
Abstract
Aldo-keto reductase family 1B10 (AKR1B10) exhibits more restricted lipid substrate specificity (including farnesal, geranylgeranial, retinal and carbonyls), and metabolizing these lipid substrates has a crucial role in promoting carcinogenesis. Overexpression of AKR1B10 has been identified in smoking-related carcinomas such as lung cancer. As development of pancreatic cancer is firmly linked to smoking, the aim of the present study was to examine the expression and oncogenic role of AKR1B10 in pancreatic adenocarcinoma. AKR1B10 expression was analyzed in 50 paraffin-embedded clinical pancreatic cancer samples using immunohistochemistry. Oncogenic function of AKR1B10 was examined in pancreatic carcinoma cells in vitro using western blotting and siRNA approaches, mainly on cell apoptosis and protein prenylation including KRAS protein and its downstream signals. Immunohistochemistry analysis revealed that AKR1B10 overexpressed in 70% (35/50) of pancreatic adenocarcinomas and majority of pancreatic intraepithelial neoplasia, but not in adjacent morphologically normal pancreatic tissue. Compared with a normal pancreatic ductal epithelial cell (HPDE6E7), all of the six cultured pancreatic adenocarcinoma cell lines had an overexpression of AKR1B10 using immunoblotting, which correlated with increase of enzyme activity. siRNA-mediated silencing of AKR1B10 expression in pancreatic cancer cells resulted in (1) increased cell apoptosis, (2) increased non-farnesyled HDJ2 protein and (3) decreased membrane-bound prenylated KRAS protein and its downstream signaling molecules including phosphorylated ERK and MEK and membrane-bound E-cadherin. Our findings provide first time evidence that AKR1B10 is a unique enzyme involved in pancreatic carcinogenesis possibly via modulation of cell apoptosis and protein prenylation.
Similar content being viewed by others


Main
Aldo-keto reductases (AKRs) are a gene superfamily that are monomeric, soluble, NADPH-dependent oxidoreductases that give functionality to the carbonyl groups present on aldehydes and ketones to form alcohols in eliminating reactions. AKRs consist of 14 families, with the AKR1 family composed of many isoforms. The AKR1B subfamily is a group of aldolase reductases, and aldo-keto reductase family 1B10 (AKR1B10) shares 70% sequence homology with AKR1B1. AKR1B1 is globally expressed in human tissues, whereas AKR1B10 is highly expressed in the small and large intestine as well as the adrenal glands, and very low levels of expression have been found in the liver. Expression of AKR1B10 in the adrenal glands is consistent with its role in steroid metabolism and in the small and large intestine for the detoxification of aldehydes within the digestive tract.1, 2 Overexpression of AKR1B10 has been identified in hepatocellular carcinoma and non-small cell lung carcinomas. Further studies suggest that AKR1B10 is a unique biomarker of smoking-related cancer and that AKR1B10 can be activated by tobacco-related carcinogenic transcription factors.3, 4, 5 As smoking is the most clearly established risk factor for pancreatic cancer and is associated with KRAS mutations,6 it is reasonable to postulate the AKR1B10 is upregulated in human pancreatic cancer.
AKR1B10 exhibits more restricted substrate specificity than that of general human aldo-ketoreductases; only farnesal, geranylgeranyl, retinal and carbonyls are its specific substrates.7, 8, 9, 10, 11 Metabolizing these specific substrates has a crucial role in promoting carcinogenesis. The reduced products of farnesal and geranylgeranyl are intermediates of cholesterol synthesis involved in protein prenylation.12 Protein prenylation is a process of lipid modification, which involves the covalent addition of either farnesyl (15-carbon) or geranylgeranyl (20-carbon) isoprenoids to C-terminal cysteines of the target protein. This post-translational modification allows the prenylated protein to localize to cellular membranes, aid in protein-protein interactions and exert its effects on a wide range of signal transduction pathways, some of which include cell growth, differentiation, maintenance of the cellular cytoskeleton and vesicle trafficking.12, 13, 14, 15 Prenylation appears to be a crucial event in carcinogenesis. Proteins that undergo prenylation include Ras and Ras-related GTP-binding proteins (G proteins), protein kinases, fungal mating factors and nuclear lamins.13, 14 More than 95% of human pancreatic cancers carry a KRAS gene mutation and the KRAS protein requires prenylation for its activity.16 Recent studies demonstrate that conversion of the protein–cysteine acceptor site in oncogenic Ras prevents prenylation and abolishes its malignant transforming ability.17 Many reports have demonstrated the central role of Ras activation in pancreatic cancer, yet the role of AKR1B10 expression in protein prenylation in pancreatic cancer have not been studied to date.
With respect to retinal, AKR1B10 is the most efficient retinal reductase.7, 9, 18, 19, 20 The conversion to retinol results in suppression of the conversion of retinal to retinoic acid—the major active anti-neoplastic metabolite. Retinol can be reversibly transformed to retinal, which in turn is irreversibly oxidized to retinoic acid. Retinoic acid is an important signaling molecule affecting cell differentiation. Several recent studies have demonstrated that AKR1B10 can influence carcinogenesis by controlling the retinoid signaling pathway.7, 8, 18 An increased level of AKR1B10 expression in neoplastic cells results in an enzymatic reduction of retinal to retinol, a concomitant decrease in retinoic acid, and a shift in signaling from differentiation to unopposed cellular proliferation.7, 8, 18
With respect to carbonyl metabolism, conversion of highly reactive aldehyde and ketone groups into hydroxyl groups by AKR1B10 in neoplastic cells protects these neoplastic cells against highly active carbonyl-induced apoptosis. Recent data demonstrates that AKR1B10 affects cell survival through modulating lipid synthesis, mitochondrial function and oxidative status, as well as carbonyl levels, and therefore as being an important cell survival protein.21 A separate study revealed that carbonyl groups in some anticancer drugs are converted by AKR1B10 to their corresponding alcohols and that this carbonyl reducing activity decreases the anticancer drug's effectiveness.22
The aims of the present studies are to determine whether the AKR1B10 expresses in human pancreatic adenocarcinomas and what is its potential functional role in pancreatic carcinoma cells. Using immunohistochemical and western blot approaches as well enzyme activity assay, we evaluated AKR1B10 expression and enzyme activity in vitro and ex vivo human pancreatic adenocarcinoma samples. To investigate the oncogenic role of AKR1B10 in pancreatic cancer, we further analyzed if knockdown of AKR1B10 expression via a siRNA approach resulted in the reduction of protein prenylation and the induction of cell apoptosis in pancreatic carcinoma cells overexpressing AKR1B10.
Materials and methods
Cell Lines
Six human pancreatic adenocarcinoma cell lines (MiaPANC, Panc10.05, CAPAN-2, CAPAN-1, CD18 and Panc2.03) and the human hepatocellular carcinoma (HepG2) cell line were obtained from ATCC (Manassas, VA). The immortalized human pancreatic duct epithelial cell line (called HPDE6E6E7) was established in Dr Ming-Sound Tsao's lab (Ontario Cancer Institute; Ontario, Canada). All cell lines were cultured in the appropriate medium obtained from Mediatech (Manassas, VA) containing 10% fetal bovine serum (ThermoScientific, Waltham, MA) and maintained at 37 °C and 5% CO2.
Protein Extraction and Immunoblot Analysis
Cultured cells representing normal and adenocarcinoma were washed with 1 × DPBS buffer, scraped with sterile cell lifters and pelleted using a table-top centrifuge at 1000 rpm for 5 min. Cells were then lysed on ice by vortexing every 5 min for 30 min using RIPA buffer containing 10 mM Tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 0.004% sodium azide, 10 mg/ml leupeptin, 10 mg/ml aprotinin, 1% phosphatase inhibitor cocktail 1 and 2 (Sigma, St Louis, MO), 2% protease inhibitor cocktail and 1% PMSF (Sigma). Membrane proteins from cultured cells were extracted for detecting the GTP-bound/active form prenylated KRAS protein (anchored on the membrane) using Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (Thermo Scientific, IL) according to manufacturer's instructions. The extracts were collected by centrifugation at 10 000 rpm for 3 min at 4 °C, followed by transfer of the supernatant to a new tube. The supernatant was incubated at 37 °C for 20 min in order to isolate the hydrophobic, membrane protein fraction. All protein concentrations were determined using Bradford reagent (Bio-Rad, Hercules, CA).
A protein (35 μg) was loaded on a 10% SDS-polyacrylamide gel and transferred to a Immun-Blot PVDF Membrane (Bio-Rad), blocked using 1 × TBST containing 0.05% Tween-20 and 5% non-fat powdered milk and followed by membrane incubation with the primary antibody solution overnight at 4 °C. The antibodies used were as follows: polyclonal rabbit anti-human AKR1B10 (1:300; LifeSpan BioSciences, Seattle, WA), cleaved caspase-8 (1:1000, Cell Signaling Technology, Danvers, MA), MEK1/2 (1:1000, Cell Signaling Technology), Caspase-3 (1:200, Cell Signaling Technology) and mouse monoclonal anti-c-KRAS (Ab-1) (2 μg/ml; Calbiochem), ERK (1:1000; Cell Signaling Technology), E-cadherin (1:2000, BD Biosciences), as well as anti-human HDJ-2/DNAJ (1 μg/ml; Thermo Scientific) and anti-human Flotillin-2 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA). The membrane was then washed with 1 × TBST and incubated with HRP-linked anti-rabbit (1:2000) or anti-mouse (1:2000) IgG and HRP-linked anti-biotin antibodies (1:1000, Cell Signaling Technology) for 1 h at room temperature. The protein–antibody complexes were detected using the 1 × LumiGLO chemiluminescent substrates (Cell Signaling Technology) according to the manufacturer's instructions and the emitted light was captured on X-ray film. Mouse anti-human β-actin (1:5000: Sigma-Aldrich, St Louis, MO) served as an internal control.
Small-Interfering RNA (siRNA) Knock-Down Assay
Sense (5′-AGAGGAAUGUGAUUGUCAUTT-3′) and anti-sense (5′-AUGACAAUCACAUUCCUCUGG-3′) oligonucleotides were purchased from Ambion (Austin, TX) for ABR1B10. The oligonucleotides were annealed following the manufacturer's protocol to generate the double-stranded siRNAs at the final concentration of 20 μM. Cells (2 × 106) were cultured in six-well plastic plates in 1 ml of DMEM without serum and transfected at ∼40% of confluence by adding 4 μl of oligofectamine (Invitrogen) and 10 μl of 20 μM stock siRNAs. Cells were incubated at 37 °C for 4 h in a CO2 incubator, followed by the addition of growth medium containing 3 × the normal concentration of serum. Cells were maintained in culture for an additional 32 h before western blot or enzyme activity analysis.
Enzyme Activity Assay
Cells were lysed on ice in buffer containing 50 mM Tris-HCl (pH 7.0), 1% protease inhibitor (Sigma-Aldrich), 1% β-mercapethanol (Sigma-Aldrich), 10% RIPA buffer (Santa Cruz Biotechnology) by vortexing briefly every 5 min over a 30 min period, followed by centrifugation at 10 000 rpm at 4 °C for 10 min. Soluble proteins in the supernatant (50 μg) were used for AKR1B10 activity assays in a reaction buffer containing 125 mM sodium phosphate (pH 7.0), 50 mM KCl and 20 mM DL-glyceraldehyde in a 35 °C water bath for 10 min. The reaction is carried out using 0.3 mM NADPH as a substrate for AKR1B10 enzymatic activity. The change/decrease in optical density at 340 nm is monitored every minute for 10 min at room temperature in a SmartSpec Plus spectrophotometer (Bio-Rad) and the difference in absorbance from the start to the end of the assay (10 min period) was used to determine AKR1B10 activity.
Immunohistochemistry and Microscopic Analysis
A total of 50 formalin-fixed, paraffin-embedded pancreatic adenocarcinomas were available for the study. The use of these specimens for research was approved by the Institutional Review Board of Northwestern University and Northwestern Memorial Hospital. Immunohistochemical analysis for AKR1B10 was performed on these paraffin sections. The sections were deparaffinized in xylene, washed in ethanol and rehydrated in 1 × TBST. Antigen retrieval was performed using a microwave technique in citric acid-based Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA). Endogenous peroxidase activity was quenched using 3% hydrogen peroxide for 10 min. Nonspecific binding was blocked by incubating the tissue sections with diluted normal horse serum for 60 min. AKR1B10 antibody (6 μg/ml; Abnova Corporation, Taiwan) was applied overnight at 4° C hour followed by secondary staining using Vector Laboratories anti-mouse ImmPRESS Detection kit with bound antibody detected using SigmaFAST DAB for 5 min. All sections were counterstained with Mayer's hematoxylin for 30 s. The hepatocellular carcinomas were used as positive control and the negative control was established with the replacement of anti-AKR1B10 antibody by normal serum. Diffuse (>75%) granular, cytoplasmic staining in epithelial cells was considered as AKR1B10 positive.
Statistical Analysis
Summary statistics such as percent, means, median, s.d. values and ranges have been determined for all outcome measures. For continuous measures, analysis of variance methods have been used. Continuous variables were compared with the Student's t-test, whereas categorical variables were compared with χ2-test. All statistical tests were two-sided, and data were considered statistically significant when P<0.05.
Results
Distinct Expression Pattern of AKR1B10 in Pancreatic Adenocarcinoma
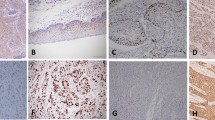
AKR1B10 expression was determined in 50 pancreatic carcinoma tissues using an immunohistochemical approach. No positive staining of AKR1B10 was observed in the morphologically normal pancreatic parenchyma (including acinar, ductal and islet cells), as seen in Figure 1a. Positive expression of AKR1B10 was identified in the cytoplasm of neoplastic cells in the invasive pancreatic ductal adenocarcinomas (Figure 1b), but not in adjacent morphologically normal ductal epithelial cells (Figure 1b; open black arrow). In all, 35 of 50 (70%) pancreatic adenocarcinomas exhibited overexpression of AKR1B10. Of the tumors expressing AKR1B10, the majority of cases were well- and moderately differentiated adenocarcinomas (carcinomas that showed well-formed glandular structure or a cribiform pattern), in which ∼71% (20/28) of well-differentiated carcinomas and 74% (14/19) of moderately differentiated carcinomas displayed AKR1B10 expression, whereas only ∼33% (1/3) of poorly differentiated carcinomas exhibited positive staining for AKR1B10. When further examining the social history of these patients, 60% of those with AKR1B10 overexpression were smokers (defined as more than 10 year smoking history).

Immunohistochemical analysis of AKR1B10 expression in human pancreatic tissues: (a) No detectable expression was observed in normal pancreatic acini or ducts. (b) Well-differentiated pancreatic ductal adenocarcinoma with high expression of AKR1B10, but negligible expression in an adjacent normal duct (open black arrow). (c) Precancerous pancreatic epithelial neoplasia also overexpressed AKR1B10 as demonstrated in pancreatic intraepithelial neoplasia-3 (large duct in center) and pancreatic intraepithelial neoplasia-1 lesions (open black arrows).
Precancerous pancreatic lesions known as pancreatic intraepithelial neoplasia were frequently observed in the adjacent morphologically normal pancreatic tissues of resected pancreatic carcinoma specimens. The expression of AKR1B10 in pancreatic intraepithelial neoplasia lesions was further analyzed. A similar positive staining intensity for AKR1B10 was observed in the pancreatic intraepithelial neoplasia lesions (Figure 1c) when compared with that observed in pancreatic carcinoma (Figure 1b). All pancreatic intraepithelial neoplasia-1 (18/18) and pancreatic intraepithelial neoplasia-3 lesions (6/6), as well as the majority of pancreatic intraepithelial neoplasia-2 lesions (10/12; 83.3%) had AKR1B10 overexpression.
Over-Expression and Increased Enzyme Activity of AKR1B10 in Cultured Pancreatic Adenocarcinoma Cell Lines
Using a quantitative western blot approach, the level of AKR1B10 expression was determined in six human pancreatic carcinoma cell lines using the immortalized normal pancreatic ductal epithelial cell line (HPDE6E6E7) as baseline control and human hepatocellular carcinoma cell line (Hep2G) as a positive control. Western blot analysis revealed overexpression of AKR1B10 protein in all of the six pancreatic adenocarcinoma cell lines as compared with a normal pancreatic ductal epithelial cell line (HPDE6E6E7), and the levels of AKR1B10 expression in these pancreatic carcinoma cell lines were either comparable or slightly lower to that observed in the Hep2G cell line (Figure 2a).

Analysis of AKR1B10 expression along with corresponding enzyme activity. (a) Using normal pancreatic ductal epithelial cell line (HPDE6E6E7) as a baseline control, and using the human hepatocellular carcinoma cell line (HepG2) as a positive control, western blot analysis revealed that AKR1B10 was overexpressed in all of the six pancreatic adenocarcinoma cell lines, and that all of six cell lines exhibited either comparable or lower levels of expression to the HepG2 positive-control cell line. (b) Oxidized NADPH was used as a monitor at 340 nm for measuring AKR1B10 enzymatic activity in these pancreatic adenocarcinoma cell lines, and showed that enzyme activity for all six cell lines paralleled AKR1B10 protein expression levels (a).
Oxidized NADPH was used as a monitor at 340 nm for measuring AKR1B10 enzymatic activity in these pancreatic adenocarcinoma cell lines.9 As shown in Figure 2b, AKR1B10 enzyme activity in the pancreatic carcinoma cell lines was significantly higher (2–3 fold) than that in the normal pancreatic ductal epithelial cell line (HPDE6E6E7) and had comparable or slightly lower activity to that seen in Hep2G cells. Of note, the enzyme activity correlated with the levels of protein expression (Figures 2a and b).
Small-Interfering RNA (siRNA)-Mediated AKR1B10 Silencing Induces Apoptotic Cell Death in Pancreatic Adenocarcinoma Cell Line
To further demonstrate the biologic role of AKR1B10 in pancreatic cancer, silencing of AKR1B10 dynamic expression in the human pancreatic adenocarcinoma cell line Panc10.05 was achieved using a siRNA approach. As seen in Figure 3 (lower panel), compared with parental Panc10.05 pancreatic carcinoma cells, Panc10.05 pancreatic carcinoma cells transfected with siRNA showed a significant downregulation of AKR1B10 expression as detected by western blot assay. Enzyme activity assay further confirmed that AKR1B10 activity was significantly lower when compared with the scrambled siRNA control Panc10.05 cell line (Figure 3, upper graph).

siRNA-mediated silencing of AKR1B10 expression and enzymatic activity. Western blot analysis demonstrates markedly decreased expression of AKR1B10 in knock-down cells (lower panel) with concomitant decrease in enzymatic activity (upper graph) that parallels AKR1B10 expression levels.
As active carbonyls are one of the specific substrates of AKR1B10, overexpression of AKR1B10 could be a mechanism of neoplastic cell resistance to active carbonyl-mediated cell death. Western blot analysis revealed that silencing of AKR1B10 expression resulted in increased apoptosis-related cleaved caspase-8 expression (Figure 4a). Immunocytochemical analysis of activated-caspase-3-labeled cell apoptosis was performed and showed that compared with the scrambled siRNA control (Figure 4b1, top image), siRNA silencing of AKR1B10 expression increased activated-caspase-3-labeled cell apoptosis (Figure 4b2, bottom image). The percentage of activated-caspase-3 labeled apoptotic cells was 1.5±1.3% in the scrambled siRNA control, and a significant increase in cell apoptosis was observed in Panc10.05 carcinoma cells with silencing of AKR1B10 expression (6.7±2.3%, P=0.029). Cell proliferation was further determined based on the clone size (number of cells per clone), and revealed that number of cells per clone in the scrambled siRNA control was 32±11, whereas a significant decrease in the number of cells per clone was identified in Panc10.05 carcinoma cells with AKR1B10 silencing (18±9, P=0.048).

AKR1B10 silencing resulted in induction of apoptosis and inhibition of cell proliferation. (a) Western blot analysis showed that increased level of cleaved caspase-8 protein, a marker of apoptosis, in Panc10.05 cells with AKR1B10 silencing. (b) Immunocytochemical analysis of cell apoptosis using activated caspase-3 in the scambled siRNA control (b1) and those with siRNA silencing of AKR1B10 expression (b2). Silencing of AKR1B10 expression resulted in increased activated-caspase-3-labeled cell apoptosis as demonstrated by the arrows in the bottom image as well as the small size of the cell clones. The number of cells per clone was further counted and revealed that a significant decrease in the number of cells per clone were identified in Panc10.05 carcinoma cells with AKR1B10 silencing when compared with the parental control (18±9 vs 32±11, P=0.048).
Silencing of AKR1B10 Expression Results in Decreased Key Prenylated Protein Expression Including Membrane-Bound KRAS Protein and its Downstream Signal Activities
The human DNA-J homolog, HDJ2, is a co-chaperone containing a cysteine-rich zinc-finger domain and a mitochondrial protein. HDJ2 is also a heat-induced and farnesylated protein that is apparently involved in mitochondrial protein import in mammals.23 HDJ2 protein is commonly used as a biomarker for the evaluation of protein prenylation. To determine if AKR1B10 is involved in the process of protein prenylation, farnesylated and non-farnesylated HDJ2 proteins were analyzed in siRNA AKR1B10-silencing cells and in the scrambled siRNA control using a western blot approach. Silencing of AKR1B10 expression resulted in an increased level of non-farnesyled HDJ2 protein (Figure 5, black upper arrow).

Western blot analysis of prenylated proteins including HDJ2 and KRAS as well its downstream signals. Increased non-farnesylated HDJ2 protein (upper black arrow) and decreased membrane-bound KRAS protein were observed in the siRNA AKR1B10 pancreatic cancer cell line when compared with the scrambled siRNA control. Increased levels of membrane-bound E-cadherin (lower black arrow) and decreased levels of KRAS downstream effectors, phosphor-ERK and phosphor-MEK1/2 were also found. Flottin-2 was used as a loading control for membrane-bound proteins, whereas β-actin is used for cytosolic proteins.
KRAS protein is another key protein, which requires prenylation for its membrane localization and activity. Like other small GTPases, KRAS regulates molecular events by cycling between an inactive GDP-bound form and an active GTP-bound form. The basis of its active state (GTP-bound) is a membrane-bound KRAS protein or the prenylated form of the KRAS protein. Immunoblot analysis of membrane protein extracts revealed markedly decreased levels of membrane-bound KRAS in Panc10.05 cells with siRNA silencing of AKR1B10 compared with the scrambled siRNA control and using the membrane protein Flotillin-2 as a quantitative loading control (Figure 5); however, the level of cytosolic/soluble KRAS protein was unchanged.
Downregulation of E-cadherin expression or function appears be an important step in carcinogenesis and cell differentiation and its expression has been shown to be regulated by the KRAS pathway.24, 25, 26, 27 Immunoblot assay further revealed that membrane-bound E-cadherin was significantly increased in Panc10.05 cells with AKR1B10 silencing (Figure 5, black lower arrow). In addition, KRAS downstream signals were also evaluated and showed that the phosphorylation of ERK and MEK was downregulated in Panc10.05 cells with AKR1B10 silencing (Figure 5).
Discussion
Pancreatic cancer is one of the most lethal malignant neoplasms in the United States.28 The risk factors for pancreatic carcinoma are strongly related to life and dietary style, environment, genetic factors and genetic–environment interaction.29, 30, 31 The identification of these factors and their interaction with the host and how this can lead to cancer development are important in developing strategies for the prevention and cure of pancreatic cancer and the reduction of its mortality.32 In humans, the most common and recognized risk factors for the development of pancreatic cancer are smoking and chronic pancreatitis.33, 34 AKR1B10 was first isolated from human hepatocellular carcinoma,1 and has been identified as a unique tumor biomarker that is overexpressed in hepatocellular carcinoma and smoking-related carcinomas such as the lung and bladder carcinomas.5, 35, 36, 37, 38 Here, we first demonstrated that AKR1B10 is overexpressed in resected human pancreatic adenocarcinomas and in vitro cultured adenocarcinoma cell lines; and further demonstrated its unique oncogenic function in activating KRAS via the process of protein prenylation. By further considering the patient's social history, overexpression of AKR1B10 was highly associated with a history of smoking.
As summarized in Figure 6, AKR1B10 is involved in regulating cellular fatty acid synthesis, lipid and isoprenoids metabolism, regulating retinal homeostasis and in the metabolization of highly active carbonyls.7, 8, 9, 10, 11 All of these unique metabolizing processes contribute to carcinogenesis as they are involved in protein prenylation, cell proliferation and apoptosis (Figure 6). Farnesol and geranylgeraniol, the reduced products of AKR1B10 from farnesyl and geranylgeranyl, are phosphorylated into farnesyl and geranylgeranyl pyrophosphates, respectively, which are the intermediates of cholesterol synthesis involved in protein prenylation.12 Thus, it has been postulated that AKR1B10 is possibly involved in the process of protein prenylation via metabolizing farnesyl and geranylgeranyl.39 Using in vitro cultured human pancreatic adenocarcinoma cell lines, we identified the upregulation of AKR1B10 in these pancreatic adenocarcinoma cell lines. We have first demonstrated that using siRNA-mediated AKR1B10 silencing, dynamic knockdown of AKR1B10 expression as well its enzymatic activity resulted in decreased level of the farnesylated human DNA-J homolog (HDJ2) chaperone, a prototypical biomarker of prenylated protein.

The proposed interactive role of AKR1B10 in carbonals, farnesyl and lipid metabolism as in maintaining the homeostasis of retinol to retinal that is involved in cellular apoptosis, protein prenylation and proliferation and contributes to pancreatic carcinogenesis.
Activating point mutations in the KRAS gene are present in ∼95% of pancreatic cancers40, 41, 42 and represent the earliest molecular events involved in pancreatic carcinogenesis.43, 44 Membrane-bound KRAS protein is another crucial biomarker of protein prenylation, which is required for anchoring of proteins to the cytosolic membrane and for their activity.13, 14 In this study, we demonstrate that the amount and activity of membrane-bound KRAS protein in the pancreatic adenocarcinoma cell line was significantly decreased when AKR1B10 expression was silenced. In addition, KRAS downstream signals including phosphorylated MEK and ERK were suppressed. These results further confirmed that AKR1B10 was involved in the process of protein prenylation and indicated that AKR1B10 could be a critical target for shutting down KRAS oncogenic function.
E-cadherin is a glycoprotein present in the adherens junctions of epithelial cells. Disruption of the proteins comprising these junctions leads to changes in cell adhesion and dedifferentiation. E-cadherin expression is frequently downregulated or absent in highly invasive, poorly differentiated carcinomas and increased expression of the E-cadherin protein in vitro can decrease tumor cell invasiveness. Recent studies have demonstrated that introduction of oncogenic KRAS into pancreatic ductal epithelial cells results in an increase in cell size and decrease/loss of epithelial cell E-cadherin expression.24 Additionally, in vitro and in vivo studies demonstrated that knockdown of KRAS results in a marked increase in E-cadherin transcript and protein expression.27 In addition, downregulation of AKR1B10 results in a conversion of retinol to retinal and further to retinoic acid that leads to cell differentiation and increase of E-cadherin transcript and protein expression. Our finding further demonstrates that silencing of AKR1B10 expression results in marked decreases in membrane-bound KRAS expression, significant increases in E-cadherin expression and inhibition of cell proliferation. These data demonstrate that alterations in KRAS expression elicit mitogenic and morphogenic cellular responses via an E-cadherin-mediated pathway.
For carbonyl metabolism, conversion of highly reactive aldehyde and ketone groups into hydroxyl groups by AKR1B10 in neoplastic cells protects against carbonyl-induced apoptosis.21 It has been shown that silencing AKR1B10 in human colonic carcinoma by siRNA approach induces cell apoptosis by regulating lipid synthesis and reducing carbonyls metabolism.21 Similar results were observed in the present studies that knocking down AKR1B10 expression in a human pancreatic adenocarcinoma cell line (Panc10.05) induced cell apoptosis and inhibited cell proliferation, suggesting AKR1B10 could be a therapeutic target for pancreatic cancer.
Another key finding was that AKR1B10 was not only overexpressed in pancreatic adenocarcinoma, but also in pancreatic intraepithelial neoplasia lesions, indicating that AKR1B10 is involved in the early stage of pancreatic carcinogenesis. AKR1B10 is the most efficient retinal reductase;7, 9, 16, 18, 19, 20 the conversion of retinal to retinol by AKR1B10 results in suppression of the conversion of retinal to retinoic acid—the major active antineoplastic metabolite. Chronic pancreatitis is a well-recognized risk factor of pancreatic cancer.9, 30, 33, 34 Chronic pancreatitis-associated malabsorption is common clinically. Malabsorption leads to a fat-soluble vitamin deficiency including vitamin A,45, 46, 47, 48 which further promotes carcinogenesis. Clinical trials have indicated that the retinol esters and carotenoids have efficacy against carcinogenesis in populations deficient in sources of vitamin A.49 As AKR1B10 is overexpressed in precancerous pancreatic intraepithelial neoplasia lesions, targeting of AKR1B10, particularly the combination of fat-soluble vitamin supplementation, could be an efficient approach for the prevention of pancreatic cancer.
In conclusion, AKR1B10 expression was found to be significantly increased in precursor lesions and infiltrating adenocarcinomas of the pancreas. Silencing of AKR1B10 expression was associated with increased apoptosis, decreased protein prenylation, and decreased activation of KRAS and several of its downstream effectors. Taken together, these findings strongly suggest that inhibition of AKR1B10, and subsequent inhibition of protein prenylation including KRAS and its downstream pathway, as well as the induction of cell apoptosis, may serve as a highly promising target for future studies focusing on the development of treatment modalities and prevention strategies of the malignant and highly lethal neoplasm-pancreatic adenocarcinoma.
References
Cao D, Fan ST, Chung SS . Identification and characterization of a novel human aldose reductase-like gene. J Biol Chem 1998;273:11429–11435.
Hyndman DJ, Flynn TG . Sequence and expression levels in human tissues of a new member of the aldo-keto reductase family. Biochem Biophys Acta 1998;1399:198–202.
Liu Z, Zhong L, Krishack PA, et al. Structure and promoter characterization of aldo-keto reductase family 1 B10 gene. Gene 2009;437:39–44.
Penning TM, Drury JE . Human aldo-keto reductases: functions, gene regulation, and single nucleotide polymorphisms. Arch Biochem Biophys 2007;464:241–250.
Fukumoto S, Yamauchi N, Moriguchi H, et al. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers' non-small cell lung carcinomas. Clin Cancer Res 2005;11:1776–1785.
Blackford A, Parmigiani G, Kensler TW, et al. Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res 2009;69:3681–3688.
Gallego O, Ruiz F, Ardèvol A, et al. Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc Natl Acad Sci USA 2007;104:20764–20769.
Ma J, Yan R, Zu X, et al. Aldo-keto reductase family 1 B10 affects fatty acid synthesis by regulating the stability of acetyl-CoA carboxylase-alpha in breast cancer cells. J Biol Chem 2008;283:3418–3423.
Quinn AM, Harvey RG, Penning TM . Oxidation of PAH trans-dihydrodiols by human aldo-keto reductase AKR1B10. Chem Res Toxicol 2008;21:2207–2215.
Endo S, Matsunaga T, Mamiya H, et al. Kinetic studies of AKR1B10, human aldose reductase-like protein: endogenous substrates and inhibition by steroids. Arch Biochem Biophys 2009;487:1–9.
Martin HJ, Maser E . Role of human aldo-keto-reductase AKR1B10 in the protection against toxic aldehydes. Chem Biol Interact 2009;178:145–150.
Casey P . Lipid modifications of G proteins. Curr Opin Cell Biol 1994;6:219–225.
Marshall CJ . Protein prenylation: a mediator of protein-protein interactions. Science 1993;259:1865–1866.
Zhang FL, Casey PJ . Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem 1996;65:241–269.
Sebti SM, Hamilton AD, (eds). Farnesyltransferase Inhibitors in Cancer Therapy. No. 8. Humana Press: Totowa, 2001;296.
Gao J, Liao J, Yang G-Y . CAAX-box protein, prenylation process and carcinogenesis. Am J Transl Res 2009;1:312–325.
Lowy DR, Willumsen BM . Function and regulation of Ras. Annu Rev Biochem 1993;62:851–891.
Ruiz FX, Gallego O, Ardevol A . et al. Aldo-keto reductases from the AKR1B subfamily: retinoid specificity and control of cellular retinoic acid levels. Chem Bio Interact 2009;178:171–177.
Crosas B, Hyndman DJ, Gallego O, et al. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: consequences for retinoid metabolism. Biochem J 2003;373:973–979.
Gallego O, Belyaeva OV, Porté S, et al. Comparative functional analysis of human medium-chain dehydrogenases, short-chain dehydrogenases/reductases and aldo-keto reductases with retinoids. Biochem J 2006;399:101–109.
Wang C, Yan R, Luo D, et al. Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J Biol Chem 2009;284:26742–26748.
Balendiran GK, Martin HJ, El-Hawary Y, et al. Cancer biomarker AKR1B10 and carbonyl metabolism. Chem Bio Interact 2009;178:134–137.
Tzankov S, Wong MJ, Shi K, et al. Functional divergence between co-chaperones of Hsc70. J Biol Chem 2008;283:27100–27109.
Agbunag C, Bar-Sagi D . Oncogenic K-ras drives cell cycle progression and phenotypic conversion of primary pancreatic duct epithelial cells. Cancer Res 2004;64:5659–5663.
Wang X-Q, Li H, Van Putten V, et al. Oncogenic K-ras regulates proliferation and cell junctions in lung epithelial cells through induction of cyclooxygenase-2 and activation of metalloproteinase-9. Mol Biol Cell 2009;20:791–800.
Li JN, Li X, Qian JM, et al. Effects of K-ras gene mutation on colon cancer cell line Caco-2 metastasis by regulating E-cadherin/beta-catenin/p120 protein complex formation and RhoA protein activity. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2010;32:46–50.
Rachagani S, Senapati S, Chakraborty S, et al. Activated KrasG12D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br J Cancer 2011;104:1038–1048.
Siegel R, Ward E, Brawley O, et al. Cancer statistics, 2011. CA Cancer J Clin 2011;61:212–236.
Hidalgo PM, Ferrero HE, Castillo FMJ, et al. Epidemiology and diagnosis of the pancreatic cancer. Rev Esp Enferm Dig 2004;96:714–722.
Michaud DS . Epidemiology of pancreatic cancer. Minerva Chir 2004;59:99–111.
Chang KJ, Parasher G, Christie C, et al. Risk of pancreatic adenocarcinoma: disparity between African Americans and other race/ethnic groups. Cancer 2005;103:349–357.
Lowenfels AB, Maisonneuve P . Epidemiology and prevention of pancreatic cancer. Jpn J Clin Oncol 2004;34:238–244.
Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med 1993;328:1433–1437.
Malka D, Hammel P, Maire F, et al. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 2002;51:849–852.
Penning TM . AKR1B10: A new diagnostic marker of non-small cell lung carcinoma in smokers. Clin Cancer Res 2005;11:1687–1690.
Wang R, Wang G, Ricard MJ, et al. Smoking-induced up-regulation of AKR1B10 expression in the airway epithelium of healthy individuals. Chest 2010;138:1402–1410.
Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 2004;101:6062–6067.
Kim B, Lee HJ, Choi HY, et al. Clinical validity of the lung cancer biomarkers identified by bioinformatics analysis of public expression data. Cancer Res 2007;67:7431–7438.
Endo S, Matsunaga T, Ohta C, et al. Roles of rat and human aldo–keto reductases in metabolism of farnesol and geranylgeraniol. Chem Biol Interact; published online 25 December 2010 [E-pub ahead of print].
Hruban RH, van Mansfeld AD, Offerhaus GJ, et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol 1993;143:545–554.
Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988;53:549–554.
Bos JL . Ras oncogenes in human cancer: a review. Cancer Res 1989;49:4682–4689.
Fernandez C, Chentaille B, Tasei AM, et al. From pancreatic intraepithelial neoplasia to cancer: a dramatic progression with K-ras status analysis. Gastroenterol Clin Biol 2005;29:465–468.
Löhr M, Klöppel G, Maisonneuve P, et al. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia 2005;7:17–23.
Dutta SK, Bustin MP, Russell RM, et al. Deficiency of fat-soluble vitamins in treated patients with pancreatic insufficiency. Ann Intern Med 1982;97:549–552.
Nakamura TTK, Imamura K, Tando Y, et al. Fat-soluble vitamins in patients with chronic pancreatitis (pancreatic insufficiency). Acta Gastroenterol Belg 1996;59:10–14.
Nakamura TTT . Pancreatic steatorrhea, malabsorption, and nutrition biochemistry: a comparison of Japanese, European, and American patients with chronic pancreatitis. Pancreas 1997;14:323–333.
Dutta SK, Bustin MP, Russell RM, et al. Deficiency of fat-soluble vitamins in treated patients with pancreatic insufficiency. Ann Intern Med 1982;97:549–552.
Alberts DS, Garcia DJ . An overview of clinical cancer chemoprevention studies with emphasis on positive phase III studies. J Nutr 1995;125 (3 Suppl):692S–697S.
Acknowledgements
This study is partially supported by CA122514, CA137467 and Adrian Mayer, MD Pancreatic Cancer Research Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Chung, Y., Matkowskyj, K., Li, H. et al. Overexpression and oncogenic function of aldo-keto reductase family 1B10 (AKR1B10) in pancreatic carcinoma. Mod Pathol 25, 758–766 (2012). https://doi.org/10.1038/modpathol.2011.191
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.191
Keywords
This article is cited by
-
Serum AKR1B10 as an indicator of unfavorable survival of hepatocellular carcinoma
Journal of Gastroenterology (2023)
-
A novel model based on disulfidptosis-related genes to predict prognosis and therapy of bladder urothelial carcinoma
Journal of Cancer Research and Clinical Oncology (2023)
-
AKR1B10 accelerates the production of proinflammatory cytokines via the NF-κB signaling pathway in colon cancer
Journal of Molecular Histology (2022)
-
CBX7 suppresses urinary bladder cancer progression via modulating AKR1B10–ERK signaling
Cell Death & Disease (2021)
-
The expression and significance of AKR1B10 in laryngeal squamous cell carcinoma
Scientific Reports (2021)
