
Abstract
IgG4-related tubulointerstitial nephritis (IgG4-TIN), the renal parenchymal lesion of IgG4-related sclerosing disease, is characterized, among other things, by the presence of numerous IgG4-positive plasma cells (IgG4+PC) in the kidney infiltrate. The specificity of this finding for IgG4-TIN has not been addressed. To address this we examined 100 consecutive renal biopsy samples with active interstitial inflammation for the presence of IgG4+PC, and correlated the findings with principal diagnosis, the available clinical histories, and the findings in four biopsy samples of IgG4-TIN. Eleven of the survey biopsy samples contained an average of more than 10 IgG4+PC per × 200 field, including two with IgG4+PC in numbers comparable to those in two of the IgG4-related tubulointerstitial disease biopsy samples. The principal pathological diagnoses in the IgG4+PC-rich cases included anti-neutrophil cytoplasmic antibody-positive necrotizing glomerulonephritis (five cases), diabetic nephropathy (two cases), idiopathic interstitial nephritis (two cases), membranous glomerulonephritis (one case), and lupus nephritis (one case). There was no reason, based on histology or clinical history, to believe that any of these cases represented previously unsuspected IgG4-related tubulointerstitial disease. We conclude that the presence of numerous IgG4+PC is essential to, but not sufficient for, the diagnosis of IgG4-TIN.
Similar content being viewed by others

Main
IgG4-related tubulointerstitial nephritis (IgG4-TIN) is one of a growing list of conditions attributed to the multiple system inflammatory disorder, IgG4-related sclerosing (or systemic) disease (IgG4-SD).1, 2, 3 The first of the IgG4-SD lesions to be described was IgG4-related sclerosing pancreatitis, also known as autoimmune pancreatitis.1 Once thought to be rare, the lesions of IgG4-SD are recognized with increasing frequency in many sites (Table 1).2, 3, 4, 5
The presence of numerous IgG4-positive plasma cells (IgG4+PC) in the inflammatory infiltrate is a histological hallmark of IgG4-SD.1, 2, 3, 4 Case reports consistently emphasize this finding in IgG4-TIN,6, 7 but there have been no detailed studies of plasma cell IgG isotype-labeling in other settings of interstitial nephritis to determine the specificity of this finding. Furthermore, little is known about whether or to what extent unrecognized variants of IgG4-TIN might account for some cases of idiopathic interstitial nephritis seen in a renal biopsy service. To help address these questions, we determined the prevalence and numbers of IgG4+PC in a survey of 100 kidney biopsy samples exhibiting active interstitial inflammation, regardless of the principal pathological diagnoses. We then correlated the findings with other histological and clinical features of the cases, and with cases of IgG4-TIN.
Materials and methods
This study was approved by the Institutional Review Board at Oregon Health & Sciences University (IRB00006441). The renal biopsy pathology reports prepared by either of the two authors from 1 January 2009 through 30 June 2010 (18 months) were searched for cases exhibiting sufficient acute or chronic active interstitial nephritis to be identified in the diagnosis or to warrant comment. Hematoxylin and eosin (H&E)-stained slides from these cases were reviewed to confirm that they showed one or more of the features of active inflammation listed below. Samples were excluded that were processed in an outside laboratory, or which provided fewer than 10 × 400 fields in the original sections or in the re-cuts prepared for immunohistochemistry studies. A total of 100 cases were identified. Four biopsy samples from three patients with IgG4-TIN, and documented IgG4-SD, were also studied. These included two cases provided for this purpose by Dr Kelly Smith at the Department of Pathology, University of Washington School of Medicine. The clinical findings of IgG4-SD in these patients, besides IgG4-TIN, included plasma cell-rich bilateral sialadenitis, lymphadenopathy, and membranous glomerulonephritis (MGN), in one patient, painless submandibular gland enlargement and chronic pancreatitis in another, and polyclonal gammopathy and hypocomplementemia in the third.
All samples were reviewed and scored without reference to the indications for biopsy or other clinical information, and without reference to the original pathology report. This blinded approach was not possible in the review of the IgG4-TIN cases, which were already familiar to both reviewers. The evaluations were conducted independently by two observers using microscopes with identical optics; differences were resolved by consensus at a double-headed microscope. After scoring the histopathology and immunostains, the findings were correlated with the diagnoses and clinical histories.
Histological Review
Inflammatory infiltrates were evaluated in sections stained with H&E. Criteria for active interstitial nephritis included the prominence of neutrophils, eosinophils, plasma cells, or macrophages, or a combination, in the infiltrate, accompanied by acute tubular injury, tubulitis, and/or edema. The estimated proportion of plasma cells in each infiltrate was judged to be rare or absent, moderate (1 to 25%), or high (more than 25%). The presence of active medullary inflammation, obliterative or sclerosing lesions, and evidence of a sharp lesional margin were also noted.
Immunohistochemistry Studies
Re-cut paraffin sections from each of the study cases were stained for IgG4 to identify IgG4+PC (IgG4 primary antibody: The Binding Site #MC011 (San Diego, CA, USA) dilution 1:1500, after cc1 mild pretreatment on Ventana XT Instruments (Tucson, AZ, USA), followed by Ventana Ultraview detection), and for the pan-plasma cell marker, CD138 (CD138 primary antibody: Cell Marque clone B-A38 predilute, after cc1 mild pretreatment, with detection as above). Each of these sections was evaluated for the numbers and distribution patterns of the labeled cells. All IgG4+PC in each biopsy sample were counted and expressed as an average number per 20 × 10 ( × 200) field, rounded to the nearest integer. Each case was then classified, based on the average IgG4+PC counts, as rare (fewer than two labeled cells per × 200 field), moderate (2 to 10 labeled cells per × 200 field), or numerous (more than 10 labeled cells per × 200 field). Individual IgG4+PC became difficult to resolve in two IgG4+PC-rich survey cases with cell densities exceeding 100 cells per × 200 field. In these cases, an estimate of >100 IgG4+PC per × 200 field is recorded. IgG4+PC numbers were also counted in a 40 × 10 ( × 400) field in a high-density area in the IgG4+PC-rich survey cases, and in the four IgG4-TIN cases. Even at × 400, the IgG4+PC were too dense to be counted accurately in two of the IgG4-TIN cases. Each case was also classified into one of three groups based on the average numbers of CD138+ cells: low (fewer than 20 labeled cells per × 200 field); moderate (20 to 100 labeled cells per × 200 field); and high (more than 100 labeled cells per × 200 field).
Results
Altogether, 100 consecutive native kidney biopsy samples with active interstitial nephritis were identified for this survey. Table 2 summarizes the results of IgG4+PC classifications by principal pathological diagnosis. Among these cases, IgG4+PC were absent or rare in 63 cases; were present in moderate numbers (2 to 10 cells per × 200 field) in 26 cases, and were numerous (13 to more than 100 cells per × 200 field) in 11 cases. Two cases were judged to contain at least 100 IgG4+PC per × 200 field. Only two biopsy samples in this group, both from lupus patients, exhibited tubulointerstitial deposits of IgG by immunofluorescence microscopy (IF) at the time of the original biopsy evaluation. Subsequent testing documented that these deposits did not include IgG4. The principal pathological diagnoses in the 11 cases with numerous IgG4+PC included: necrotizing and crescentic glomerulonephritis (five cases), diabetic nephropathy (two cases), active lupus nephritis (one case), MGN (one case), and idiopathic chronic active interstitial nephritis (two cases). As described below, we concluded that it is unlikely that any of these IgG4+PC-rich interstitial nephritides represented a previously unrecognized case of IgG4-TIN.
Necrotizing and Crescentic Glomerulonephritis
A total of 43 biopsy samples exhibiting NCGN were evaluated in our laboratory during the study period. Sixteen cases had interstitial nephritis and otherwise met criteria for inclusion in the study, five of which contained numerous IgG4+PC, with cell counts ranging from 16 to more than 100 per × 200 field. Each of these five patients had an anti-neutrophil cytoplasmic antibody (ANCA). These included a 19-year-old man with a ventriculoperitoneal shunt (49 IgG4+PC per × 200 field) and a 63-year-old man with pneumonia (30 IgG4+PC per × 200 field), both with granular glomerular IgG deposits by IF and anti-myeloperoxidase ANCA (MPO-ANCA). Both patients were felt likely to have forms of post-infectious glomerulonephritis. A 66-year-old woman (75 IgG4+PC per × 200 field) had NCGN with linear glomerular capillary IgG deposits by IF, and both an MPO-ANCA and an anti-glomerular basement membrane antibody. Two patients, 57- and 72-year-old men (16 and >100 IgG4+PC per × 200 field), had pauci-immune NCGN by IF, the first with an anti-proteinase 3 ANCA (PR3-ANCA), and the other an MPO-ANCA. Neither had evidence of vasculitis.
In the remaining 11 NCGN survey cases, the numbers of IgG4+PC ranged from 0 to 5 per × 200 field. They included two biopsy samples with granular deposits, both in patients who were ANCA-negative. One was a 66-year-old man with clinical Henoch-Schönlein purpura and IgA-dominant mesangial deposits, and the other was an 11-year-old girl with mesangial deposits of IgG, C3, and C1q who presented with an acute viral syndrome. She was ultimately judged to have post-infectious glomerulonephritis. Among the other biopsy samples in this group, three exhibited linear capillary wall IgG deposits; one with an MPO-ANCA and two who were ANCA-negative, including one with necrotizing arteritis. Six biopsy samples demonstrated pauci-immune necrotizing glomerulonephritis without arteritis. Four of these patients had an MPO-ANCA; one had a PR3-ANCA; and one was ANCA-negative.
Membranous Glomerulonephritis
Among 55 cases of MGN seen during the study period, eight met inclusion criteria. On the basis of the available clinical histories, a concurrent drug hypersensitivity reaction was suspected in four of these cases. MGN with interstitial nephritis was also observed in a patient with hepatitis B. The patient with MGN and IgG4+PC-rich interstitial nephritis (13 per × 200 field) was an 81-year-old man with rheumatoid arthritis, a monoclonal gammopathy, and a normal bone marrow biopsy. He had received gold therapy in the remote past, but was not taking prescribed medications during the time he developed nephrotic syndrome. A review of his past medical history and symptoms with his nephrologist revealed nothing to suggest an IgG4-related disorder. Shortly after the biopsy he moved his household and sought the care of a physician in another community, and subsequently died in unknown clinical circumstances.
Diabetic Nephropathy
Diabetic nephropathy was the principal finding in 93 biopsy samples seen during the study period. Mild interstitial inflammation was present in most of these samples, but eight were found to have an interstitial nephritis prominent enough to warrant a separate diagnosis, including two with high IgG4+PC numbers (16 and 17 per × 200 field). Both patients had long histories of diabetes and hypertension, and related complications, and neither had known medical histories to suggest IgG4-SD.
Interstitial Nephritis
Of the 100 biopsy samples in the study, 24 had active interstitial nephritis as the principal finding. As noted in Table 2, these cases represented several clinicopathological groups: probable drug hypersensitivity reaction; reflux nephropathy, Sjogren's syndrome, tubulointerstitial nephritis-uveitis syndrome, and idiopathic acute or chronic active interstitial nephritis. Only two biopsy samples with numerous IgG4+PC were identified among these 24 cases, both of them from the idiopathic group. Both exhibited extensive, dense interstitial infiltrates with conspicuous fibrosis and numerous plasma cells. Because IgG4-TIN was considered during the original work-ups of both cases, brief clinical summaries are provided below.
Case 1
This 55-year-old woman, a long-haul truck driver, presented with a creatinine of 3.0 mg/dl (266 μmol/l), pyuria and hematuria, and 3 g of proteinuria per 24 h. Her renal function had been normal 6 years earlier. In addition to the infiltrates, her biopsy sample exhibited severe arterionephrosclerosis and was suspicious for focal and segmental glomerulosclerosis. Her plasma cell infiltrate included 20 IgG4+PC per × 200 field. Immunofluorescence microscopy was negative for tubulointerstitial immune deposits. She had not been taking prescription medications, but had used a variety of unspecified proprietary drugs, and herbal medications and supplements for many years. Aristolochia exposure could not be documented. She offered no history or complaints suggestive of pancreatic or salivary gland inflammatory disease or other lesions of IgG4-SD, and serum IgG subclass levels were normal before treatment. A course of steroid treatment was initiated, but she failed to keep appointments or respond to telephone enquiries afterward, and she was lost to follow-up until recently, when she presented to her physician with advanced chronic kidney disease.
Case 2
This 80-year-old woman's serum creatinine was found to have risen to 3.3 mg/dl (293 μmol/l) from a baseline of 0.8 mg/dl (71 μmol/l) six months earlier. During that period she had experienced increasing weakness, fatigue, and abdominal pain, and she had become anemic. Her regular medications were losartin, lorazepam, and omeprazole. Abdominal computerized tomography (CT) was negative, except for a suspected renal infarct. A bone marrow biopsy sample was non-diagnostic. She had a low-titer ANA and a negative anti-double-stranded DNA. A kidney biopsy sample at that time demonstrated diffuse, dense lymphoplasmacytic inflammation, with numerous eosinophils. Immunofluorescence microscopy was negative for tubulointerstitial immune deposits. The infiltrate included more than 100 IgG4+PC per × 200 field (Figures 1a and b). The principal diagnostic considerations at the time were an adverse drug reaction and IgG4-related TIN, but a single small knot of extracapillary cells in one glomerulus and the suspected renal infarct also prompted suspicion of vasculitis. Studies for serum IgG subclasses shortly after the biopsy demonstrated low IgG3 and IgG4 levels. She was found to have a PR3-ANCA and she was treated empirically for a microscopic vasculitis with steroids and cyclophosphamide. Her renal function stabilized during treatment, but further details of her clinical history are not available.

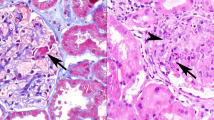
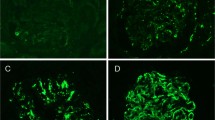
Illustrative histology and immunohistochemistry findings. (a) Sclerosing idiopathic interstitial nephritis with a plasma cell-rich interstitial infiltrate. As described in the text, this patient was subsequently treated for anti-neutrophil cytoplasmic antibody (ANCA)+microscopic vasculitis. Note that inspite of dense sclerosis, tubular elements are still identifiable in most areas. (b) IgG4 immunohistochemical stain of the biopsy in panel (a) showing IgG4-positive plasma cells (IgG4+PC) distributed in focal, dense clusters. (c) IgG4-related tubulointerstitial nephritis (IgG4-TIN) with sclerosing lymphoplasmacytic inflammation. Note the uniformity of the process and apparent obliteration of the tubules. (d) IgG4 immunohistochemical stain of the biopsy illustrated in panel (c) showing moderate numbers of IgG4+PC scattered throughout the tissue. (e) The interface between IgG4-TIN and adjacent tissue forms a sharp margin. Only a few inflammatory cells extend into the intact parenchyma. (f) IgG4 immunohistochemical stain of another biopsy of IgG4-TIN showing a dense infiltrate of IgG4+PC compared with the biopsy illustrated in panel (d). Note the uniform distribution the IgG4+PC in this tissue.
Lupus Nephritis
Sixty-one biopsy samples were received for evaluation of lupus nephritis during the study time frame. Most of these exhibited some degree of interstitial inflammation, but only six warranted a separate diagnosis of active interstitial nephritis, and otherwise met inclusion criteria for the study. Among these, one contained numerous interstitial IgG4+PC (50 per × 200 field). This case was one of three in this group with granular tubulointerstitial immune IgG deposits by IF, which were shown not to include IgG4 deposits.
Correlation of Histological Findings with IgG4+PC Counts
Each sample was evaluated for the presence of acute tubular injury, tubulitis, composition of the infiltrate (prominence of eosinophils, neutrophils, or macrophages, in addition to plasma cells), edema, medullary inflammation, and dense tubulointerstitial fibrosis. Neither the presence nor the absence of any of these features distinguished the IgG4+PC-rich cases among the 100 cases in the survey sample, overall. Acute tubular necrosis, tubulitis, and edema were notable in three, two, and one of the IgG4+PC-rich biopsy samples, respectively, whereas the same features were observed in 18, 17, and 8 of the 100 survey sample cases overall. Neutrophils, eosinophils, and macrophages were particularly prominent in one, four, and none of the IgG4+PC-rich cases, respectively, and in 17, 22, and 2 cases in the entire survey. One of the IgG4+PC-rich cases demonstrated medullary involvement, and four of them exhibited dense fibrosis, whereas similar findings were seen in 5 and 18 cases, respectively, among those in the entire survey. Moreover, except for the presence of numerous eosinophils in three of the four IgG4-TIN biopsy samples, the principal features of active interstitial inflammation in these cases were numerous plasma cells and variable edema.
The numbers and proportions of plasma cells in the survey cases, whether determined in H&E-stained sections or in CD138-stained sections, correlated only modestly with the numbers of IgG4+PC. Among the 11 cases with highest IgG4+PC counts, plasma cells numbers seen in the H&E-stained sections were judged to be rare in three (27%), moderate in five (46%), and high in three (27%), compared with 64%, 30%, and 6%, respectively, in the 100 study cases, overall. When the 11 IgG4+PC-rich cases were examined using the CD138 stain, plasma cell numbers were low in two (18%), moderate in two (18%), and high in seven (64%) cases, compared with 52%, 30%, and 18%, respectively, in the study cases, overall. Plasma cells were judged to be numerous in all of the IgG4-TIN cases by both methods. The proportions of IgG4+PC in plasma cell infiltrates varied considerably in both survey cases and IgG4-TIN cases, although they were never judged to comprise more than half of any plasma cell population. Because of this variation, the CD138 stain added little to the modest information provided by H&E-stained sections alone in distinguishing IgG4+PC-rich infiltrates.
Comparisons with IgG4-TIN Cases
Striking histological differences distinguished the reference IgG4-TIN cases from the IgG4+PC-rich interstitial infiltrates identified in this survey. In the IgG4-TIN cases, sclerosis consisted of a uniform, virtually uninterrupted, scar-like matrix with obliteration of most tubules, and isolation of glomeruli (Figures 1c and e). The densities of the infiltrates varied among these four biopsy samples, but in all instances, the inflammatory cells were more or less evenly distributed through the lesional tissue, typically including plasma cell clusters. Geographic or zonal involvement in the kidney was evident by the presence of both wholly affected and entirely normal tissue cores in one case, and by a sharp line of demarcation between lesional tissue and normal parenchyma in the same core in another (Figure 1e). Eosinophils were numerous in three of the four IgG4-TIN biopsy samples. In contrast to these findings, the fibrosis and inflammation in the IgG4+PC-rich survey cases were distributed in patchy or reticular patterns, typically forming streaks that separated tubules and segregated islands of normal parenchyma. Even where inflammation and fibrosis were diffuse, they did not entirely obscure or replace the atrophic tubules (Figures 1a and b). There was no evidence of sharp zonal or geographic renal involvement in any of these non-IgG4-TIN cases.
IgG4+PC were generally, though not always, much more numerous and more densely distributed in the IgG4-TIN cases than in the IgG4+PC-rich survey cases. All, but two, of the IgG4+PC-rich survey biopsy samples contained focal IgG4+PC aggregates ranging from 10 to 65 cells per × 400 field, whereas all of the IgG4-TIN cases exhibited a relatively uniform density of IgG4+PC, with at least 100 cells per × 400 field. The least cellular of the IgG4-TIN cases, however, and the two most cellular IgG4+PC-rich survey cases had IgG4+PC infiltrates with comparable maximum density, all slightly in excess of 100 cells per × 400 field (Figures 1b and d). By contrast, the most cellular of the IgG4-TIN cases were estimated to contain several hundreds of IgG4+PC per × 400 field (Figure 1f).
Discussion
In published reports, most cases of IgG4-TIN accompany other lesions of IgG4-SD5, 6, 7, 8, 9, 10, 11 (Table 1), particularly those in pancreas, hepatobiliary system, lymph nodes, and/or salivary glands. This has been our experience as well, but in all of the IgG4-TIN reference biopsy samples reviewed in this study, IgG4-SD first came to light as a result of the kidney biopsy. In IgG4-TIN, renal biopsy is usually contemplated to evaluate an asymptomatic rise in serum creatinine, proteinuria, renal enlargement, and/or focal renal parenchymal lesions.6, 7, 8, 9, 10, 11 As with other forms of IgG4-SD, IgG4-TIN is often accompanied by serological findings consistent with an immune complex disorder, including marked elevation of serum IgG, particularly IgG4,6, 8, 9, 10, 11 the presence of ANA and other autoantibodies,6, 8, 11 and hypocomplementemia.6, 8, 10, 11 When examined by enhanced CT, the renal parenchymal lesions are typically focal or multifocal and expansile, sometimes mimicking cancer.5, 6 Histologically, IgG4-TIN is characterized by the presence of IgG4+PC-rich lymphoplasmacytic infiltrates, obliterative, expansile sclerosis,6, 7, 8 and often by peritubular and/or interstitial immune deposits, including IgG4 deposits.6 Numerous IgG4+PC are virtually always present in the infiltrates of IgG4-TIN,6 as they are in other IgG4-SD lesions,2, 3 but the criteria and specificity of this finding for IgG4-TIN has not been examined. Strehl et al12 have recently provided indirect evidence that IgG4+PC counts may not be specific by documenting the presence of numerous IgG4+PC in several inflammatory lesions in non-renal sites, unrelated to IgG4-SD. We therefore sought to determine the incidence of an IgG4+PC-rich infiltrate in a series of renal biopsy samples chosen only for the presence of an active interstitial nephritis, regardless of clinical setting or other pathological diagnoses.
The results of our survey indicate that the presence of numerous IgG4+PC is not specific for IgG4-TIN. Eleven of the biopsy samples in the survey, or more than 10% of these interstitial nephritides, contained infiltrates with 13 or more IgG4+PC per × 200 field. We concluded that none of these cases is likely to be an example of IgG4-TIN because their histology differed substantially from that in the IgG4-TIN reference cases, and none had clinical findings suggestive of IgG4-TIN or other lesions of IgG4-SD. IgG4+PC were much more abundant in IgG4-TIN cases than in most of the survey cases, but there was some overlap between the two groups. For this reason, it may prove to be unproductive to try to establish a threshold density of IgG4+PC to distinguish IgG4-TIN from non-IgG4-TIN cases. Rather, the findings indicate that the significance of numerous IgG4+PC must be evaluated in the context of the histological and clinical findings of the case, as summarized in Table 3.
IgG4-rich infiltrates were found in 5 of 16 NCGN cases in this survey, all of them in patients with either MPO-ANCA or PR3-ANCA, and including pauci-immune glomerular lesions and those with either granular or linear immune deposits. This finding is noteworthy in view of recent interest in the possible role of IgG4 ANCA in the pathogenesis of small-vessel vasculitis,13, 14, 15 but our IgG4+PC-rich cases were not clearly different, based on ANCA status or immunopathological findings, from the 11 NCGN cases in the survey with few or no IgG4+PC. Despite the frequent finding of autoantibodies in patients with IgG4-SD, we are not aware of reports of ANCA in this setting.
Only 2 of 25 biopsy samples showing interstitial nephritis alone were found to have numerous IgG4+PC. These two cases could not be confidently classified as to probable cause, but neither presented with clinical evidence indicative of IgG4-SD, nor exhibited additional histological or IF findings suspicious for IgG4-TIN. One of these patients was found to have a PR3-ANCA following the biopsy, and an unsampled ANCA-associated NCGN was subsequently favored clinically.
The finding of IgG4+PC-rich interstitial inflammation in two patients with diabetic nephropathy is intriguing, given recent onset diabetes sometimes complicates IgG4-related sclerosing pancreatitis.3, 16 However, neither case in this survey had clinical or pathological features to suggest pancreatitis or other lesions of IgG4-SD, and both patients had long histories of complicated diabetes by the time of their biopsies.
The presence of an IgG4+PC-rich interstitial infiltrate in a biopsy sample otherwise exhibiting MGN is interesting in light of the described association of IgG4-SD and MGN,7, 8, 11, 17, 18 and the observation that IgG4 may be important in the pathogenesis of primary MGN.19 The IgG4+PC-rich infiltrate in this case was unaccompanied by other pathological features of IgG4-TIN, and the patient had had no known clinical evidence to suggest IgG4-SD.
The clinical and pathological events that occur during the early stages of the IgG4-TIN lesion are not known. Although we cannot exclude it entirely, we have no basis to believe that any of the IgG4+PC-rich cases identified in this survey is a mild or atypical form of IgG4-TIN. This suggests that IgG4-TIN is not often the cause of idiopathic nephritis, but additional studies will be necessary to determine its incidence in this population, and to better define the earliest form of this disorder. It has been suggested that IgG4-SD lesions evolve from a highly cellular inflammatory process toward dense sclerosis,3, 6 and that immune complex deposits are a relatively late manifestation of the disorder.6 The pathogenesis of IgG4-SD is not known, including whether IgG4 production is a primary or secondary phenomenon, or even whether its role is to mitigate rather than instigate the inflammatory process.6, 20 It is plausible, therefore, that the earliest IgG4-TIN lesion contains relatively few IgG4+PC.
In conclusion, the presence of an IgG4+PC-rich interstitial infiltrate is a relatively common finding in various settings of interstitial nephritis, particularly in NCGN, and is not specific for IgG4-TIN. Evaluation of interstitial inflammatory infiltrates for the presence of IgG4+PC is a useful ancillary study primarily when there are other clinical and pathological features suggestive of IgG4-TIN (Table 3), but may be unrevealing in the majority of biopsy samples with active interstitial nephritis.
References
Kamisawa T, Okamoto A . IgG4-related sclerosing disease. World J Gastroenterol 2008;14:3948–3955.
Bateman AC, Deheragoda MG . IgG4-related systemic sclerosing disease – an emerging and under-diagnosed condition. Histopathol 2009;55:373–383.
Cheuk W, Chan JKC . IgG4-related sclerosing disease. A critical appraisal of an evolving clinicopathological entity. Adv Anat Pathol 2010;17:303–332.
Zamboni G, Capelli P, Scarpa A, et al. Nonneoplastic mimickers of pancreatic neoplasms. Arch Pathol Lab Med 2009;133:439–453.
Takahashi N, Kawashima A, Fletcher JG, et al. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology 2007;242:791–801.
Cornell LD, Chicano SL, Deshpande V, et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol 2007;31:1586–1597.
Watson SJW, Jenkins DAS, Bellamy COS . Nephropathy in IgG4-related systemic disease. Am J Surg Pathol 2006;30:1472–1477.
Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int 2010;78:1016–1023.
Murashima M, Tomaszewski J, Glickman JD . Chronic tubulointerstitial nephritis presenting as multiple renal nodules and pancreatic insufficiency. Am J Kid Dis 2007;49:E7–E10.
Yoneda K, Murata K, Katayama K, et al. Tubulointerstitial nephritis associated with IgG4-related autoimmune disease. Am J Kid Dis 2007;50:455–462.
Hill P, Russell P, Sammartino C, et al. Acute kidney injury and proteinuria in a patient with diabetes and a submandibular mass. Am J Kid Dis 2009;54:375–380.
Strehl JD, Hartman A, Abbas A . Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol 2011;64:237–243.
Brouer E, Cohen Tervaert JW, Horst G, et al. Predominance of IgG1 and IgG4 subclasses of anti-neutrophil cytoplasmic autoantibodies (ANCA) in patients with Wegener's granulomatosis and clinically related disorders. Clin Exp Immunol 1991;83:379–386.
Holland M, Hewins P, Goodall M, et al. Anti-neutrophil cytoplasmic antibody IgG subclasses in Wegner's granulomatosis: a possible pathogenic role for the IgG4 subclass. Clin Exp Immunol 2004;138:183–192.
Hussain A, Pankhurst T, Goddall M, et al. Chimeric IgG4 PR3-ANCA induces selective inflammatory responses from neutrophils through engagement of Fcγ receptors. Immunology 2009;128:236–244.
Uchida K, Okazaki K, Konishi Y, et al. Clinical analysis of autoimmune pancreatitis. Am J Gastroenterol 2000;95:2788–2794.
Saeki T, Imai N, Ito T, et al. Membranous nephropathy associated with IgG4-related systemic disease and without autoimmune pancreatitis. Clin Nephrol 2009;71:173–178.
Uchiyama-Tanaka Y, Mori Y, Kimura T, et al. Acute tubulointerstitial nephritis associated with autoimmune-related pancreatitis. Am J Kid Dis 2004;43:E13–E25.
Beck LH, Bonegio RGB, Lambeau G, et al. M-Type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2009;361:11–21.
Aalberse RC, Schuurman J . IgG4: breaking the rules. Immunology 2002;105:9–19.
Acknowledgements
We are grateful to Lihn Matsamura for her excellent immunohistochemistry preparations for this study, and to Dr Kelly Smith of the Department of Pathology, University of Washington School of Medicine for providing two of the reference biopsy samples of IgG4-related tubulointerstitial nephritis we used in the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Houghton, D., Troxell, M. An abundance of IgG4+ plasma cells is not specific for IgG4-related tubulointerstitial nephritis. Mod Pathol 24, 1480–1487 (2011). https://doi.org/10.1038/modpathol.2011.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.101
Keywords
This article is cited by
-
ANCA-associated kidney disease preceded by orbital pseudotumor
Pediatric Nephrology (2024)
-
Utility of CD138/syndecan-1 immunohistochemistry for localization of plasmacytes is tissue-dependent in B6 mice
BMC Research Notes (2022)
-
Clinicopathological analysis of ANCA-associated glomerulonephritis focusing on plasma cell infiltrate
Clinical and Experimental Nephrology (2019)
-
Evaluation of diagnostic criteria for IgG4-related tubulointerstitial nephritis
Diagnostic Pathology (2015)
