic myelomonocytic leukemia-like morphology changes in the marrow after the development of monocytosis showed frequent abnormal monocytes in the peripheral blood smears, comparable to those observed in cases of de novo chronic myelomonocytic leukemia.
Cytogenetic Analysis
For 8 of 10 patients concurrent cytogenetic results were available both before and after the onset of monocytosis. Three of eight patients (43%) showed a normal karyotype. In the remaining five cases, cytogenetic abnormalities included del(13q) (n=3), trisomy 9 and 21 (n=1), t(1;15) (n=1) (Table 2). All cases were negative for t(9;22) by conventional cytogenetics and fluorescence in situ hybridization confirmed the result in four cases. There was no cytogenetic evolution or change in BCR-ABL1 status associated with the development of monocytosis.
Molecular Analysis
All cases were investigated for JAK2 mutational status. Five cases (50%) were JAK2V617F mutation-positive and five cases were JAK2V617F mutation-negative. There was no cytogenetic evolution or change in JAK2 status associated with the development of monocytosis.
The cases were studied for NRAS and KRAS mutational status, using DNA extracted from trephine bone marrow biopsies or archival bone marrow aspirate cytospins. Owing to the acid decalcification, not all cases were evaluable for both genes. None of the nine cases showed evidence of NRAS mutations in exons 12, 13 and 61 by sequencing. Four cases (no. 1, 2, 3, and 5), including the two cases with chronic myelomonocytic leukemia-like evolution in the bone marrow, revealed KRAS exon 12 (twice G12D, once G12C) or exon 13 (G13D) mutations, using the clamped probe assay. Case 2 showed the same G12D mutation in both marrow samples before and after the development of monocytosis. In the remaining mutated cases, only one sample was analyzable. Of interest, conventional PCR and sequencing in the cases with sufficient DNA failed to demonstrate the presence of the mutation, indicating a low prevalence of the mutated allele.
The same type of analysis was performed on a control group, constituted of 13 cases of classical primary myelofibrosis in fibrotic phase. None of these cases showed evidence of KRAS mutations in exons 12 and 13, using the sensitive clamped probe assay. Moreover, in nine of these cases NRAS mutational status was also investigated, and none showed evidence of NRAS mutations in exons 12, 13 and 61 by sequencing.
Discussion
Prognostic evaluation in primary myelofibrosis has been primarily based on clinical features and blood count results obtained at the time of diagnosis. Huang et al,15 who studied risk factors present at the time of diagnosis specifically related to leukemic transformation, showed that a peripheral blood blast count >3% or a platelet count of <100 × 109/l were strong and independent predictors of leukemic transformation in patients with primary myelofibrosis. More recently, dynamic prognostic models able to assess patient prognosis at different times during the course of disease have been proposed by different groups.16, 17 The one published by Morel et al16 on behalf of the International Working Group for Myeloproliferative Neoplasms Research and Treatment was based on data available for 525 primary myelofibrosis patients and showed that the development of leukocytosis (>25 × 109) independently predicts poor survival. However, WBC differential counts values were not reported in the study.
Our data show that patients with established primary myelofibrosis may develop persistent significant monocytosis during the course of their disease. By comparing these patients with the ones that did not develop monocytosis, it is clear that there is no association between prior type of therapy and the development of monocytosis. In the majority of the patients monocytosis persisted unchanged even after subsequent therapy and was associated with worsening disease with decreasing hemoglobin and platelet counts and transfusion requirement. Half of them died soon after the development of monocytosis.
In three of the patients, the bone marrow samples obtained after the onset of monocytosis showed significant changes in morphology, which shifted from the original primary myelofibrosis appearance to a chronic myelomonocytic leukemia-like marrow. These included a predominance of small hypolobated megakaryocytes more consistent with a myelodysplastic/myeloproliferative neoplasm such as chronic myelomonocytic leukemia, instead of the large hyperlobated forms that are typically seen in primary myelofibrosis. In two of the three patients the morphological changes included a parallel decrease in the amount of marrow fibrosis. The findings observed in these three cases would suggest the possibility of evolution from primary myelofibrosis to a truly ‘secondary’ chronic myelomonocytic leukemia. In the more numerous group of primary myelofibrosis patients who developed monocytosis, the bone marrow retained a largely ‘intact’ primary myelofibrosis morphology following the development of monocytosis. In two of these patients, however, an increased number of marrow monocytes was identified by immunostaining with CD14, a finding which is typically seen in chronic myelomonocytic leukemia and in a proportion of cases of acute myeloid leukemia but not in myeloproliferative neoplasms.18, 19, 20 The variable marrow characteristics seen in cases of primary myelofibrosis associated with monocytosis would suggest that different pathogenetic mechanisms are involved in this phenomenon and that it may be genetically heterogeneous.
The presence of additional cytogenetic abnormalities in myeloproliferative neoplasms during the course of disease is considered evidence of disease progression. In our cases, although the frequency of cytogenetic abnormalities was higher (57%) than that expected in primary myelofibrosis (30%),1, 21 there was no association between the development of monocytosis and the type or occurrence of cytogenetic abnormalities. Thus, in our series evidence of a parallel cytogenetic progression was not detected. This is similar to what has been reported by others in the setting of chronic myelomonocytic leukemia secondary to myelodysplastic syndromes.6
RAS mutations have been reported to occur as an acquired genetic event in the course of chronic myelomonocytic leukemia.22 In these cases, the presence of the mutation has been associated with the transition from a myelodysplastic type of chronic myelomonocytic leukemia to a myeloproliferative variant of the disease.22 A similar effect has been reported for the occurrence of an acquired JAK2 mutation in patients with chronic myelomonocytic leukemia.23 However, in contrast to chronic myelomonocytic leukemia, no evidence of RAS or FLT3 mutations were found in cases of myelodysplastic syndromes with secondary ‘monocytic evolution’.6 In our series, a KRAS mutation in exons 12 or 13 was detected in 4/9 cases at some point, including the two patients with chronic myelomonocytic leukemia-like marrow appearance. In contrast, none of the thirteen cases included in the control group of primary myelofibrosis that never developed monocytosis showed any mutation of KRAS. The mutational load was low, as the mutations were only detectable by the more sensitive clamped probe assay and not by conventional sequencing in cases analyzed by both techniques. The relevance of this finding for the development of monocytosis is unclear, as we did not have enough paired samples before and after development of monocytosis to satisfactorily address this issue. Regarding the frequency of JAK2V617F mutation in our cases, this was similar to what has been described in general in primary myelofibrosis.1 There was no change in JAK2 status after the development of monocytosis.
In view of the absence of cytogenetic evidence of clonal progression, the most likely pathogenetic mechanism involved in these cases is that the monocytic expansion may be due to clonal heterogeneity, perhaps due to the development of a new clone during the late stage of the disease. The concept of clonal heterogeneity in myeloproliferative neoplasms has received attention in recent years. It has been shown to be the main mechanism responsible for two different types of leukemic transformation seen in JAK2 mutation-positive myeloproliferative neoplasms, one with JAK2 mutation-positive blasts and one with JAK2 wild-type blasts.24 Clonal heterogeneity has also been implicated in rare cases of myeloproliferative neoplasms with combined BCR-ABL1 and JAK2 mutation-positivity.25, 26 In these cases, a BCR-ABL1-negative clone carrying JAK2 mutation appears during the course of imatinib treatment. In addition, clonal heterogeneity has been demonstrated in cases of myeloproliferative neoplasms with del(20q)27 and myelodysplastic syndromes with isolated 5q deletion associated with JAK2 mutation.28 TET2 mutations are the most common genetic abnormality found in chronic myelomonocytic leukemia, being present in around 40% cases.29, 30 Although TET2 mutations can represent early molecular events in myeloproliferative neoplasms, in a recent study of JAK2 mutation-positive myeloproliferative neoplasms based on colony assays, genotyping for TET2 and JAK2 mutations, it was demonstrated that TET2 can be a late event in the progression of myeloproliferative neoplasms and may represent an acquired abnormality in a separate (JAK2 mutation-negative) clone.31 It is intriguing to speculate that the acquisition of KRAS, TET2 or other mutagenic events could have played a role in the phenotypic shift observed in our cases. One could hypothesize that possibly either owing to clonal defect present at the disease onset in only a minority of the cells and its subsequent expansion, or because of a new genetic event acquired later in the course of the disease, some patients with myeloproliferative neoplasms have a monocytic proliferation capacity that eventually manifests as a chronic myelomonocytic leukemia or chronic myelomonocytic leukemia-like phenotype.
Regardless of the mechanisms involved in its occurrence, the development of monocytosis and/or the shift from primary myelofibrosis to a chronic myelomonocytic leukemia-like bone marrow morphology has important clinical implications. Our study not only confirms the results of an earlier study on the prognostic relevance of monocytosis in primary myelofibrosis,9 but also expands it by showing that an acquired monocytic proliferation in the course of disease may be equivalent to an ‘accelerated phase’. Our data further suggest that monocytosis is an easy to follow, useful dynamic prognostic parameter, which may indicate clinical progression in cases in which the blast count or cytogenetics have remained ‘stable’. However, in view of the lack of adequate clinical information and inhomogeneity in the treatment received, a direct statistical comparison between the cases who developed monocytosis and our whole group of primary myelofibrosis patients could not be performed in this study. Additional studies using carefully selected matched population of primary myelofibrosis cases with and without monocytosis would be needed to further confirm these findings and to elucidate the reasons for the heterogeneity observed in the marrow results.
In conclusion, because of their rapid disease progression after the development of monocytic proliferation, this group of primary myelofibrosis patients should be considered separately. For these patients, it might be conceivable to consider treatments similar to those used to treat patients with chronic myelomonocytic leukemia (eg, decitabine or other hypomethylating agents).
References
Thiele J, Kvasnicka HM, Tefferi A et al Primary myelofibrosis In: Swerdlow SH, Campo E, Harris NL, et al(eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Lyon, 2008, pp 44–47.
Cervantes F, Dupriez B, Pereira A et al New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009;113:2895–2901.
Orazi A, Chiu R, O'Malley DP et al Chronic myelomonocytic leukemia: The role of bone marrow biopsy immunohistology. Mod Pathol 2006;19:1536–1545.
Orazi A, Germing U . The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia 2008;22:1308–1319.
Orazi A, Bennett JM, Germing U et al Chronic myelomonocytic leukaemia In:Swerdlow SH, Campo E, Harris NL, et al(eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Lyon, 2008, pp 76–79.
Wang SA, Galili N, Cerny J et al Chronic myelomonocytic leukemia evolving from preexisting myelodysplasia shares many features with de novo disease. Am J Clin Pathol 2006;126:789–797.
Takasaki Y, Iwanaga M, Tsukasaki K et al Impact of visceral involvements and blood cell count abnormalities on survival in adult T-cell leukemia/lymphoma (ATLL). Leuk Res 2007;31:751–757.
Chen MH, Chang PM, Chen PM et al Prognostic significance of a pretreatment hematologic profile in patients with head and neck cancer. J Cancer Res Clin Oncol 2009;135:1783–1790.
Elliott MA, Verstovsek S, Dingli D et al Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk Res 2007;31:1503–1509.
Thiele J, Kvasnicka HM, Orazi A . Bone marrow histopathology in myeloproliferative disorders--current diagnostic approach. Semin Hematol 2005;42:184–195.
Thiele J, Kvasnicka HM, Facchetti F et al European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005;90:1128–1132.
Shaffer LG, Slovak ML, Campbell LJ, (eds) ISCN (2009): an International System for Human Cytogenetic Nomenclature. Karger Basel, 2009.
Chen Q, Lu P, Jones AV et al Amplification refractory mutation system, a highly sensitive and simple polymerase chain reaction assay, for the detection of JAK2 V617F mutation in chronic myeloproliferative disorders. J Mol Diagn 2007;9:272–276.
Dabritz J, Hanfler J, Preston R et al Detection of Ki-ras mutations in tissue and plasma samples of patients with pancreatic cancer using PNA-mediated PCR clamping and hybridisation probes. Br J Cancer 2005;92:405–412.
Huang J, Li CY, Mesa RA et al Risk factors for leukemic transformation in patients with primary myelofibrosis. Cancer 2008;112:2726–2732.
Morel P, Duhamel A, Hivert B et al Identification during the follow-up of time-dependent prognostic factors for the competing risks of death and blast phase in primary myelofibrosis: a study of 172 patients. Blood 2010;115:4350–4355.
Cervantes F, Pereira A . Prognostication in primary myelofibrosis. Curr Hematol Malig Rep 2012;7:43–49.
Qubaja M, Marmey B, Le Tourneau A et al The detection of CD14 and CD16 in paraffin-embedded bone marrow biopsies is useful for the diagnosis of chronic myelomonocytic leukemia. Virchows Arch 2009;454:411–419.
Dunphy CH . Comparative analysis of detecting monocytic cells and their aberrancy. Appl Immunohistochem Mol Morphol 2011;19:336–340.
Rollins-Raval MA, Roth CG . The value of immunohistochemistry for CD14, CD123, CD33, myeloperoxidase and CD68R in the diagnosis of acute and chronic myelomonocytic leukaemias. Histopathology 2012;60:933–942.
Hussein K, Van Dyke DL, Tefferi A . Conventional cytogenetics in myelofibrosis: literature review and discussion. Eur J Haematol 2009;82:329–338.
Ricci C, Fermo E, Corti S et al RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res 2010;16:2246–2256.
Pich A, Riera L, Sismondi F et al JAK2V617F activating mutation is associated with the myeloproliferative type of chronic myelomonocytic leukaemia. J Clin Pathol 2009;62:798–801.
Beer PA, Delhommeau F, LeCouedic JP et al Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010;115:2891–2900.
Hussein K, Bock O, Seegers A et al Myelofibrosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABL translocation and JAK2V617F mutation. Blood 2007;109:4106–4107.
Hussein K, Bock O, Theophile K et al Biclonal expansion and heterogeneous lineage involvement in a case of chronic myeloproliferative disease with concurrent MPLW515L/JAK2V617F mutation. Blood 2009;113:1391–1392.
Schaub FX, Jager R, Looser R et al Clonal analysis of deletions on chromosome 20q and JAK2-V617F in MPD suggests that del20q acts independently and is not one of the predisposing mutations for JAK2-V617F. Blood 2009;113:2022–2027.
Sokol L, Caceres G, Rocha K et al JAK2(V617F) mutation in myelodysplastic syndrome (MDS) with del(5q) arises in genetically discordant clones. Leuk Res 2010;34:821–823.
Schaub FX, Looser R, Li S et al Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 2010;115:2003–2007.
Smith AE, Mohamedali AM, Kulasekararaj A et al Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood 2010;116:3923–3932.
Kohlmann A, Grossmann V, Klein HU et al Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol 2010;28:3858–3865.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work has been presented in part at the 100th Annual Meeting of the United States and Canadian Academy of Pathology, San Antonio, TX, USA, February 26-March 4, 2011.
Rights and permissions
About this article
Cite this article
Boiocchi, L., Espinal-Witter, R., Geyer, J. et al. Development of monocytosis in patients with primary myelofibrosis indicates an accelerated phase of the disease. Mod Pathol 26, 204–212 (2013). https://doi.org/10.1038/modpathol.2012.165
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.165
Keywords
This article is cited by
-
Myeloproliferative Neoplasms with Monocytosis
Current Hematologic Malignancy Reports (2022)
-
Philadelphia-negative myeloproliferative neoplasms display alterations in monocyte subpopulations frequency and immunophenotype
Medical Oncology (2022)
-
The ABNL-MARRO 001 study: a phase 1–2 study of randomly allocated active myeloid target compound combinations in MDS/MPN overlap syndromes
BMC Cancer (2022)
-
Progression, transformation, and unusual manifestations of myelodysplastic syndromes and myelodysplastic-myeloproliferative neoplasms: lessons learned from the XIV European Bone Marrow Working Group Course 2019
Annals of Hematology (2021)
-
Laboratory Evaluation and Pathological Workup of Neoplastic Monocytosis — Chronic Myelomonocytic Leukemia and Beyond
Current Hematologic Malignancy Reports (2021)
