
Abstract
Inflammatory myofibroblastic tumor is a rare mesenchymal neoplasm that harbors an anaplastic lymphoma kinase (ALK) gene rearrangement in the majority of cases. It is composed of fibroblastic–myofibroblastic cells with a characteristic inflammatory infiltrate that consists predominantly of plasma cells. In contrast, IgG4-related sclerosing disease is a recently described multisystem disorder with a histological appearance similar to inflammatory myofibroblastic tumor. The plasma cell infiltrate is characteristic in IgG4-related sclerosing disease and has been studied as a tool to render this diagnosis. Histologically, the two disorders overlap, although there are significant clinical differences. This study analyzes the histological appearance of 36 inflammatory myofibroblastic tumors, compares them with IgG4-related sclerosing disease, and assesses the plasma cell profile using immunohistochemistry to determine the range and proportion of IgG4 plasma cells. The majority of patients were children and young adults, mainly with solitary masses and no clinical manifestations of IgG4-related sclerosing disease. ALK-1 positivity was present in 23 cases (64%). None showed obliterative phlebitis or prominent lymphoid aggregates. Of 36 inflammatory myofibroblastic tumors, 15 cases showed an IgG4/IgG ratio ≥0.10, a cutoff described in the literature as supportive of IgG4-related sclerosing disease and up to 33 IgG4-positive plasma cells per high-power field indicating a mild-to-moderate increase as compared with IgG4-related sclerosing disease. Currently, the diagnostic recognition of inflammatory myofibroblastic tumor is based on clinicopathological features and diagnostic adjuncts, such as ALK-1 reactivity and genetic tests. Although inflammatory myofibroblastic tumor and IgG4-related sclerosing disease are distinct entities, a subset of inflammatory myofibroblastic tumors exhibit an IgG4/IgG ratio that is within the range for IgG4-related sclerosing disease. Therefore, the ratio alone cannot be used as a reliable discriminator between these two entities and other clinical and pathologic features must always be taken into account.
Similar content being viewed by others

Main
Inflammatory myofibroblastic tumor (IMT) is a rare mesenchymal neoplasm of intermediate biologic potential that typically occurs in the soft tissue or viscera of children and young adults and harbors an anaplastic lymphoma kinase (ALK) gene rearrangement in more than half of cases.1, 2, 3, 4, 5 It is composed of fibroblasts and myofibroblasts accompanied by an inflammatory infiltrate of plasma cells with variable lymphocytes and eosinophils (see Figure 1).6 A reported 15–30% of patients have a clinical and laboratory syndrome of fever, malaise, weight loss, anemia, elevated erythrocyte sedimentation rate, thrombocytosis, polyclonal hypergammaglobulinemia, and other inflammatory marker elevations.7, 8, 9, 10, 11, 12, 13

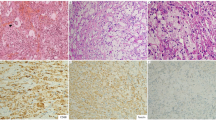
(a) Inflammatory myofibroblastic tumor showing intersecting fascicles of spindled myofibroblasts with intervening, pleomorphic ganglion-like cells and interspersed lymphocytes and plasma cells, H & E, × 20 magnification. (b) ALK immunohistochemical stain showing granular cytoplasmic staining in the spindled cells, × 40. (c) SMA immunohistochemical stain with strong, cytoplasmic positivity, × 40. (d). IgG4 immunohistochemical stain highlighting plasma cells within the inflammatory infiltrate, × 40.
IgG4-related sclerosing disease (IgG4SD) is a steroid-responsive multiorgan system disorder that encompasses a spectrum of clinical conditions, including autoimmune pancreatitis, sclerosing cholangitis, chronic sclerosing sialadenitis, tubulointerstitial nephritis, cutaneous pseudolymphoma, and sclerosing processes in the lung and pleura, liver, breast, and meninges.5, 14, 15, 16, 17 The histological findings of fibrosis and chronic inflammation are relatively nonspecific, and there are no known unique genetic markers. A large proportion of the plasma cells in IgG4SD express IgG4 antibody, a finding that in recent literature has been the subject of interest as a pathologic discriminator for IgG4SD. The morphological overlap between IMT and IgG4SD with abundant plasma cells has raised the question of whether the two disorders might be closely related in their pathogenesis or part of a clinicopathological continuum rather than separate entities.
This study of 36 IMT assesses the plasma cell profile with IgG and IgG4 immunohistochemistry in the context of the clinical and pathological features, outcome, and ALK-1 expression. The results are compared with published reports of IgG4SD to investigate the extent of overlap and potential distinguishing features of the two diseases.
Materials and methods
A total of 36 IMTs were studied. Cases that met the WHO criteria for a diagnosis of IMT6 were obtained from consultation files of the authors and from surgical pathology archives at Vanderbilt University Medical Center, 2002–2009. A total of 26 cases had been published previously.18, 19 All available surgical pathology reports, consultation reports, microscopic slides, and cytogenetic results were reviewed. Medical records and follow-up information were reviewed when available. Immunohistochemistry was performed on formalin-fixed paraffin-embedded tissue sections using standard techniques. Table 1 lists the antigen or antibody, clone, source, dilutions, and antigen retrieval conditions. All slides were counterstained with hematoxylin. Appropriate positive and negative controls were used for each antibody. All stains were interpreted as positive or negative, with positive staining noted as focal or diffuse. Positive ALK-1 staining cases were further assessed for the pattern: cytoplasmic, granular, or nuclear.
IgG and IgG4 plasma cells were counted per high-power field (HPF, × 40 objective, × 10 eyepiece) in the area of highest density of positive cells. A total of six HPF were counted and averaged to one HPF. The IgG4/IgG plasma cell ratio was calculated. As in the previous literature about IgG4SD, an IgG4/IgG plasma cell ratio equal to or >0.10 was used as a discriminator between low and high IgG4 plasma cells.20
FISH for ALK rearrangement was not performed for this study. Results from cytogenetics reports done as part of a previous study were reviewed.3
Institutional Review Board approval was obtained from Vanderbilt University.
Results
The clinicopathological features and outcome of 36 IMTs are shown in Table 2. The 17 male and 19 female patients ranged in age at diagnosis from 6 months to 41years, with a mean age of 13.3 years. In all, 44% (16 cases) occurred between birth and 9 years, 31% (11 cases) between 10 and 19 years, the remaining cases occurred at 20 years of age or later. The anatomic sites were widely distributed, with 47% (17 cases, including two multinodular IMTs) in the mesentery, 25% in the lung (7 cases), chest (1 case), or upper respiratory tract (1 case), 14% in the pelvis (3 cases) or peritoneum (2 cases), 11% (4 cases) in the urinary bladder, and 3% in the distal extremity (1 case). A total of 11 patients had concurrent clinical and laboratory features of the inflammatory syndrome associated with IMT, 5 patients lacked these syndromic manifestations, and 20 cases had incomplete information about its presence or absence. A total of 30 patients (83%) were treated with surgery only, 4 (11%) with surgery plus chemotherapy, 1 (3%) with radiation therapy in addition to surgery and chemotherapy, and 1 (3%) with prednisone. Information about outcome was available for 29 patients at follow-up intervals of 0.1 to 18 years (mean 3 years). In all, 62% (18 cases) had no evidence of recurrence. In all, 31% (11 cases) showed recurrence, of which two patients later developed metastases and one of those succumbed to the disease.
Pathologically, the tumor diameter ranged from 2 to 41 cm, with a mean of 7.5 cm, and 86% (31 cases) ≤10 cm in diameter. Two mesenteric IMTs had a multinodular growth pattern confined to the mesentery. Histologically, all met morphological criteria for IMT, with sheets and fascicles of spindled and polygonal ganglion-like cells intermingled with an inflammatory infiltrate of plasma cells, lymphocytes, variable eosinophils, and rare neutrophils. Acute inflammation occurred mainly in tumors associated with mucosal ulceration. Obliterative phlebitis and prominent lymphoid aggregates were absent.
Immunohistochemical analysis revealed ALK-1 positivity in 64% (23 cases), with an approximately even distribution among cytoplasmic and granular staining patterns. None of the cases had a nuclear membrane pattern of staining. Smooth muscle actin reactivity was present in 76% (25 of 33 cases tested). Information about FISH for ALK was available for 19 cases, with 74% showing an ALK split.
IgG and IgG4 immunohistochemical results were both available for 30 cases, with the remaining 6 cases showing undetectable or equivocal reactivity. The number of IgG-positive plasma cells per HPF ranged from 3 to 85 in 30 cases. The overall mean was 22 IgG-positive plasma cells per HPF. IgG4-positive plasma cells ranged from 1 to 33 per HPF in 23 cases, with an overall mean of 7 per HPF. A total of 14 cases had no IgG4 plasma cells. The IgG4/IgG ratio ranged from 0.02 to 0.94 in 21 cases. A total of 10 other cases had a ratio of 0 and one case was excluded for technical reasons. In addition, five other cases were excluded from calculation (Table 3). Among the 20 cases with positive ratios, 50% had an IgG4/IgG ratio ≥to 0.10 and 17% had a ratio lt&;0.10. There was no significant difference in the IgG4/IgG ratio between ALK-1-positive versus ALK-1-negative IMT (P=0.64). Serum IgG4 levels were not available on any cases.
Discussion
IMT and IgG4SD are both fibro-inflammatory proliferations that are included in the larger constellation of heterogenous lesions often known as the so-called ‘inflammatory pseudotumors’. Inflammatory myofibroblastic tumor is currently classified as an intermediate, rarely metastasizing neoplasm composed of myofibroblasts and fibroblasts accompanied by an inflammatory infiltrate of plasma cells, variable lymphocytes, and eosinophils. Approximately 50–70% of the tumors harbor an ALK gene rearrangement or ALK aneuploidy, which is detectable by immunohistochemistry, conventional cytogenetics, fluorescence in situ hybridization, or reverse transcriptase polymerase chain reaction.1, 2, 3, 6, 21, 22, 23 Most patients with IMT are children, adolescents, or young adults, although the tumor can occur throughout life.6, 7 Inflammatory myofibroblastic tumor originates anywhere in the body, but has a predilection for the abdominal cavity, pelvis, retroperitoneum, lungs, mediastinum, and upper respiratory tract.14, 15, 16, 17, 24
In contrast to IMT, IgG4SD is an inflammatory disorder that is accompanied by an elevated serum IgG4 and is histologically characterized by an infiltrative inflammatory fibro-sclerosing process that usually contains abundant IgG4 positive plasma cells. According to current understanding, IgG4SD affects various organs such as the lungs, liver, thyroid, pancreas, and biliary tract, and soft tissues, including the mediastinum, retroperitoneum, and orbit.15, 25, 26, 27 The concept of IgG4SD was introduced a decade ago following observation of high serum IgG4 concentrations in patients with sclerosing (autoimmune) pancreatitis. Since the early reports, the diagnostic spectrum of hyper-IgG4 disease or IgG4SD and its clinical variants have evolved and expanded to include inflammatory fibrosclerosing lesions in many different organs and in soft tissue. The current understanding of IgG4SD is that it can occur in solitary or multiple sites, that it affects middle age to elderly adults, and that steroid therapy is an effective treatment.15, 25, 26, 27, 28 Pathologically IgG4SD is a diffusely infiltrative process that entraps surrounding structures, has a hypocellular collagenized and fibrous appearance accompanied by abundant plasma cells and prominent lymphoid aggregates, and contains foci of obliterative phlebitis.15, 20, 27, 28, 29 Although elevated serum IgG4 levels are considered characteristic, particularly in Asian patients, the criteria for diagnosing IgG4SD on the basis of IgG4 plasma cell counts or ratios in tissue samples or histopathology are still evolving. Minimum IgG4 plasma counts for a diagnosis of IgG4SD in various studies have ranged from 10–30 IgG4 positive plasma cells per HPF with ‘highly suggestive’ counts in the range of 60–100 IgG4 positive plasma cells per HPF.28, 29, 30 In contrast, the IMT in the present series all had IgG4 counts less than 33 per HPF with a mean of 7 per HPF, using methodology similar to Yamamoto and colleagues.30 The IgG4 to IgG plasma cell ratio for IgG4SD has ranged from 0.1 to 0.4 or 0.5 in the literature, although a ratio of 0.4 or 0.5 is currently favored as more supportive of the diagnosis.20 Although in most cases, inflammatory myofibroblastic tumor and IgG4SD are distinguishable by their clinical, radiologic, and pathologic features, a subset can display overlapping features that lead to diagnostic challenges, especially in small biopsies. This has raised the question of whether the two entities have a common pathogenesis or might represent a diagnostic continuum.
This study evaluated the clinicopathologic features of 36 IMT including ALK-1 immunohistochemistry, and investigated the inflammatory infiltrate for plasma cell IgG and IgG4 expression and other markers. Among the 36 IMT, the male to female ratio was approximately equal, the mean age at diagnosis was 13.3 years with a range of 6 months to 41 years, and the most frequent sites were the mesentery, peritoneum, pelvis, and lung. The local recurrence rate was 31% and one patient had metastases. With immunohistochemistry, 64% were ALK-1 positive.
In comparison with published reports of IgG4SD, the IMT in this series had lower numbers of IgG4 positive plasma cells and lower IgG4 to IgG ratios with a few exceptions. In 30 of the 36 cases, paired immunohistochemical samples with adequate results for both IgG and IgG4 in the plasma cells were analyzed. All 36 cases had unequivocally positive or negative IgG4 immunohistochemical results, which permitted counting of IgG4 positive plasma cells. The number of IgG4 positive plasma cells in the 36 cases ranged from zero to 33 per HPF, with a mean of 7 per HPF. Thirteen cases had no detectable IgG4 positive plasma cells. When these cases were excluded, the mean IgG4 positive plasma cell count was 7 per HPF. For the 30 cases with an IgG4 to IgG ratio of less than or equal to 1, those ratios ranged from zero to 0.94. Fifteen cases had a ratio less than 0.10 including a subset of 10 cases with an IgG4 to IgG ratio of zero. Ten additional cases had a ratio between 0.10 and 0.40 and 5 cases had a ratio between 0.40 and 1.0. In the five cases with an IgG4 to IgG ratio of 0.4–1.0, the IgG4 positive plasma cell counts ranged from 19 to 33 per HPF. There was no significant difference in IgG or IgG4 plasma cell count or ratio between the ALK-1 positive and ALK-1 negative inflammatory myofibroblastic tumors. Serum IgG4 levels were not available for the 36 patients with inflammatory myofibroblastic tumors.
When the findings for the IMT analyzed in this study are compared with the literature on IgG4SD, several key differences are evident. Clinically, IMT affects younger patients and is less likely to arise within an organ, with the exception of the lung. Pathologically, IMT is a more circumscribed mass rather than an infiltrative process, and typically lacks the obliterative phlebitis and abundant lymphoid aggregates of IgG4SD, as previously noted by Yamamoto.30 The IgG4 positive plasma cell counts and IgG4 to IgG positive plasma cell ratios are generally lower in IMT than in IgG4SD, although overlap can occur, especially in the range of the lower cutoff values. The presence or absence of ALK over-expression or rearrangement in the IMT in this series did not appear to have any relationship to IgG4 plasma cell counts, nor did age appear to be an influence. ALK gene rearrangements and protein over-expression are reportedly absent in IgG4SD, which can be helpful to support the diagnosis of IMT.30 Previous studies of cellular atypia, mitotic rate, proliferative activity, and necrosis in IMT have shown a broad spectrum of morphologic findings and a lack of prognostic or diagnostic significance.18
In conclusion IMT is clinically, pathologically, and genetically distinct from IgG4SD. This and previous studies indicate that the two diseases are nosologically separate. Despite some morphologic similarities to IgG4SD, IMT is distinguished pathologically by circumscribed mass formation, lower levels of IgG4 plasma cells in the tumoral inflammatory infiltrate, relative paucity of lymphoid aggregates, absence of obliterative phlebitis, and ALK-1 over- expression in the majority of cases. The question of whether IgG4SD and ALK-negative IMTs have similarities inevitably arises in the context of this study. There are many diagnostic pitfalls to consider in ALK-negative IMTs, especially in older patients for whom carcinomas, melanomas, dendritic neoplasms, and sarcomas with spindle cell morphology and an inflammatory infiltrate are in the differential diagnosis. These can be addressed diagnostically with adjuncts such as immunohistochemistry, cytogenetics, and molecular genetics. As the data in this study shows, ALK-negative IMT have similar IgG4 plasma cell counts and ratios to ALK positive IMT. There is no clear utility in evaluation of the IgG4/IgG ratio in such cases if the lesion fulfills the above-mentioned characteristic features of IMT.
References
Griffin CA, Hawkins AL, Dvorak C, et al. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res 1999;59:2776–2780.
Lawrence B, Perez-Atayde A, Hibbard MK, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol 2000;157:377–384.
Coffin CM, Patel A, Perkins S, et al. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol 2001;14:569–576.
Debelenko LV, Arthur DC, Pack SD, et al. Identification of CARS-ALK fusion in primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab Invest 2003;83:1255–1265.
Debiec-Rychter M, Marynen P, Hagemeijer A, et al. ALK-ATIC fusion in urinary bladder inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 2003;38:187–190.
Coffin CM, Fletcher JA . Inflammatory myofibroblastic tumour. In: Fletcher CDM, Unni KK, Mertens F, (eds). Pathology and genetics of tumours of soft tissue and bone. World health organization classification of tumours. IARC Press: Lyon, 2002, pp 91–93.
Tang TT, Segura AD, Oechler HW, et al. Inflammatory myofibrohistiocytic proliferation simulating sarcoma in children. Cancer 1990;65:16–34.
Kutluk T, Emir S, Karnak I, et al. Mesentric inflammatory psuedotumor: usual presentation with leukemoid reaction and massive calcified mass. J Pediatr Hematol Oncol 2002;24:158–159.
Sciot R, Dal Cin P, Fletcher CD, et al. Inflammatory myofibroblastic tumor of bone: report of two cases with evidence of clonal chromosomal changes. Am J Surg Pathol 1997;21:1166–1172.
Ramachandra S, Hollowood K, Bisceglia M, et al. Inflammatory pseudotumour of soft tissues: a clinicopathological and immunohistochemical analysis of 18 cases. Histopathology 1995;27:313–323.
Hausler M, Schaade L, Ramaekers VT, et al. Inflammatory pseudotumors of the central nervous system: report of 3 cases and a literature review. Hum Pathol 2003;34:253–262.
Rabban JT, Zaloudek CJ, Shekitka KM, et al. Inflammatory myofibroblastic tumor of the uterus: a clinicopathologic study of 6 cases emphasizing distinction from aggressive mesenchymal tumors. Am J Surg Pathol 2005;29:1348–1355.
Gleason BC, Hornick JL . Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol 2008;61:428–437.
Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003;38:982–984.
Kamisawa T, Nakajima H, Egawa N, et al. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006;6:132–137.
Cheuk W, Lee KC, Chong LY, et al. IgG4-related sclerosing disease: a potential new etiology of cutaneous pseudolymphoma. Am J Surg Pathol 2009;33:1713–1719.
Zen Y, Inoue D, Kitao A, et al. IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 2009;33:1886–1893.
Coffin CM, Hornick JL, Fletcher CDM . Inflammatory myofibroblastic tumor comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 2007;31:509–520.
Coffin CM, Watterson J, Priest JR, et al. Extrapulmonary myofibroblastic tumor (inflammatory pseudotumor): a clinicopathologic and immunohitochemical study of 84 cases. Am J Surg Pathol 1995;19:859–872.
Sepehr A, Mino-Kenudson M, Ogawa F, et al. IgG4+ to IgG+ plasma cells ratio of ampulla can help differentiate autoimmune pancreatitis from other ‘mass forming’ pancreatic lesions. Am J Surg Pathol 2008;32:1770–1779.
Bridge JA, Kanamori M, Ma Z, et al. Rusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory fibroblastic tumor. Am J Pathol 2001;159:411–415.
Ma Z, Hill DA, Collins MH, et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 2003;37:98–105.
Cole B, Zhou H, McAllister N, et al. Inflammatory myofibroblastic tumor with thrombocytosis and a unique chromosomal translocation with ALK rearrangemen. Arch Pathol Lab Med 2006;130:1042–1045.
Chan SK, Cheuk W, Chan KT, et al. IgG4-related sclerosing pachymeningitis: a previously unrecognized form of central nervous system involvement in IgG4-related sclerosing disease. Am J Surg Pathol 2009;33:1249–1252.
Gelrud A, Freedman SD . Autoimmune pancreatitis. J Gastrointestinal Surg 2005;9:2–5.
Deshpande V, Chicano S, Finkelberg D, et al. Autoimmune pancreatitis: a systemic immune complex mediated disease. Am J Surg Pathol 2006;30:1537–1545.
Zen Y, Kitagawa S, Minato H, et al. IgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of the lung. Hum Pathol 2005;36:710–717.
Kamisawa T, Tu Y, Nakajima H, et al. Usefulness of biopsying the major duodenal papilla to diagnose autoimmune pancreatitis: a prospective study using IgG4-immunostaining. World J Gastroenterol 2006;12:2031–2033.
Kitagawa S, Zen Y, Harada K, et al. Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Kuttner's tumor). Am J Surg Pathol 2005;29:783–791.
Yamamoto H, Yamaguchi H, Aishima S, et al. Inflammatory myofibroblastic tumor versus IgG4-related sclerosing disease and inflammatory pseudotumor: a comparative clinicopathologic study. Am J Surg Pathol 2009;33:1330–1340.
Acknowledgements
Kristi Kelley and Lindsey Walker assisted with manuscript preparation. This study was supported with funds from the Ernest W. Goodpasture Endowed Professorship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Saab, S., Hornick, J., Fletcher, C. et al. IgG4 plasma cells in inflammatory myofibroblastic tumor: inflammatory marker or pathogenic link?. Mod Pathol 24, 606–612 (2011). https://doi.org/10.1038/modpathol.2010.226
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.226
Keywords
This article is cited by
-
Infantile inflammatory myofibroblastic tumors: clinicopathological and molecular characterization of 12 cases
Modern Pathology (2020)
-
Successful treatment based on molecular biological assessment of invasive anaplastic lymphoma kinase-positive inflammatory myofibroblastic tumor of the lung
Surgical Case Reports (2019)
-
Can colonic inflammatory polyp with numerous immunoglobulin G4-positive plasma cells represent a colonic manifestation of immunoglobulin G4-related disease? A case report
Clinical Journal of Gastroenterology (2019)
-
Inflammatory pseudotumor-like follicular dendritic cell tumor: an underdiagnosed neoplasia
Applied Cancer Research (2017)
-
Primary Inflammatory demyelinating pseudotumor in the left frontal lobe with meningeal involvement presenting as malignant neoplasms
Chinese Neurosurgical Journal (2016)
