
Abstract
The matricellular protein CCN1 (CYR61) is known to function in wound healing and is upregulated in colons of patients with Crohn’s disease and ulcerative colitis, yet its specific role in colitis is unknown. Here we have used Ccn1dm/dm knockin mice expressing a CCN1 mutant unable to bind integrins α6β1 and αMβ2 as a model to probe CCN1 function in dextran sodium sulfate (DSS)-induced colitis. Ccn1dm/dm mice exhibited high mortality, impaired mucosal healing, and diminished interleukin-6 (IL-6) expression during the repair phase of DSS-induced colitis compared with wild-type mice, despite having comparable severity of initial inflammation and tissue injury. CCN1-induced IL-6 expression in macrophages through integrin αMβ2 and in fibroblasts through α6β1, and IL-6 promoted intestinal epithelial cell (IEC) proliferation. Administration of purified CCN1 protein fully rescued Ccn1dm/dm mice from DSS-induced mortality, restored IEC proliferation and enhanced mucosal healing, whereas delivery of IL-6 partially rectified these defects. CCN1 therapy accelerated mucosal healing and recovery from DSS-induced colitis even in wild-type mice. These findings reveal a critical role for CCN1 in restoring mucosal homeostasis after intestinal injury in part through integrin-mediated induction of IL-6 expression, and suggest a therapeutic potential for activating the CCN1/IL-6 axis for treating inflammatory bowel disease.
Similar content being viewed by others


INTRODUCTION
Crohn’s disease (CD) and ulcerative colitis, two major subtypes of inflammatory bowel disease (IBD), are chronic, relapsing, and remitting inflammatory disorders of the gastrointestinal tract that affect 1.4 million people in the US.1 These medically incurable diseases of poorly defined etiology generally begin in young adulthood and continue throughout life, often requiring lifelong management. Traditional treatment modalities have aimed at dampening inflammation in the gastrointestinal tract to alleviate symptoms in patients, and this rationale prompted the development of monoclonal antibodies (mAbs) targeting the pro-inflammatory cytokine tumor necrosis factor-α (TNF-α) for the treatment of IBD. Although blockade of TNF-α induces clinical remission and has become a critical component in the therapeutic arsenal for IBD, many patients (∼40%) do not respond, lose their response during treatment, or develop complications due to side effects.2, 3 Patients who are refractory to therapies may eventually require colectomy and are at increased risks of developing colorectal cancer, causing significant morbidity. Thus, there is an urgent need for alternative treatment options, and considerable effort has been focused on the identification of novel therapeutic targets.4
Among the targets under investigation for IBD therapy is interleukin-6 (IL-6), a multifunctional cytokine expressed by diverse cell types during inflammation.5, 6 IL-6 is induced upon intestinal injury in the blood and colonic tissue of IBD patients, and is thought to be involved in the pathogenesis of IBD by inducing T-cell activation and suppressing T-cell apoptosis.6, 7 Consistent with this notion, mAbs against IL-6 receptor (IL-6R) prevent T cell-mediated murine colitis,7, 8 and a pilot clinical trial using anti-IL-6R mAbs showed symptomatic improvement in patients with CD.9 Paradoxically, IL-6-deficient mice suffer increased mortality, morbidity, and mucosal damage in infectious and non-infectious models of colitis, suggesting that IL-6 may have pleiotropic and potentially antithetical roles in IBD.10, 11 Therefore, further elucidation of the regulation and function of IL-6 in colitis is likely needed to optimize its therapeutic potential.
CCN1 (CYR61), a secreted matricellular protein of ∼40 kDa, is emerging as an important regulator of tissue homeostasis and injury repair.12 Acting primarily through direct binding to integrin receptors, CCN1 regulates diverse cellular functions in a cell type- and context-dependent manner.12 Whereas CCN1 is essential for angiogenesis and cardiovascular development during embryogenesis,13 its expression has been linked to inflammation and tissue repair in adulthood.14, 15 However, the specific functions of CCN1 in various inflammation-associated pathologies are only beginning to be appreciated. For example, in cutaneous wound healing and in chronic liver injuries induced by hepatotoxin or cholestasis, CCN1 functions to diminish and restrict tissue fibrosis in the maturation phase of tissue repair by triggering cellular senescence in activated myofibroblasts.16, 17 CCN1 can also contribute to inflammatory damage by inducing the expression of pro-inflammatory cytokines in macrophages and enhancing the cytotoxicity of TNF family cytokines.18, 19, 20 Importantly, CCN1 expression is elevated in biopsies of patients with ulcerative colitis or CD, and in mice with experimental colitis.21 However, the precise function of CCN1 in IBD remains unknown.
Here we provide the first evidence that CCN1 has a critical role in promoting recovery and mucosal healing in colitis, in part through integrin-mediated induction of IL-6 expression during the repair phase. Moreover, administration of CCN1 protein accelerated recovery and mucosal healing in wild–type (WT) and Ccn1 mutant mice. Our findings reveal CCN1 as a critical regulator of mucosal healing in colitis, uncover the importance of CCN1-induced IL-6 in intestinal–epithelial restitution, and suggest a therapeutic potential in activating the CCN1/IL-6 axis for the treatment of IBD.
RESULTS
Ccn1dm/dm mice suffer increased mortality as well as impaired recovery and mucosal healing upon DSS challenge
Immunohistochemical analysis showed that CCN1 protein was mainly associated with the surface epithelial cells in the normal colon, but was detected in the entire mucosal epithelium when mice were challenged with 5% DSS to induce colitis22 (Supplementary Figure S1A online). This expression pattern was confirmed in CCN1-EGFP (enhanced green fluorescent protein) mice23 in which Ccn1 expression was visualized by anti-GFP staining, with GFP restricted to terminally differentiated intestinal epithelial cells (IECs) in the normal colon but expanded to the entire crypt upon DSS exposure (Supplementary Figure S1B). Consistently, Ccn1 mRNA was increased in the colon of DSS-challenged mice compared with untreated mice (P<0.01; Supplementary Figure S1C), suggesting that CCN1 is actively regulated during colitis. CCN1 exerts diverse effects in various cell types through distinct integrins, among them αMβ2 in macrophages and α6β1 in fibroblasts.12 To elucidate the role of CCN1 in IBD, we have used Ccn1dm/dm knockin mice in which the Ccn1 genomic locus is replaced by an allele encoding a CCN1 double mutant protein (DM-CCN1) disrupted in its overlapping binding sites for integrins αMβ2 and α6β1.24, 25 This genetic model circumvents the embryonic lethality of Ccn1-null mice,13 and avoids potential limitations of cell type-specific deletions as CCN1 may be secreted by multiple cell types in the colonic tissue microenvironment. Ccn1dm/dm mice are viable, fertile, morphologically, and behaviorally normal,16, 18 and display a normal intestinal histology in the absence of pathogenic insults.
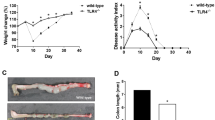
WT, Ccn1wt/wt and Ccn1dm/dm mice were subjected to a single or repeated cycles of DSS challenge, which induced acute or chronic colitis, respectively, in an established protocol for the induction of intestinal inflammation.22 When challenged with 5% DSS (w/v), only 60 and 20% of Ccn1dm/dm mice survived through the first and third cycles of DSS feeding, respectively, compared with 95 and 80% of WT mice (P<0.05 and P<0.001, respectively; Figure 1a), suggesting that CCN1 serves an important protective role in both acute and chronic DSS-induced colitis. However, increased lethality of Ccn1dm/dm mice was not due to exacerbated disease as judged by body weight loss, disease activity index (DAI), and colonic shortening (Figure 1b–d). The intestinal barrier function after DSS feeding, as evaluated by detection of gavaged fluorescein isothiocyanate-conjugated dextran in the serum, was also similar between the two genotypes (Figure 1e). Histological analyses of the distal colon showed comparable crypt loss and epithelial damage (Figure 1f) with similar numbers of apoptotic cells (Figure 1g). Lastly, myeloperoxidase activity in the colonic tissue was comparable in WT and Ccn1dm/dm mice, indicating similar extent of neutrophil infiltration into the mucosal epithelium and lamina propria (Figure 1h). These results show that CCN1 functions mediated through its α6β1/αMβ2 binding sites are critical for animal survival in DSS-induced colitis, although loss of these functions did not result in exacerbated morbidity or inflammatory damage.

Higher mortality but not morbidity in DSS-challenged Ccn1dm/dm mice. (a) Wild-type and Ccn1dm/dm mice (n=20 per genotype) were given 5% (w/v) of DSS water for 5 days followed by 14 days of recovery with normal drinking water as indicated. Survival was monitored through three cycles of DSS challenge. *P<0.05, **P<0.001 (log-rank test). The disease course of acute colitis was monitored by measuring (b) body weight change, (c) DAI, and (d) colon lengths on indicated days after initiation of DSS exposure. (e) Orally gavaged FITC particles were detected in the serum to evaluate intestinal barrier integrity (n=3). (f) Mucosal damage was assessed by H&E staining of paraffin-embedded sections of the distal colon; histological scores are shown on right (n=4–6). Scale bar = 100 μm. (g) Cell death in the distal colon was measured by TUNEL, and cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI). Quantitation is shown on the right (n=3). Scale bar = 50 μm. (h) MPO activity from colonic tissue (n=3–6). DAI, disease activity index; DSS, dextran sodium sulfate; FITC, fluorescein isothiocyanate; MPO, myeloperoxidase.
To test the hypothesis that CCN1 may function to promote recovery and mucosal healing in colitis, we lowered the dose of DSS to 3.5% (w/v) to avoid death of Ccn1dm/dm mice, and examined recovery (as judged by DAI) and mucosal healing (as judged by epithelial restitution). When administered 3.5% DSS, Ccn1 mRNA expression was elevated throughout the recovery phase compared with the untreated mice (day 8 and 14, P<0.01; day 11, P<0.05; Supplementary Figure S1D). As expected, WT and Ccn1dm/dm mice showed comparable body weight loss, DAI, colonic shortening, and histological score during the onset of disease from days 0 to 8 (Figure 2a–d). However, as recovery commenced on day 9 and the DAI began to decline in WT, Ccn1dm/dm mice experienced a prolonged disease course with elevated DAI (Figure 2b), slow recovery of body weight and colon length (Figure 2a and c), and unrelenting epithelial erosion and ulceration through day 14 (P<0.001; Figure 2d), indicating that Ccn1dm/dm mice suffer impaired recovery and mucosal healing from colitis.

Impaired recovery and mucosal healing in DSS-challenged Ccn1dm/dm mice. Wild-type and Ccn1dm/dm mice were given DSS water (3.5% w/v) for 5 days followed by normal drinking water. (a) Changes in body weight and (b) DAI were recorded daily, and (c) colonic length was measured (n=7–8). (d) H&E staining of paraffin-embedded sections of the distal colon from mice 5, 8, 11, and 14 days after initiation of DSS feeding. Histological scores are shown on bottom (n=4–6). *P<0.05, **P<0.01, ***P<0.001. Scale bar = 100 μm. DAI, disease activity index; DSS, dextran sodium sulfate; H&E, hematoxylin and eosin.
CCN1 protein rescues Ccn1dm/dm mice from lethality and accelerates mucosal healing
To assess whether increased mortality of Ccn1dm/dm mice is indeed due to compromised CCN1 activity during recovery, we tested the effect of supplying WT-CCN1 protein during the recovery phase. After 5 days of 5% DSS feeding, Ccn1dm/dm mice were injected (intraperitoneally) daily with purified WT-CCN1 for 5 consecutive days. Strikingly, 90% of CCN1-treated Ccn1dm/dm mice survived through three cycles of DSS challenge, compared with only 20% survival in vehicle-treated controls (P<0.01; Figure 3a) and 80% in WT mice (Figure 1a). Thus, CCN1 therapy fully restored survival in Ccn1dm/dm mice to WT level. To evaluate the role of CCN1 during recovery, mice fed 3.5% DSS were similarly injected with WT-CCN1. These mice recovered their body weight (P<0.05) and colon length significantly faster than vehicle controls (day 8, P<0.05; days 11 and 14, P<0.01; Figure 3b and c), comparable to WT animals (Figure 2a and c). Histology showed that CCN1-treated Ccn1dm/dm mice completely restored the crypt architecture in the distal colon as early as day 11 after initiation of DSS exposure (P<0.001; Figure 3d), further indicating that CCN1 promotes tissue repair after DSS-induced colitis.

Treatment with WT-CCN1 promotes survival and mucosal repair. (a) Ccn1dm/dm mice (n=10) were injected i.p. with 5 μg of WT-CCN1 or vehicle control daily for 5 days after each cycle of 5% DSS feeding and monitored for survival. *P<0.05, **P<0.01 (log-rank test). (b) Mice similarly treated except with 3.5% DSS were monitored for body weight change and (c) colon lengths (n=6–8). (d) Mucosal recovery was evaluated by H&E staining of paraffin-embedded colonic tissue. Histological scores are shown on right. Data are presented as mean±s.e.m.; n=3–4; *P<0.05, **P<0.01, ***P<0.001. Scale bar = 100 μm. DSS, dextran sodium sulfate; H&E, hematoxylin and eosin; i.p., intraperitoneally; WT, wild type.
Proliferation of IECs is a critical component in intestinal tissue regeneration. Therefore, we postulated that CCN1 may have a direct role in intestinal tissue repair by promoting proliferation of colonic epithelial cells. To test this possibility, we treated young adult mouse colon (YAMC) IECs with bovine serum albumin, purified WT-CCN1 or DM-CCN1. Surprisingly, neither WT-CCN1 nor DM-CCN1 had any effect on the proliferation of YAMCs in culture as judged by cell number counts and Ki67 staining (Supplementary Figure S2), suggesting that CCN1 may indirectly mediate tissue repair after DSS-induced injury.
CCN1 induces IL-6 in vivo and in macrophages and fibroblasts
As cytokines are important in inflammation and repair, we measured cytokine expression in the WT and Ccn1dm/dm colon after 3.5% DSS challenge. Most differences were small: WT mice transiently expressed somewhat higher levels of the pro-inflammatory cytokines interferon (IFN)-γ and IL-17, but TNF-α expression was the same in both genotypes (Supplementary Figure S3A). Likewise, Ccn1dm/dm mice showed similar or higher levels of the anti-inflammatory cytokines IL-4, TGF-β, and IL-22, whereas IL-10 expression was the same (Supplementary Figure S3B). Despite the expression of reparative cytokines such as IL-22, IL-10, and TGF-β,26 Ccn1dm/dm mice showed impaired mucosal repair (Figure 2), suggesting that expression of these cytokines was insufficient to initiate efficient recovery and repair in Ccn1dm/dm mice. By contrast, whereas IL-6 expression was similar in Ccn1dm/dm and WT mice at day 5 when inflammatory damage began to be observed, it was dramatically lower in Ccn1dm/dm mice (>7-fold) by day 8 when damage had peaked and repair began (Figure 4a). Thus, Ccn1dm/dm mice suffered a large deficit in IL-6 expression in the repair phase but not the initiation phase of colitis. IL-6 expression remained induced but at a lower level at later times, and the difference between the two genotypes diminished, suggesting an exquisite control of IL-6 by CCN1 in colitis. To assess the ability of CCN1 to induce the expression of IL-6 and other cytokines in vivo, we injected WT-CCN1 intraperitoneally into WT mice in the absence of induced colitis. We found that WT-CCN1 upregulated serum IL-6 level in a time- and dose-dependent manner as judged by enzyme-linked immunosorbent assay (ELISA), whereas DM-CCN1 had no effect (P<0.05; Figure 4b and c), indicating that CCN1 acts through its α6β1/αMβ2 binding sites to induce IL-6. Moreover, we did not detect an increase in systemic levels of IL-1α, IL-1β, IL-2, IL-4, IL-12, IL-17α, IFN-γ, TNF-α, and granulocyte-macrophage colony-stimulating factor after injection of WT-CCN1 protein by ELISA, indicating that IL-6 is a specific target of CCN1 regulation (Figure 4b). In the context of DSS-induced colitis, WT-CCN1 elevated IL-6 in blood from 18 to 787 pg/ml in Ccn1dm/dm mice on day 8 after DSS exposure (P<0.05; Figure 4d), further confirming that CCN1 can induce IL-6 during the repair phase.

WT-CCN1, but not DM-CCN1, induces IL-6 in vivo. (a) IL-6 mRNA in the colon of wild-type and Ccn1dm/dm mice treated with 3.5% DSS for 5 days was measured by qRT-PCR, and normalized to healthy wild-type colon with cyclophilin E as internal reference. Data are shown as mean±s.d.; n=3–4. **P<0.003. (b) IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-12, IL-17α, IFN-γ, TNF-α, and GM-CSF protein was measured by ELISA in the serum of wild-type mice at indicated times after a single injection i.p. of purified WT-CCN1 protein (5 or 20 μg), or (c) 24 h after i.p. injection of 10 μg of WT-CCN1, DM-CCN1 protein, or PBS (n=4). (d) IL-6 was measured from the serum of 3.5% DSS-challenged wild type and Ccn1dm/dm mice with and without CCN1 treatment by ELISA. Data are presented as mean±s.e.m.; n=7; *P<0.05. DM, double mutant; DSS, dextran sodium sulfate; ELISA, enzyme-linked immunosorbent assay; GM-CSF, granulocyte-macrophage colony-stimulating factor; H&E, hematoxylin and eosin; IFN-γ, interferon; IL, interleukin; i.p., intraperitoneally; NS, not significant; qRT-PCR, quantitative reverse transcription–PCR; TNF-α, tumor necrosis factor-α; WT, wild type.
IL-6 is thought to be produced by several cell types in the lamina propria, including macrophages and fibroblasts.27 To identify the cellular source of CCN1-induced IL-6, we first tested CCN1 functions in I-13.35 macrophages. WT-CCN1-induced IL-6 mRNA and protein levels in these cells, whereas DM-CCN1 did not (Figure 5a and b). Pre-incubation of macrophages with mAbs against integrin αM or β2 abrogated CCN1-induced IL-6 expression, showing the requirement of αMβ2 (Figure 5c). In addition, WT-CCN1, but not DM-CCN1, greatly upregulated IL-6 mRNA and protein levels in 18Co human colonic fibroblasts (Figure 5d and e). As CCN1 and TNF-α are known to be co-expressed in inflammation, we tested whether they act synergistically to regulate IL-6. Remarkably, CCN1 in combination with TNF-α induced IL-6 mRNA in 18Co cells by nearly 250-fold, or 15- and 23-fold higher than stimulation by CCN1 or TNF-α alone, respectively (Figure 5d). This synergistic effect was also observed by measuring IL-6 protein levels in the conditioned media of 18Co cells (Figure 5e). Consistent with integrin α6β1 being the principal CCN1 receptor in fibroblasts,28 pretreatment of 18Co cells with anti-α6 mAb blocked CCN1-induced IL-6 expression (Figure 5f). Although α6 can heterodimerize with β1 and β4 subunits, β4 is predominantly localized to hemidesmosomes and is not express in fibroblasts. Together, these results show that CCN1 upregulates IL-6 mRNA and protein in macrophages through αMβ2 and in fibroblasts through α6β1, consistent with the deficiency in IL-6 expression in Ccn1dm/dm mice (Figure 4b) and inability of DM-CCN1 to induce IL-6 in vivo (Figure 4d).

WT-CCN1, but not DM-CCN1, induces IL-6 in macrophages and fibroblasts. (a) IL-6 mRNA was measured by qRT-PCR and (b) IL-6 protein from conditioned media was measured by ELISA in serum-starved (overnight) I13.35 macrophages treated with 5 μg ml−1 of purified WT-CCN1 or DM-CCN1, or BSA for 24 h. (c) I13.35 macrophages were incubated with blocking mAbs against integrin αM (50 μg ml−1) or β2 (50 μg ml−1) 1 h prior to treatment with WT-CCN1 for 24 h. IL-6 mRNA was measured by qRT-PCR. (d) IL-6 mRNA was measured by qRT-PCR and (e) IL-6 protein from conditioned media was measured by ELISA in serum-starved 18Co fibroblasts treated with 5 μg ml−1 of purified WT-CCN1, DM-CCN1, BSA, or 25 ng ml−1 of TNF-α for 24 h. (f) 18Co fibroblasts were incubated with blocking mAb against integrin α6 for 1 h, then treated with WT-CCN1, TNF-α, or BSA for 24 h as above. IL-6 mRNA was measured by qRT-PCR. Data shown as mean±s.d. of triplicate experiments. BSA, bovine serum albumin; DM, double mutant; ELISA, enzyme-linked immunosorbent assay; H&E, hematoxylin and eosin; IFN-γ, interferon; IL, interleukin; mAbs, monoclonal antibodies; qRT-PCR, quantitative reverse transcription–PCR; TNF-α, tumor necrosis factor-α; WT, wild type.
CCN1-induced IL-6 promotes intestinal epithelial healing
To determine whether deficits in IL-6 expression contributed to impaired healing in Ccn1dm/dm mice, we delivered five daily injections of recombinant IL-6 (rIL-6) to Ccn1dm/dm mice following 5% DSS feeding. Remarkably, rIL-6 treatment increased survival from 60 to 95% in the first cycle of DSS challenge (P<0.01) and from 20 to 65% by the third cycle (P<0.01; Figure 6a). In Ccn1dm/dm mice fed 3.5% DSS, rIL-6 treatment also enhanced body weight recovery, although the numbers did not reach statistical significance (Figure 6b). Histological evaluation showed that rIL-6 treatment accelerated mucosal restitution in Ccn1dm/dm mice compared with vehicle controls, although improvements in histological scores still lagged behind WT mice (Figures 2d and 6c). These results indicate that IL-6 treatment partially rescued lethality and accelerated intestinal repair in Ccn1dm/dm mice following DSS-induced colitis.

Delivery of IL-6 enhances survival and mucosal repair of Ccn1dm/dm mice by restoring IEC proliferation. (a) Ccn1dm/dm mice (n=20) were injected i.p. with rIL-6 (100 ng) or vehicle control daily for 5 consecutive days after each cycle of 5% DSS feeding and were monitored for survival. **P<0.01 (log-rank test). (b) Body weight change was monitored daily in 3.5% DSS-challenged Ccn1dm/dm mice treated with 100 ng of rIL-6 or vehicle control for 5 consecutive days after DSS feeding. (c) Mucosal damage in mice treated in b was evaluated by H&E staining of paraffin-embedded sections of the distal colon. Histological scores are shown on the right (n=3–4; *P<0.05). Scale bar = 100 μm. (d) Wild-type and Ccn1dm/dm mice were challenged with 3.5% DSS, and Ccn1dm/dm mice were further treated with WT-CCN1 (5 μg) or rIL-6 (100 ng) as indicated. PCNA and E-cadherin was detected by immunofluorescence microscopy to assess IEC proliferation. DAPI was used for counterstaining. Quantitation of PCNA staining is shown on right (n=3–4; *P<0.05, **P<0.01). Scale bar = 50 μm. (e) YAMC cells were treated with BSA, IL-6 (50 or 100 ng ml−1), or EGF (100 ng/ ml−1) for 24 h and stained for Ki67 by immunofluorescence and quantitated. Where indicated, YAMC cells were pre-incubated with blocking polyclonal antibodies for IL-6 receptor-α (IL-6Rα) or IgG for 1 h before IL-6 addition. Data are shown as mean±s.d.; *P<0.05, **P<0.01. Scale bar = 300 μm. BSA, bovine serum albumin; DAPI, 4,6-diamidino-2-phenylindole; DSS, dextran sodium sulfate; EGF, epidermal growth factor; H&E, hematoxylin and eosin; IEC, intestinal epithelial cell; IFN-γ, interferon; IgG, immunoglobulin G; IL, interleukin; i.p., intraperitoneally; PCNA, proliferating cell nuclear antigen; qRT-PCR, quantitative reverse transcription–PCR; rIL-6, recombinant IL-6; TNF-α, tumor necrosis factor-α; WT, wild type; YAMC, young adult mouse colon.
To examine whether CCN1-induced IL-6 stimulated cell proliferation during tissue repair, we counted the number of proliferating cells in tissue sections of WT, Ccn1dm/dm, rIL-6-treated and CCN1-treated Ccn1dm/dm mice stained with antibodies against proliferating cell nuclear antigen, a proliferation marker. There were significantly fewer proliferating cells in Ccn1dm/dm mice compared with WT mice (P<0.01), and treatment with rIL-6 or WT-CCN1 partially or completely restored cell proliferation, respectively, to WT levels (Figure 6d). Furthermore, proliferating cell nuclear antigen-positive cells co-localized with E-cadherin, an epithelial cell marker, in WT and rIL-6 or CCN1-treated Ccn1dm/dm mice, but not in Ccn1dm/dm mice, indicating that there was a deficit in IECs undergoing proliferation in Ccn1dm/dm mice that was rectified by treatment with rIL-6 or CCN1.
To assess the activity of IL-6 in IEC proliferation directly, YAMC cells were treated with bovine serum albumin, rIL-6, or epidermal growth factor as a positive control and stained for Ki67. Treatment of YAMCs with rIL-6 increased Ki67-positive cells in a dose-dependent manner and to the same extent as epidermal growth factor, a known mitogen for IECs, when both were applied at 100 ng ml−1 (Figure 6e). Pretreatment of cells with anti-IL-6Rα antibody 1 h before rIL-6 addition blocked the increase in Ki67-positive cells, indicating that rIL-6 acts directly through its specific receptor to enhance IEC proliferation.
CCN1 promotes recovery and mucosal healing in DSS-challenged WT animals
To determine whether the healing effect of CCN1 in colitis is limited to mice with deficiency in CCN1 activity or IL-6 expression, and to test whether CCN1 may have therapeutic value in treating colitis, we delivered five daily injections of purified WT-CCN1 (10 μg intraperitoneally) into WT animals with colitis induced by 5% DSS. WT-CCN1 protein accelerated body weight recovery and attenuated DAI from days 8 to 11 in WT mice with colitis (Figure 7a and b). To investigate whether the improved clinical outcome was a result of enhanced intestinal mucosal repair, we harvested the distal colon on day 11 and evaluated for tissue injury. Histology of the distal colon showed significantly fewer areas of ulceration and a higher number of organized crypts compared with vehicle controls (P<0.01; Figure 7c). Thus, CCN1 therapy enhances recovery and promotes mucosal healing in colitis even in WT mice with a fully functional CCN1.

CCN1 treatment accelerates recovery and tissue repair in wild-type mice. Wild-type mice were challenged with 5% DSS for 5 days, then injected i.p. with vehicle control or 10 μg of purified WT-CCN1 daily for 5 consecutive days post-DSS feeding. (a) Body weight change and (b) DAI were monitored. (c) Histological evaluation by H&E staining of paraffin-embedded sections of the distal colon. Histological scores are on the right. Data are represented as mean±s.e.m.; n=6; *P<0.05, **P<0.01, ***P<0.001. Scale bar = 100 μm. DAI, disease activity index; DSS, dextran sodium sulfate; H&E, hematoxylin and eosin; i.p., intraperitoneally; WT, wild type.
DISCUSSION
Traditional treatment modalities for IBD have aimed at dampening inflammation in the gastrointestinal tract to alleviate symptoms in patients. Recent studies have shown that IBD patients who achieve and maintain mucosal healing have more favorable long-term outcomes than patients who do not, and thus mucosal healing is emerging as a critical endpoint in clinical trials and practice.6, 29 Although anti-TNF-α therapy for IBD was initially developed to reduce inflammation, it was found to improve symptoms as well as induce endoscopic remission and sustained mucosal healing.30, 31 In this report, we provide the first evidence that CCN1 promotes mucosal healing in murine colitis. Administration of exogenous CCN1 can accelerate mucosal restitution from colitis in both WT and Ccn1 mutant mice, underscoring a therapeutic potential for CCN1 in IBD.
As a multifunctional matricellular protein, secreted CCN1 is associated with the extracellular matrix and regulates diverse cellular functions through interaction with distinct integrins in various cell types.12 Although gene array studies have linked aberrant CCN1 expression with a multitude of inflammation-related diseases, the specific functions of CCN1 in various pathological contexts are only now beginning to emerge.14 Interestingly, different aspects of CCN1 activities may be revealed depending on the specific tissue and nature of injury. For example, in skin wound healing and in chronic inflammatory hepatic injuries that induce liver fibrosis, CCN1 functions to dampen fibrosis by acting through integrin α6β1 to induce cellular senescence and expression of the antifibrotic senescence-associated secretory phenotype in myofibroblasts.16, 17 In experimental hepatitis induced by concanavalin A, anti-CD95 antibody, or alcohol gavage, CCN1 exacerbates hepatocyte apoptosis by acting in synergy with TNF-α and FasL.18, 19 Additionally, a role for CCN1 in bone fracture repair has been attributed to its angiogenic activity.32
Ccn1dm/dm mice suffer deficient IL-6 expression in the repair phase of colitis, culminating in increased mortality and impaired mucosal healing. These defects can be substantially rescued by delivery of exogenous IL-6, indicating that CCN1 promotes mucosal healing in part through IL-6 (Figure 6). DSS-induced colitis is linked to damage in the intestinal epithelium, leading to an exuberant inflammatory response due to invasion of the intestinal mucosa by the intraluminal microbiota. Pattern recognition receptors that respond to invading bacteria such as toll-like receptors and the nucleotide-binding oligomerization domain-containing protein-like receptors can induce IL-6 through activation of NFκB, and both receptor systems have been implicated in the pathogenesis of IBD.1, 33, 34 Furthermore, transcriptional factor IFN regulatory factor 4 has been demonstrated to drive the expression of IL-6 in T lymphocytes and contribute to the pathogenesis of the colitis.35 Thus, it is not surprising that upregulation of IL-6 expression in the initiation phase (∼4-fold; day 5) of colitis is independent of CCN1 (Figure 4a). Remarkably, CCN1 exerts exquisite control on IL-6, and a large upregulation of IL-6 expression in the repair phase (∼140-fold; day 8) is largely CCN1-dependent. Therefore, Ccn1dm/dm mice provide a unique model in which IL-6 expression in the initiation and repair phases of colitis can be dissociated. CCN1 and TNF-α can act synergistically to induce a high level of IL-6 expression (Figure 5d). It is of interest to note that Ccn1dm/dm mice suffered impaired healing even though they expressed a higher level of IL-22 (Supplementary Figure S3B), a cytokine known to participate in mucosal repair,36 suggesting that CCN1 and IL-6 activities are indispensable for efficient mucosal healing.
A rare though significant adverse effect in patients undergoing anti-IL-6 therapy for rheumatoid arthritis37 and CD (ClinicalTrail.gov identifier NCT01545050)5 is intestinal perforation. This observation is consistent with our finding that downregulation of IL-6 compromises gastrointestinal mucosal healing. However, we have not observed direct evidence of intestinal perforation in Ccn1dm/dm mice, as they suffered similar initial inflammatory damage as WT mice as measured by body weight loss, DAI, colonic length, and intestinal barrier function (days 0–8, Figure 1b–e). Furthermore, bacterial culture of serum and peritoneal lavage fluid from Ccn1dm/dm mice also did not show evidence of intestinal perforation (data not shown). The overwhelming number of deaths in Ccn1dm/dm mice occurred (days 9–10) when loss of body weight exceeded that in WT mice in the recovery phase (Figure 2a), suggesting that acute volume depletion might be a significant factor contributing to mortality.
CCN1 regulates diverse cellular functions in a cell type- and integrin-dependent manner.12 Here we show that CCN1 induces IL-6 in macrophages through integrin αMβ2 and in fibroblasts through α6β1 (Figure 5). These activities are abrogated in DM-CCN1, consistent with impaired IL-6 expression in Ccn1dm/dm mice. CCN1 may also induce the expression of genes other than IL-6 or provide additional functions that promote mucosal repair, as delivery of CCN1 fully rescued defects in Ccn1dm/dm mice whereas IL-6 was only partially effective (Figures 6 and 7). However, it is also possible that the IL-6 treatment regimen has not been fully optimized. CCN1 is also known to induce angiogenesis through integrin αVβ3 in endothelial cells,38 although the binding site for αVβ3 is unaffected in DM-CCN1 and we did not observe angiogenic defects in the colon of Ccn1dm/dm mice (data not shown).
Several lines of evidence indicate that IL-6 is a pro-inflammatory cytokine required for the establishment of IBD,7, 8 and a pilot clinical trial showed symptomatic improvements in CD patients treated with anti-IL-6R antibodies.9 Thus, IL-6 has been identified as a therapeutic target for IBD and clinical trials to evaluate the efficacy of anti-IL-6 therapies are in progress.4, 5, 6 However, accumulating evidence also indicates that IL-6 can serve important protective and homeostatic functions in the intestinal mucosa by promoting IEC proliferation and survival, and regulating intestinal barrier function in mice.39, 40, 41, 42 Existing results and findings presented herein are consistent with the interpretation that IL-6 has a dual role in IBD: it is important for the development of colitis as a pro-inflammatory cytokine, but also critical for mucosal repair. Thus, IL-6-deficient mice showed reduced inflammation during colitis induction,43, 44 but suffered impaired mucosal healing during recovery.10, 45 These findings suggest that although anti-IL-6 therapy may afford benefit by dampening the pro-inflammatory functions of IL-6, it may also impede mucosal healing. Therefore, further optimization of anti-IL-6 treatment regimens and/or selective induction of IL-6 during mucosal healing, potentially through the CCN1/IL-6 axis, may enhance the therapeutic value of targeting IL-6 in IBD.
Methods
Cell culture, proteins, antibodies, and reagents. Immortalized splenic macrophage cell line I-13.35 (CRL-2471, ATCC, Manassa, VA) derived from TLR4-defective C3H/HeJ strain and human 18Co colonic fibroblasts (CCL-228, ATCC) were maintained in Dulbecco’s modified Eagle’s medium containing 20% LADMAC conditioned media and Eagle’s minimum essential medium, respectively, supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) at 37 °C with 10% CO2. The conditionally immortalized YAMC cell line (a gift of R. Whitehead, Vanderbilt University Medical Center), which carries a heat-labile SV40 large T antigen under the control of an IFN-γ-dependent promoter,46 was cultured on rat tail collagen (BD Biosciences, San Jose, CA)-coated plates in RPMI 1640 medium with 5% fetal bovine serum, 2 mM L-gluatamine, 5 U ml−1 IFN-γ (Peprotech, Rocky Hill, NJ), and 5 μg ml−1 of insulin (Gibco, Grand Island, NY) at 32 °C (permissive condition). Cells were transferred to RPMI 1640 containing 1% fetal bovine serum without IFN-γ and insulin at 37 °C (nonpermissive condition) 24 h prior to experiments. WT-CCN1 and DM-CCN1 proteins were purified as described.25, 47 Recombinant mouse IL-6 was from Peprotech (Rocky Hill, NJ) and recombinant TNF-α was from Apotech (Epalinges, Switzerland), and epidermal growth factor from Sigma-Aldrich (St Louis, MO). Neutralizing mAbs against intergrin αM (clone M1/70.15) was from Abd Serotec (Raleigh, NC), integrin β2 (clone M18/2.a.12.7) from Santa Cruz Biotechnology (Dallas, TX), integrin α6 (clone GoH3) from Beckman Coulter (Indianapolis, IN), and IL-6Rα from R&D (Minneapolis, MN).
Mice and DSS-induced colitis. Ccn1dm/dm mice were backcrossed 20 × into the C57BL/6 background.16, 17, 18 Transgenic CCN1(CYR61)-EGFP mice in FVB/N-Swiss webster background harboring a modified bacterial artificial chromosome with an EGFP inserted upstream of the initiating ATG codon of the first coding exon of the Cyr61 gene was obtained from the Mutant Mouse Regional Resource Centers at University of California, Davis.23 Female WT C57BL/6, Ccn1dm/dm, CCN1-EGFP mice (12–16 weeks) were given 5% or 3.5% (w/v) DSS (36–50 kDa; MP Biomedicals, Solon, OH) in the drinking water for 5 days followed by 14 days of regular water. This cycle was repeated 3 × in survival experiments. Where indicated, mice were injected intraperitoneally with CCN1 or IL-6 in 200 μl of phosphate-buffered saline. All animal procedures were approved by the University of Illinois Animal Care Committee.
DAI and histological analysis. Mice were examined daily and DAI was assessed based on symptom severities as described.48 Distal colons were formalin fixed and paraffin embedded; sections were stained with hematoxylin and eosin and photomicrographed. Histological score was based on published criteria49 with each parameter multiplied by percentage of tissue involvement for a maximum score of 10.
RNA isolation, quantitative reverse transcription–PCR, and ELISA. RNA was extracted using TRIzol (Invitrogen, Grand Island, NY) and RNeasy Mini Kit (Qiagen, St. Louis, MO), and reverse transcribed to complementary DNA using SuperScript Reverse Transcriptase III following the manufacturer’s protocol (Invitrogen). Quantitative reverse transcription–PCR was performed with iCycler thermal cycler (Bio-Rad, Hercules, CA) using iQ SYBR Green supermix. Cyclophilin E was used as an internal standard. List of primer sequences used for quantitative reverse transcription–PCR is provided in Supplementary Table S1. ELISA was conducted using IL-6 Quantikine ELISA kit (R&D); serum protein levels were quantified using Inflammatory Cytokine and Chemokine Multi-Analyte ELISArray and IL-6 single analyte ELISA kits (Qiagen) with manufacturer’s protocol.
In vivo permeability assay. Mice were gavaged with fluorescein isothiocyanate-dextran (MW 4000; Sigma-Aldrich) at 60 mg kg−1 of body weight 4 h prior to sacrifice. Blood was collected via cardiac puncture, and fluorescence intensity in the serum was measured (excitation, 485 nm; emission, 520 nm) using Fluostar Omega multimode microplate reader. Fluorescein isothiocyanate-dextran concentrations were determine from a standard curve generated by a serial dilution of fluorescein isothiocyanate-dextran.
TUNEL and myeloperoxidase assays. TUNEL assay was performed using ApopTag Red detection kit (Millipore, Billerica, MA) according to manufacturer’s protocol. Four randomly selected high-power microscopic fields of each sample were examined using Leica DM 4,000B microscope mounted with QI Click digital CCD camera. Number of TUNEL-positive cells were scored and normalized by total number of cells.
Myeloperoxidase activity was measured as previous described.50 Enzymatic activity was measured at 450 nm using 96-well Multiskan microplate reader and determined from a standard curve generated by a serial dilution of purified myeloperoxidase enzyme (Enzo Life Sciences, Farmingdale, NY).
Immunofluorescence microscopy and cell proliferation assay. Paraffin-embedded colon sections were stained with anti-E-cadherin antibody (Santa Cruz Biotechnology), anti-GFP (Abcam, Cambridge, MA), or mAb against proliferating cell nuclear antigen (Abcam), and visualized with anti-rabbit immunoglobulin G Texas Red and anti-mouse immunoglobulin G Alexa Fluor 488 (Invitrogen). For cell proliferation, Ki67 was detected using rabbit polyclonal antibodies (Abcam) and anti-rabbit immunoglobulin G Alexa Fluor 488 (Invitrogen). All assays were done in triplicates, and >300 cells were counted in each sample from random fields.
Statistical analysis. Data are expressed as mean±s.e.m. All experiments were performed in triplicates unless stated otherwise. Statistical significance was determined by Student’s t test. Survival curves were calculated using Kaplan–Meier method. Statistical significance for survival between populations was analyzed by log-rank test.
References
Abraham, C. & Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 361, 2066–2078 (2009).
Nielsen, O.H. & Ainsworth, M.A. Tumor necrosis factor inhibitors for inflammatory bowel disease. N. Engl. J. Med. 369, 754–762 (2013).
Leal, R.F. et al. Identification of inflammatory mediators in patients with Crohn's disease unresponsive to anti-TNFalpha therapy. Gut 64, 233–242 (2015).
Scharl, M., Vavricka, S.R. & Rogler, G. Review: new anti-cytokines for IBD: what is in the pipeline? Curr. Drug Targets 14, 1405–1420 (2013).
Allocca, M., Jovani, M., Fiorino, G., Schreiber, S. & Danese, S. Anti-IL-6 treatment for inflammatory bowel diseases: next cytokine, next target. Curr. Drug Targets 14, 1508–1521 (2013).
Neurath, M.F. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal. Immunol. 7, 6–19 (2014).
Atreya, R. et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat. Med. 6, 583–588 (2000).
Yamamoto, M., Yoshizaki, K., Kishimoto, T. & Ito, H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J. Immunol. 164, 4878–4882 (2000).
Ito, H. et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology 126, 989–996 (2004).
Tebbutt, N.C. et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 8, 1089–1097 (2002).
Dann, S.M. et al. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 180, 6816–6826 (2008).
Lau, L.F. CCN1/CYR61: the very model of a modern matricellular protein. Cell. Mol. Life Sci. 68, 3149–3163 (2011).
Mo, F.E. et al. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol. Cell Biol. 22, 8709–8720 (2002).
Jun, J.I. & Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 10, 945–963 (2011).
Emre, Y. & Imhof, B.A. Matricellular protein CCN1/CYR61: a new player in inflammation and leukocyte trafficking. Semin. Immunopathol. 36, 253–259 (2014).
Jun, J.I. & Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685 (2010).
Kim, K.H., Chen, C.C., Monzon, R.I. & Lau, L.F. The matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell Biol. 33, 2078–2090 (2013).
Chen, C.-C. et al. Cytotoxicity of TNFα is regulated by integrin-mediated matrix signaling. EMBO J. 26, 1257–1267 (2007).
Juric, V., Chen, C.C. & Lau, L.F. Fas-mediated apoptosis is regulated by the extracellular matrix protein CCN1 (CYR61) in vitro and in vivo. Mol. Cell Biol. 29, 3266–3279 (2009).
Bai, T., Chen, C.-C. & Lau, L.F. The matricellular protein CCN1 activates a pro-inflammatory genetic program in murine macrophages. J. Immunol. 184, 3223–3232 (2010).
Koon, H.W. et al. Substance P-mediated expression of the pro-angiogenic factor CCN1 modulates the course of colitis. Am. J. Pathol. 173, 400–410 (2008).
Wirtz, S., Neufert, C., Weigmann, B. & Neurath, M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2, 541–546 (2007).
Gong, S. et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925 (2003).
Schober, J.M., Lau, L.F., Ugarova, T.P. & Lam, S.C. Identification of a novel integrin αMβ2 binding site in CCN1 (CYR61), a matricellular protein expressed in healing wounds and atherosclerotic lesions. J. Biol. Chem. 278, 25808–25815 (2003).
Leu, S.-J. et al. Targeted mutagenesis of the matricellular protein CCN1 (CYR61): selective inactivation of integrin α6β1-heparan sulfate proteoglycan coreceptor-mediated cellular activities. J. Biol. Chem. 279, 44177–44187 (2004).
Mizoguchi, A. Healing of intestinal inflammation by IL-22. Inflamm. Bowel Dis. 18, 1777–1784 (2012).
Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 14, 329–342 (2014).
Chen, N., Chen, C.C. & Lau, L.F. Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin α6β1 and cell surface heparan sulfate proteoglycans. J. Biol. Chem. 275, 24953–24961 (2000).
Pineton de, C.G., Peyrin-Biroulet, L., Lemann, M. & Colombel, J.F. Clinical implications of mucosal healing for the management of IBD. Nat. Rev. Gastroenterol. Hepatol. 7, 15–29 (2010).
Rutgeerts, P. et al. Adalimumab induces and maintains mucosal healing in patients with Crohn's disease: data from the EXTEND trial. Gastroenterology 142, 1102–1111 (2012).
Hebuterne, X. et al. Endoscopic improvement of mucosal lesions in patients with moderate to severe ileocolonic Crohn's disease following treatment with certolizumab pegol. Gut 62, 201–208 (2013).
Athanasopoulos, A.N. et al. Vascular endothelial growth factor (VEGF)-induced up-regulation of CCN1 in osteoblasts mediates proangiogenic activities in endothelial cells and promotes fracture healing. J. Biol. Chem. 282, 26746–26753 (2007).
Henckaerts, L. et al. Mutations in pattern recognition receptor genes modulate seroreactivity to microbial antigens in patients with inflammatory bowel disease. Gut 56, 1536–1542 (2007).
Liu, J., Wang, Y. & Ouyang, X. Beyond toll-like receptors: Porphyromonas gingivalis induces IL-6, IL-8, and VCAM-1 expression through NOD-mediated NF-kappaB and ERK signaling pathways in periodontal fibroblasts. Inflammation 37, 522–533 (2014).
Mudter, J. et al. The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J Clin. Invest. 118, 2415–2426 (2008).
Pickert, G. et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 206, 1465–1472 (2009).
Gout, T., Ostor, A.J. & Nisar, M.K. Lower gastrointestinal perforation in rheumatoid arthritis patients treated with conventional DMARDs or tocilizumab: a systematic literature review. Clin. Rheumatol. 30, 1471–1474 (2011).
Chen, N., Leu, S.-J., Todorovic, V., Lam, S.C.T. & Lau, L.F. Identification of a novel integrin αvβ3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J. Biol. Chem 279, 44166–44176 (2004).
Becker, C. et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21, 491–501 (2004).
Grivennikov, S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 (2009).
Jin, X., Zimmers, T.A., Zhang, Z., Pierce, R.H. & Koniaris, L.G. Interleukin-6 is an important in vivo inhibitor of intestinal epithelial cell death in mice. Gut 59, 186–196 (2010).
Wang, L., Srinivasan, S., Theiss, A.L., Merlin, D. & Sitaraman, S.V. Interleukin-6 induces keratin expression in intestinal epithelial cells: potential role of keratin-8 in interleukin-6-induced barrier function alterations. J. Biol. Chem. 282, 8219–8227 (2007).
Naito, Y. et al. Reduced intestinal inflammation induced by dextran sodium sulfate in interleukin-6-deficient mice. Int. J. Mol. Med. 14, 191–196 (2004).
Gay, J., Kokkotou, E., O'Brien, M., Pothoulakis, C. & Karalis, K.P. Interleukin-6 genetic ablation protects from trinitrobenzene sulfonic acid-induced colitis in mice. Putative effect of antiinflammatory cytokines. Neuroimmunomodulation 13, 114–121 (2006).
Fattori, E. et al. Defective inflammatory response in interleukin 6-deficient mice. J. Exp. Med. 180, 1243–1250 (1994).
Whitehead, R.H., VanEeden, P.E., Noble, M.D., Ataliotis, P. & Jat, P.S. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc. Natl. Acad. Sci. USA 90, 587–591 (1993).
Kireeva, M.L., Mo, F.-E., Yang, G.P. & Lau, L.F. Cyr61, product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol. Cell Biol. 16, 1326–1334 (1996).
Gommeaux, J. et al. Colitis and colitis-associated cancer are exacerbated in mice deficient for tumor protein 53-induced nuclear protein 1. Mol. Cell Biol. 27, 2215–2228 (2007).
Vowinkel, T. et al. Apolipoprotein A-IV inhibits experimental colitis. J. Clin. Invest. 114, 260–269 (2004).
Yamamoto, S. et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 14, 738–747 (2008).
Acknowledgements
We thank Robert H. Whitehead for a generous gift of YAMC, Chih-Chiun Chen for helpful suggestions, and Seungwon Shin and Guoqiang Yan for excellent technical assistance. JSC was supported by the UIC Medical Scientist Training Program and NIH grant T32 DK07739. This work was supported by grants from the NIH (R01AR61791 and R01GM78492) to LFL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Choi, J., Kim, KH. & Lau, L. The matricellular protein CCN1 promotes mucosal healing in murine colitis through IL-6. Mucosal Immunol 8, 1285–1296 (2015). https://doi.org/10.1038/mi.2015.19
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2015.19
This article is cited by
-
CCN1 interacts with integrins to regulate intestinal stem cell proliferation and differentiation
Nature Communications (2022)
-
Expression of Cyr61 is associated with clinical course in patients with Crohn’s disease
BMC Gastroenterology (2021)
-
Cyr61 Alleviates Cholangitis by Inhibiting Cytotoxic Effects of CD8+ T Cells on Biliary Epithelial Cells
Current Medical Science (2021)
-
Correlation between serum cysteine-rich protein 61 and disease activity of antineutrophil cytoplasmic antibody–associated vasculitis
Clinical Rheumatology (2021)
-
A mutation that blocks integrin α4β7 activation prevents adaptive immune-mediated colitis without increasing susceptibility to innate colitis
BMC Biology (2020)
