
Abstract
Hypercapnic acidosis may attenuate ventilator-induced lung oxidative stress injury and alveolar cell apoptosis, but the underlying mechanisms are poorly understood. We examined the effects of hypercapnic acidosis on the role of apoptosis signal-regulating kinase 1 (ASK1), which activates the c-Jun N-terminal kinase (JNK) and p38 cascade in both apoptosis and oxidative reactions, in high-pressure ventilation stimulated rat lungs. Rats were ventilated with a peak inspiratory pressure (PIP) of 30 cmH2O for 4 h and randomly given FiCO2 to achieve normocapnia (PaCO2 at 35–45 mm Hg) or hypercapnia (PaCO2 at 80–100 mm Hg); normally ventilated rats with PIP of 15 cmH2O were used as controls. Lung injury was quantified by gas exchange, microvascular leaks, histology, levels of inflammatory cytokines, and pulmonary oxidative reactions. Apoptosis through the ASK1-JNK/p38 mitogen-activated protein kinase (MAPK) cascade in type II alveolar epithelial cells (AECIIs) were evaluated by examination of caspase-3 activation. The results showed that injurious ventilation caused significant lung injury, including deteriorative oxygenation, changes of histology, and the release of inflammatory cytokines. In addition, the high-pressure mechanical stretch also induced apoptosis and caspase-3 activation in the AECIIs. Hypercapnia attenuated these responses, suppressing the ASK1 signal pathways with its downstream kinase phosphorylation of p38 MAPK and JNK, and caspase-3 activation. Thus, hypercapnia can attenuate cell apoptosis and oxidative stress damage in rat lungs during injurious ventilation, at least in part, due to the suppression of the ASK1-JNK/p38 MAPK pathways.
Similar content being viewed by others


INTRODUCTION
Mechanical ventilation can result in overdistension of the alveoli, leading to pulmonary edema, the release of cytokines and deterioration in oxygenation, which are described as ventilator-induced lung injury (VILI).1, 2 The primarily biological mechanisms include the release of inflammatory mediators and oxygen radicals, leading to end-organ epithelial cell apoptosis and organ dysfunction.3, 4, 5
In a previous study, a high tidal volume in mouse lung could induce the activation of c-Jun N-terminal kinase (JNK), an increase in macrophage inflammatory protein-2 (MIP-2) expression, and the migration of neutrophil into the lung.6 Activated JNK is dependent on the activation of apoptosis signal-related kinase 1 (ASK1), which is identified as a member of the mitogen-activated protein kinase (MAPK) family, in cultured bronchial epithelium.7, 8 Activation of ASK1 in turn leads to the activation of other components of MAPKs, such as JNK and p38, and results in an initiation of cell apoptosis.9 Another studies also demonstrated that large tidal volume ventilation-induced apoptosis was dependent, at least in part, in the ASK1/JNK/AP-1 pathway10 and a p38 MAP kinase-xanthine oxidoreductase-dependent pathway.11
There is an evolving body of evidence suggesting that hypercapnic acidosis exerts biologically important beneficial effects in experimental models. Laffey et al12, 13 showed that therapeutic hypercapnia was applied to the animals by the deliberated elevation of CO2, which also markedly reduced the extent of ischemia-reperfusion injuries. Subsequent studies have demonstrated hypercapnia’s protective effects against VILI in both isolated perfused lungs and an animal model.14, 15, 16 The mechanism by which hypercapnic acidosis affects acute lung injury, includes the attenuation of NF-κB activation,17 the inhibition of endogenous xanthine oxidase18 and oxygen radical formation,19 and the reducing release of the cytokines.12, 20, 21 However, it is still unclear whether hypercapnic acidosis may affect cell apoptosis and oxidative stress injury of the alveolar by MAPK pathways in VILI.
Therefore, the objectives of this study were to determine whether hypercapnia may attenuate the formation of mechanical stretch-induced oxygen radical, cell apoptosis, and the changes of a MAPK signal transduction of lungs in a rat model of high-pressure ventilation (HPV).
MATERIALS AND METHODS
Adult male Sprague Dawley (s.d.) rats, body weight 250–300 g, were bought from (The Depth of animals experiment, Harbin Medical University, China). The experimental protocols were approved by the Harbin Medical University Animal Care and Use Committee, Harbin, China.
Experimental Protocol
A rat model of VILI was established according the previous study.22 In brief, a tracheostomy was performed with an intraperitoneal injection of 30 mg/kg pentobarbital sodium (Abbott, North Chicago, IL, USA). The internal carotid artery was cannulated to take the blood samples for the blood gas analysis and to monitor the arterial pressure. Body temperature was maintained at 36.5–38.0 °C, anesthesia and supplemental fluids were administered intravenously for the requirement throughout the experiment. Mechanical ventilation was initiated in the pressure-controlled mode (Kent Scientific Ventilator-Dual Mode, USA) with a peak inspiratory pressure (PIP) of 15 cmH2O, a positive end-expiratory pressure (PEEP) of 2 cm H2O, a frequency of 30 breaths/min to maintain PaCO2 at 35–45 mm Hg, an inspiration-to-expiration (I:E) ratio of 1:2, and an inhaled oxygen fraction (FiO2) of 0.7 for 15 min, after which the baseline data were collected and the animals were randomly allocated to three ventilation strategies, each for 4 h, the groups were described as follows: (1) The control ventilation group (PIP 15 cmH2O, PEEP 2 cmH2O, frequency 30 min); n=18, normocapnia (FiO2 0.7, FiCO2 0, balance N2). (2) The injurious ventilation group (PIP 30 cmH2O, PEEP 2 cmH2O, frequency 30 min); n=18, PaCO2 was maintained in the normal range through inhaling the gas mixture (FiO2 0.7, FiCO2 4∼5%, balance N2). (3) The injurious ventilation/hypercapnia group (PIP 30 cmH2O, PEEP 2 cmH2O, frequency 30 min); n=18, PaCO2 was maintained in the target range of 80 mm Hg–100 mm Hg through inhaling the gas mixture (FiO2 0.7, FiCO2 11∼12%, balance N2). Tidal volumes (VT) were determined every 60 min with a VT Plus HF Gas Flow Analyzer (Fluke Corporation, USA). Inhaled and exhaled CO2 and O2 were tested using a gas monitor (DATEX Instrumentarium, Helsinki, Finland).
Tissue Sampling and Assays
At the end of the experiment, the abdomen and chest of each animal was opened under anesthetic. The left lung was isolated with surgical silk tied around the left main bronchus. The right lobe bronchus was lavaged with 4 ml of saline (0.9%, 4 °C) and 2–3 ml of bronchoalveolar lavage fluid (BALF) was collected. A 1.0-ml aliquot was used for differential cell counts. The remainder of the fluid was immediately centrifuged (3000 × g for 10 min) at 4 °C. The supernatant was stored at –20 °C for measurement of the inflammatory mediators and total protein concentration (BCA; Pierce, Rockford, IL, USA). The upper sections of the left lungs were excised, weighted, and desiccated at 70 °C for 1 week to measure the wet-to-dry weight ratio (W/D) for quantifying pulmonary edema formation. The one-third of left side lung tissue was homogenized and then centrifuged (4 °C), and the supernatant was stored frozen (–80 °C) to measure reactive oxygen species. The right side of rat lung tissue was snap frozen and used for protein extraction. Subsequently, the two-third of left side rat lung tissues were used for histology. Slices were embedded in paraffin, and sections (5 μm) were used for hematoxylin and eosin (H&E) and immunohistochemical staining.
Isolation of Rat Type II Alveolar Epithelial Cells
Type II alveolar epithelial cells (AECIIs) were isolated from four rats’ lung tissues in each group according to established procedures.23, 24 Briefly, the lungs were lavaged three times via the trachea with 5 ml of Dulbecco’s PBS, and then were digested by instilling them with elastase (Worthington Biochem, Freehold, NJ, USA) via the trachea at a specific activity of 4.3 units/ml in JMEM mixed with 100 mg/ml DNase I (Sigma Chemical St Louis, MO, USA) and incubated at 37.8 °C for 35 min. The lungs were minced and the suspension was agitated on ice and filtered sequentially through nylon mesh with pore sizes of 150 μm and 70 μm. The AECIIs were separated from the macrophages by panning on rat IgG-coated Petri dishes and were identified using histochemical staining with alkaline phosphatase,25, 26 and >90% of the cells were blue positive on day 2. Experiments were performed on the second day after the isolation.
Apoptosis Assay by FACS
The isolated cells were stained with FITC-conjugated Annexin V and propidium iodide (PI) using the Apoptosis Detection Kit (BD Biosciences Pharmingen, USA). Duplicate in each sample were run according to the manufacturer’s instructions and analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA).
Measurement of Malondialdehyde Content, Activity of Superoxide Dismutase, Lactate Dehydrogenase, and Myeloperoxidase in the Supernatant of Lung Tissues
The malondialdehyde (MDA) content in the lung tissue was measured by the previous method27 and expressed as nanomoles per milligram of protein. The superoxide dismutase (SOD) activity in the lung tissue was determined according to the previous27 and expressed as units per milligram of protein. Serum lactate dehydrogenase (LDH) activity was measured by the pyruvic acid method28 according to the manufacturer’s instructions (Jiancheng Bio-Technology, Nanjing, China). The myeloperoxidase (MPO) activity of the lung tissue used a reagent kit (Jiancheng Bio-Technology, Nanjing, China) and the results were expressed as units per gram of wet lung tissue (U/g). Each measurement was performed in triplicate.
Histology and Immunohistochemistry
The lung sections were stained with H&E and then examined by a pathologist who was blinded to the protocol. Samples were assigned an injury score in each of four categories (interstitial edema, alveolar edema, neutrophil infiltration, and hyaline membrane formation) based on severity (0=not present, 4=severe, and present throughout). The overall histologic injury score (HIS) was evaluated according to a previous study.14
Immunostaining with an antibody against activated caspase-3 (Cell Signaling Technology, USA) was used to quantify the apoptosis in the lung sections.29 The number of cleaved-caspase-3-positive cells was counted at the surface of the airway epithelial cells in 10 fields using the × 40 objective lens.
Enzyme-Linked Immunoassays for Inflammatory Mediators and ASK1
To examine the levels of inflammation in the rat lung tissues, the levels of tumor necrosis factor (TNF)-α and MIP-2 were measured in the BALF by ELISA assay (R&D Systems and BioSource, USA).
The ASK1 phosphorylation expression of rat lung tissues was measured with a Phospho-ASK1 (S966) Sandwich ELISA Kit (ImmunoWay Biotechnology Company, USA). Duplicate in each sample were run according to the manufacturer’s instructions.
Western Blot Analysis of JNK, p38 MAPK, and Cleaved Caspase-3
The AECIIs were harvested. The total cellular proteins were extracted by RIPA buffer (Radio-Immunoprecipitation Assay Buffer) containing 0.2% Triton X-100, 5 mmol/l EDTA, 1 mmol/l PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin, supplemented with 100 mmol/l NaF, 2 mmol/l Na3VO4), and lysed for 30 min on ice. The total numbers of protein were quantified by using the Bradford method. The proteins in each sample were separated with 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and then were transferred onto polyvinylidene fluoride (PVDF) transfer membranes. The transfer membranes were semidried for 20 min at 15 V. The membranes were washed with PBS three times. After blocking, the membranes were placed into 1 × PBS with an anti-phosphorylated JNK, or an anti-phosphorylated p38 MAPK antibody (rabbit anti-rat) (1:1000), or an anti-cleaved caspase-3 antibody (Cell Signaling Technology) and incubated overnight at 4 °C in a slow shaker. The membranes were washed three times with PBS, and then, incubated the secondary antibody (goat anti-rabbit) (1: 2500) for 2 h. After incubation, the membranes were washed three times with 0.1% Tween in PBS. The membranes were incubated with the substrate for 5 min. The specific brand was quantitatively analyzed by using the Quantity one ChemiDocXRS Imagine Collect System and analysis software (Bio-Rad, USA).
Statistical Analysis
All data were expressed as mean+s.d. for normally distributed data and as the median (interquartile range) if they were non-normally distributed. The statistical analyses were analyzed with SPSS statistics software (version 13.01S; Beijing Stats Data Mining, Beijing, China). Group comparisons were evaluated by a one-way ANOVA, followed by the Student–Newman–Keuls test, or the Kruskal–Wallis test, followed by the Mann–Whitney U-test with the Bonferroni correction for multiple comparisons, as appropriate. The significance was set at P<0.05.
RESULTS
Gas Exchange and Hemodynamic Parameters
There was no significant difference in the pH, PaO2, PaCO2, mean arterial pressure, and VT at the baseline (Table 1). Our data showed that the PaO2/FiO2 and MAP in the high-pressure ventilation group decreased progressively after 2 h, and they were significantly lower than those values in the other two groups at the end of the protocol.
Mechanical Ventilation Induces Lung Injury In Vivo and is Attenuated by HCA
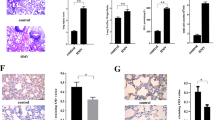
The rats were ventilated using high-pressure ventilation (PIP: 30 cmH2O, 4 h) with low PEEP (2 cmH2O) models, which impaired oxygenation and induced an acute lung inflammatory response, indicating that the levels of TNF-α and MIP-2 in the BALF were significantly higher in the HPV group compared with the control group (Table 1, P<0.05). As shown in Figures 1a and b, BALF protein concentrations and the WW: DW in the injurious group were significantly reduced compared with the control group (P<0.05). We further found that HPV resulted in an increase in the total cell count in the BALF (Figure 1c), and in significant neutrophil recruitment to the alveoli (Figure 1d). In addition, HPV resulted in histological changes marked by alveolar-septal thickening, hyaline membrane formation, and neutrophils infiltration (Figure 2). However, our data demonstrated that the damage changes were much lower in rat lungs from the hypercapnic acidosis group than the injurious group. The lung injury score was lower in the hypercapnia group (7.3±1.6) than that in the injurious group (14.8±2.7, P<0.05), and higher than the control group (4.4±1.3, P<0.05). As described above, hypercapnic acidosis exhibited significant advantages in maintaining stable hemodynamic changes and systemic oxygenation, a decreasing lung microvascular permeability (Figures 1a and b), attenuated lung-scores (Figure 2), and the decreasing inflammatory injury induced by HPV (Table 1).

Effect of hypercapnic acidosis on major markers of injury in rats’ lungs induced by high-pressure ventilation (HPV) for 4 h. (a) Bronchoalveolar lavage fluid (BALF) total protein levels (microgram/ml) were augmented by high-pressure ventilation but diminished by hypercapnia acidosis. (b) Pulmonary edema formation was quantified by measuring the wet and desiccated dry weights of the lung tissue. (c, d) The total and neutrophil cell count in the lavage group increased following HPV but were attenuated in the hypercapnia group. The data are representative of eight rats in each group. *P<0.05 vs control group (normal ventilation with a peak inspiratory pressure (PIP) of 15 cmH2O), †P<0.05 vs injurious group (ventilation with a PIP of 30 cmH2O).

Histological analysis of lungs including the lung injury score in the three groups. Formalin-fixed lung sections were stained with hematoxylin and eosin (H&E). All panels represent × 40 original magnification. The data are representative of six rats in each group. *P<0.05 vs control group; †P<0.001 in the hypercapnia group vs the injurious group.
Effect of HCA on Oxidative Stress Injury in the Rat Lung Tissues Induced by HPV
The levels of MDA was significantly increased and the level of SOD was significantly decreased in the HPV group when compared with the control group (P<0.05). HCA caused a marked high activities of SOD and a low level of MDA (Figures 3a and b). The LDH activity was also much high in the control group, whereas HCA reversed this change (Figure 3c). The neutrophil migration into the lung lavage fluid and MPO levels was significantly elevated after the HPV for 4 h compared with the control group. HCA significantly decreased the neutrophil influx and MPO levels (Figure 3d) compared with the HPV group.

Treatment of hypercapnic acidosis markedly enhanced superoxide dismutase (SOD) activity (a), decreased malondialdehyde (MDA) content (b) lactate dehydrogenase (LDH) activity (c), and myeloperoxidase (MPO) activity (d). Treatment with HCA caused a significant elevation in the SOD activities and a decrease in the MDA levels of the lung tissue. The HCA significantly decreased the LDH and MPO activity compared with the only HPV group. The data are representative of eight rats in each group. *P<0.05 vs control group (normal ventilation with a peak inspiratory pressure (PIP) of 15 cmH2O), †P<0.05 vs injurious group (ventilation with a PIP of 30 cmH2O).
Effect of HCA on Apoptosis in Rat Lung Tissues Induced by HPV
The number of caspase-3-positive cells at the surface of the airway epithelial cells and alveoli were taken as a marker of apoptosis. As shown in Figure 4, cleaved-caspase-3-positive cells were markedly increased in the HPV group compared with the control group (Figure 4a). The number of positive cells was reduced in the HCA group in comparison with the control group (Figure 4b). The level of cleaved-caspase-3 in the HPV (4 h) stimulated lung tissues was significantly higher than that in the normal ventilation group. However, the level of cleaved-caspase-3 in the lung tissues was decreased in HCA (Figure 4c).

Detection of caspase-3 protein in the lung tissues of rats. (a) Immunohistochemical staining of caspase-3 in the airway epithelial cells and the alveoli of lung tissues. The presence of brown granules in the nucleus was defined as a positive cell. (b) Scatter plot of individual caspase-3-positive (+) cells/mm2 in the lung tissue of animals ventilated at a peak inspiratory pressure (PIP) 15 cmH2O/positive end-expiratory pressure (PEEP) 2 cmH2O (control group) and PIP 30 cmH2O/PEEP 2 cmH2O with normocapnia (injurious group) or hypercapnia treatment (hypercapnia group) (n=4 per group). (c) Western blot for cleaved caspase-3 in the lung tissues. The amounts of cleaved caspase-3 proteins were quantified and are presented as the amount of cleaved caspase-3 proteins relative to the control cells (1.0). β-Actin for loading control. The data are representative of four rats in each group. *P<0.05 vs control group; †P<0.05 vs injurious group. Internal scale bar=50 μm.
Although caspase-3 activation is typical of apoptotic cell death, it is not synonymous with apoptosis and must be interpreted in conjunction with other data demonstrating apoptotic changes. The apoptosis of pulmonary AECIIs has been proven to have an important role in high tidal volume mechanical ventilation-induced lung injury.11 Therefore, we planned to isolate enriched populations of the AECIIs with sufficient viability and purity for functional studies. As shown in Figure 5b, there was a significant difference in the rate of early apoptosis or late apoptosis/necrosis between the control and the HPV groups. HCA attenuated apoptosis in the AECIIs compared with the HPV-only rat lung.

Annexin V-FITC/propidium iodide (PI) staining of type II alveolar epithelial cells (AECIIs) in the lung tissue of rats. (a) Alkaline phosphatase staining of type II cells. Blue reaction product identifies alkaline phosphatase activity (bar=50 μm). (b) Cells were stained with annexin V and PI and analyzed by flow cytometry. Annexin V-positive/PI-negative staining (lower right quadrant) represented early apoptosis; Annexin V-positive/PI-positive (upper right quadrant) represented late apoptosis/necrosis. (b) Histogram of early apoptosis and late apoptosis/necrosis in the three groups. There were significant differences in the rate of early apoptosis and late apoptosis/necrosis between the control and injured groups. HCA apparently attenuated apoptosis in AECII compared with the injury-only ventilated rat lung. For the definitions of group abbreviations, see Figure 4. The data are representative of four rats in each group. *P<0.05 vs control group; †P<0.001 in the hypercapnia group vs the injurious group.
Effect of HCA on Lung Mechanical Stretch-Induced ASK1 and JNK/ p38 MAPK Activation
We performed an ELISA to observe the potential changes in phosphorylated ASK1 levels, which were significantly increased following the HPV (30 cmH2O, 4 h) but attenuated by HCA (P<0.05) (Figure 6a).

Hypercapnic acidosis attenuated the increase apoptosis signal-regulating kinase 1 (ASK1), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK) protein activation of type II alveolar epithelial cells (AECIIs) in the lung tissue of rats following high-pressure ventilation for 4 h. ELISA was performed using an antibody that recognizes the phosphorylated ASK1 (a); Western blot was performed using an antibody that recognizes the phosphorylated JNK expressions (b) and the antibody that recognizes the phosphorylated p38 expressions (c). Arbitrary units were expressed as relative phosphorylation. For the definitions of the group abbreviations, see Figure 4. The data are representative of four rats. *P<0.05 vs control group, †P<0.05 vs injurious group.
The levels of phosphorylated JNK and p38 MAPK were also increased following mechanical ventilation in the HPV-only group when compared with the control group (Figures 6b and c). Accordingly, HCA also significantly decreased the ASK1/JNK and p38 MAPK activities in comparison with the control group (P<0.05).
DISCUSSION
Intracellular signaling pathways such as MAPK pathways have been reported to be involved in the development of VILI.30, 31, 32 However, it is still unclear that HCA attenuated the oxidative stress and inflammatory injury induced by the lung mechanical stretch, inhibited the activation of ASK1/JNK and p38 MAP kinase, which have the ability to mediate apoptosis, and contributed to increase capillary permeability. This study demonstrates that high-pressure mechanical ventilation in the rat for 4 h caused lung oxidative stress and inflammatory injury, lung edema formation, and apoptosis, which were attenuated by HCA (PaCO2 at 80–100 mm Hg) and accompanied by a decrease in the activity of ASK1/JNK and p38 MAP kinase.
JNK and p38 MAPK in pulmonary vascular endothelial cells (PVE) are activated in response to various stimuli including proinflammatory cytokines and oxidative stress.33 JNK and p38 MAPK are activated by each upstream MAPK kinase, ASK1 is one MAPKKK and has an important role in cytokines and apoptosis.34 In the present study, we examined the effect of HCA on high-pressure stretching induced apoptosis and cytokine production, and oxidative stress in the lung, with the activity of ASK1/JNK and p38 MAP kinase in the isolated type II cells of lung. To examine the potential effects of HCA on the oxidantation, we explored whether high-pressure stretching induced the content of MDA and SOD activity and MPO production in the rat lung tissue. Our data show that HCA can acutely and effectively inhibit the MPO pathway with its related oxidant reactions in the inflamed lung. These results were consistent with the previous study demonstrating that acidosis can inhibit this myeloperoxidase-dependent oxidant pathway both in vitro35, 36 and in vivo.19 The results suggest that HCA could attenuate oxidative injury and that the HCA interference might be a protective strategy for AECIIs undergoing HPV.
The signaling pathway by which hypercapnic acidosis acts as an anti-inflammatory agent is not fully understood. In this study, to determine whether the attenuation of the MAPK activation by hypercapnic acidosis actually exerts an important effect on reducing HPV-induced inflammation and apoptosis, we examined the infiltration of cytokines and inflammatory cells into pulmonary tissues. Our findings demonstrated that ventilating rats with hypercapnic gas decreased the MIP-2 and TNF-α levels in BALF, which was in accord with the study of Laffey et al37 To further characterize the potential effect of HCA on the ASK1-JNK/p38 MAPK cascade in HPV-induced injury, we isolated specific type II cells after high-pressure ventilation for 4 h with hypercapnic gas. We found that the effects of mechanical stretch could induce cell apoptosis and phosphorylation of ASK1-JNK and p38 MAPK protein in the type II cells of lung. Furthermore, we determined the activation of caspase-3 because caspase-3 is one of the key executors of apoptosis.38, 39 High-pressure ventilation activated caspase-3 and concomitantly promoted apoptosis in alveolar epithelial cells, indicating that caspase-3, at least in part, is involved in apoptosis in mechanical stretch-stimulated alveolar epithelial cells. Although several lines of evidence suggest that both caspase and the JNK/p38 MAPK cascade are key pathways resulting in apoptosis,40 the exact relationship between the caspase pathways and the MAPK pathways is still unclear. Li Fu et al6, 10 reported that the ASK1 and JNK activity inhibitor was used in an animal of VILI model and their results suggested that the ASK1-JNK pathway had a key role in both stretch-induced apoptosis and cytokine production. However, it difficult to assess the possible mechanisms without the use of specific inhibitors to these pathways. Although a specific inhibitor of ASK1 was not used in this study, our findings showed high-pressure stretch-induced injury triggers by an ASK1-mediated signaling cascade, neutrophil infiltration, MIP-2 expression, and apoptosis initiation in the model of VILI. Our study suggests that the ASK1 activation induced by high-pressure stretch is another way of the cytokine production and apoptosis. Interrogation of the hypercapnia’s influence on MAPK pathways as relates to apoptosis is important, but administration of an apoptosis inhibitor (inhaled or IP) would have been interesting to assess both the physiological and inflammatory impact to understand the relevance of apoptosis and hypercapnia.
In addition to the MAPK pathway, we have previously demonstrated the activation of NF-κB subjected to high-pressure ventilation in the rats (unpublished results). Ventilation leading to the nuclear translocation of NF-κB has been previously shown in cell culture,41 in isolated perfused rat lungs,42 and in animal lung injury induced by ventilation.43 In this study, the activation of NF-κB was markedly attenuated by HCA accompanied by the reduction of oxidative stress from neutrophil-mediated injury. It has also been reported that hypercapnic acidosis inhibits the adhesion of endotoxin-induced neutrophils to pulmonary endothelial cells through attenuating IκB-α degradation, which in turn neutralizes the DNA-binding activity of NF-κB.21 Thus, our study demonstrated that hypercapnic acidosis may also affect the VILI by mediating the pathways of ASK1-JNK and p38 MAPK, apoptosis, and inflammatory cytokines in a rat model.44
Clinical Implications
In our study, hypercapnic acidosis inhibited mechanical stretch-induced lung inflammatory injury and cell apoptosis through an underlying molecular mechanism of attenuating the phosphorylation levels of ASK1-JNK and p38 MAPK protein. Although a lot of evidence suggests that hypercapnic acidosis has a valuable role in experimental models,14, 15, 18, 37, 45 we also should pay attention to consider harmful effects of hypercapnia and/or acidosis on its potential clinical application. It is clear that hypercapnic acidosis may exert adverse hemodynamic effects in patients with ARDS.46 In addition, our previous study (unpublished data) also demonstrated that moderating hypercapnic acidosis (PaCO2 of 80–100 mm Hg) could result in the improving oxygenation, histologic characteristics, and a lessened inflammatory response, compared with severe hypercapnic acidosis (PaCO2 of 130–150 mm Hg). A previous study has also shown that the beneficial effects of moderating hypercapnia may be offset by the high level of adverse effects caused by itself.47 Thus, although acidosis induced by acute hypercapnia may be as a protective factor in acute models of ALI, the effects of hypercapnia during long-term exposure need to be further studied.
In conclusion, the present study proved that the administration of HCA significantly decreases lung vascular leakage and inflammation in a rat model of HPV-mediated VILI. The protection effect of HCA may involve the inhibition of inflammation and apoptosis via the inhibition of ASK1/JNK and p38 MAP kinase phosphorylation signaling pathway.
References
Matthay MA, Bhattacharya S, Gaver D et al. Ventilator-induced lung injury: in vivo and in vitro mechanisms. Am J Physiol Lung Cell Mol Physiol 2002;283:L678–L682.
Tsuno K, Miura K, Takeya M et al. Histopathologic pulmonary changes from mechanical ventilation at high peak airway pressures. Am Rev Respir Dis 1991;143 (5 Pt 1):1115–1120.
Hammerschmidt S, Kuhn H, Grasenack T et al. Apoptosis and necrosis induced by cyclic mechanical stretching in alveolar type II cells. Am J Respir Cell Mol Biol 2004;30:396–402.
Imai Y, Parodo J, Kajikawa O et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA 2003;289:2104–2112.
Bailey TC, Martin EL, Zhao L et al. High oxygen concentrations predispose mouse lungs to the deleterious effects of high stretch ventilation. J Appl Physiol 2003;94:975–982.
Li LF, Yu L, Quinn DA . Ventilation-induced neutrophil infiltration depends on c-Jun N-terminal kinase. Am J Respir Crit Care Med 2004;169:518–524.
Jibiki I, Hashimoto S, Maruoka S et al. Apoptosis signal-regulating kinase 1-mediated signaling pathway regulates nitric oxide-induced activator protein-1 activation in human bronchial epithelial cells. Am J Respir Crit Care Med 2003;167:856–861.
Matsuzawa A, Nishitoh H, Tobiume K et al. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid Redox Signal 2002;4:415–425.
Tobiume K, Matsuzawa A, Takahashi T et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001;2:222–228.
Li LF, Liao SK, Lee CH et al. Ventilation-induced neutrophil infiltration and apoptosis depend on apoptosis signal-regulated kinase 1 pathway. Crit Care Med 2005;33:1913–1921.
Le A, Damico R, Damarla M et al. Alveolar cell apoptosis is dependent on p38 MAP kinase-mediated activation of xanthine oxidoreductase in ventilator-induced lung injury. J Appl Physiol 2008;105:1282–1290.
Laffey JG, Jankov RP, Engelberts D et al. Effects of therapeutic hypercapnia on mesenteric ischemia-reperfusion injury. Am J Respir Crit Care Med 2003;168:1383–1390.
Laffey JG, Tanaka M, Engelberts D et al. Therapeutic hypercapnia reduces pulmonary and systemic injury following in vivo lung reperfusion. Am J Respir Crit Care Med 2000;162:2287–2294.
Sinclair SE, Kregenow DA, Lamm WJ et al. Hypercapnic acidosis is protective in an in vivo model of ventilator-induced lung injury. Am J Respir Crit Care Med 2002;166:403–408.
Broccard AF, Hotchkiss JR, Vannay C et al. Protective effects of hypercapnic acidosis on ventilator-induced lung injury. Am J Respir Crit Care Med 2001;164:802–806.
Peltekova V, Engelberts D, Otulakowski G et al. Hypercapnic acidosis in ventilator-induced lung injury. Intensive Care Med 2010;36:869–878.
O'Toole D, Hassett P, Contreras M et al. Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-kappaB dependent mechanism. Thorax 2009;64:976–982.
Shibata K, Cregg N, Engelberts D et al. Hypercapnic acidosis may attenuate acute lung injury by inhibition of endogenous xanthine oxidase. Am J Respir Crit Care Med 1998;158 (5 Pt 1):1578–1584.
Nichol AD, O'Cronin DF, Naughton F et al. Hypercapnic acidosis reduces oxidative reactions in endotoxin-induced lung injury. Anesthesiology 2010;113:116–125.
Laffey JG, Honan D, Hopkins N et al. Hypercapnic acidosis attenuates endotoxin-induced acute lung injury. Am J Respir Crit Care Med 2004;169:46–56.
Takeshita K, Suzuki Y, Nishio K et al. Hypercapnic acidosis attenuates endotoxin-induced nuclear factor-[kappa]B activation. Am J Respir Cell Mol Biol 2003;29:124–132.
Imanaka H, Shimaoka M, Matsuura N et al. Ventilator-induced lung injury is associated with neutrophil infiltration, macrophage activation, and TGF-beta 1 mRNA upregulation in rat lungs. Anesth Analg 2001;92:428–436.
Dobbs LG . Isolation and culture of alveolar type II cells. Am J Physiol 1990;258 (4 Pt 1):L134–L147.
Chen J, Chen Z, Narasaraju T et al. Isolation of highly pure alveolar epithelial type I and type II cells from rat lungs. Lab Invest 2004;84:727–735.
Beckstead JH, Halverson PS, Ries CA et al. Enzyme histochemistry and immunohistochemistry on biopsy specimens of pathologic human bone marrow. Blood 1981;57:1088–1098.
Edelson JD, Shannon JM, Mason RJ . Alkaline phosphatase: a marker of alveolar type II cell differentiation. Am Rev Respir Dis 1988;138:1268–1275.
Jiang M, Zhang J . Involvement of plasma-membrane NADPH oxidase in abscisic acid- and water stress-induced antioxidant defense in leaves of maize seedlings. Planta 2002;215:1022–1030.
Korzeniewski C, Callewaert DM . An enzyme-release assay for natural cytotoxicity. J Immunol Methods 1983;64:313–320.
Knapp S, Leemans JC, Florquin S et al. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Crit Care Med 2003;167:171–179.
Yang KY, Arcaroli JJ, Abraham E . Early alterations in neutrophil activation are associated with outcome in acute lung injury. Am J Respir Crit Care Med 2003;167:1567–1574.
Chess PR, O'Reilly MA, Sachs F et al. Reactive oxidant and p42/44 MAP kinase signaling is necessary for mechanical strain-induced proliferation in pulmonary epithelial cells. J Appl Physiol 2005;99:1226–1232.
Abdulnour RE, Peng X, Finigan JH et al. Mechanical stress activates xanthine oxidoreductase through MAP kinase-dependent pathways. Am J Physiol Lung Cell Mol Physiol 2006;291:L345–L353.
Machino T, Hashimoto S, Maruoka S et al. Apoptosis signal-regulating kinase 1-mediated signaling pathway regulates hydrogen peroxide-induced apoptosis in human pulmonary vascular endothelial cells. Crit Care Med 2003;31:2776–2781.
Wendt CH, Polunovsky VA, Peterson MS et al. Alveolar epithelial cells regulate the induction of endothelial cell apoptosis. Am J Physiol 1994;267 (4 Pt 1):C893–C900.
Berlett BS, Levine RL, Stadtman ER . Carbon dioxide stimulates peroxynitrite-mediated nitration of tyrosine residues and inhibits oxidation of methionine residues of glutamine synthetase: both modifications mimic effects of adenylylation. Proc Natl Acad Sci USA 1998;95:2784–2789.
Zhu S, Basiouny KF, Crow JP et al. Carbon dioxide enhances nitration of surfactant protein A by activated alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2000;278:L1025–L1031.
Laffey JG, Tanaka M, Engelberts D et al. Therapeutic hypercapnia reduces pulmonary and systemic injury following in vivo lung reperfusion. Am J Respir Crit Care Med 2000;162:2287–2294.
Reed JC . Mechanisms of apoptosis. Am J Pathol 2000;157:1415–1430.
Schulze-Osthoff K, Ferrari D, Los M et al. Apoptosis signaling by death receptors. Eur J Biochem 1998;254:439–459.
Mandlekar S, Yu R, Tan TH et al. Activation of caspase-3 and c-Jun NH2-terminal kinase-1 signaling pathways in tamoxifen-induced apoptosis of human breast cancer cells. Cancer Res 2000;60:5995–6000.
Pugin J, Dunn I, Jolliet P et al. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol 1998;275 (6 Pt 1):L1040–L1050.
Held HD, Boettcher S, Hamann L et al. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med 2001;163 (3 Pt 1):711–716.
Uhlig U, Haitsma JJ, Goldmann T et al. Ventilation-induced activation of the mitogen-activated protein kinase pathway. Eur Respir J 2002;20:946–956.
D'Arcangelo D, Gaetano C, Capogrossi MC . Acidification prevents endothelial cell apoptosis by Axl activation. Circ Res 2002;91:e4–12.
Laffey JG, Engelberts D, Duggan M et al. Carbon dioxide attenuates pulmonary impairment resulting from hyperventilation. Crit Care Med 2003;31:2634–2640.
Weber T, Tschernich H, Sitzwohl C et al. Tromethamine buffer modifies the depressant effect of permissive hypercapnia on myocardial contractility in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2000;162 (4 Pt 1):1361–1365.
Zhou Q, Cao B, Niu L et al. Effects of permissive hypercapnia on transient global cerebral ischemia-reperfusion injury in rats. Anesthesiology 2010;112:288–297.
Acknowledgements
We are indebted to Yan Liu (Dept of statistics, Harbin Medical University) for expert technical assistance and Professor Jiaren Liu (Harvard medical school) for assistance with English language usage. This study was supported by the National Natural Science Foundation of China (Grant No. 30772085) and graduate innovation fund of Heilongjiang Province.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Mechanical ventilation can cause pathological changes in lung tissue. This paper shows that hypercapnic acidosis signi?cantly decreases lung vascular leakage and the injury caused by in?ammatory cytokines and oxidative stress in a model of high-pressure ventilation-induced acute lung injury. The mechanism for these effects involves the reduction of inflammation and apoptosis via inhibition of the ASK1/JNK pathway and p38 MAP kinase phosphorylation.
Rights and permissions
About this article
Cite this article
Yang, WC., Song, CY., Wang, N. et al. Hypercapnic acidosis confers antioxidant and anti-apoptosis effects against ventilator-induced lung injury. Lab Invest 93, 1339–1349 (2013). https://doi.org/10.1038/labinvest.2013.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.118
Keywords
This article is cited by
-
The role of hypercapnia in acute respiratory failure
Intensive Care Medicine Experimental (2019)
-
Comparison of the effects of moderate and severe hypercapnic acidosis on ventilation-induced lung injury
BMC Anesthesiology (2015)
