
Abstract
Enamel-renal-gingival syndrome (ERGS; OMIM #204690), a rare autosomal recessive disorder caused by mutations in FAM20A, is characterized by nephrocalcinosis, nephrolithiasis, amelogenesis imperfecta, hypoplastic type, gingival fibromatosis and other dental abnormalities, including hypodontia and unerupted teeth with large dental follicles. We report three patients and their families with findings suggestive of ERGS. Mutation analysis of FAM20A was performed in all patients and their family members. Patients with homozygous frameshift and compound heterozygous mutations in FAM20A had typical clinical findings along with periodontitis. The other had a novel homozygous missense mutation in exon 10, mild gingival fibromatosis and renal calcifications. The periodontitis in our patients may be a syndrome component, and similar findings in previous reports suggest more than coincidence. Fam20a is an allosteric activator that increases Fam20c kinase activity. It is hypothesized that lack of FAM20A activation of FAM20C in our patients with FAM20A mutations might have caused amelogenesis imperfecta, abnormal bone remodeling and periodontitis. Nephrocalcinosis appears not to be a consistent finding of the syndrome and the missense mutation may correlate with mild gingival fibromatosis. Here we report three patients with homozygous or compound heterozygous mutations in FAM20A and findings that extend the phenotypic spectrum of this disorder, showing that protein truncation is associated with greater clinical severity.
Similar content being viewed by others
Introduction
Enamel-renal-gingival syndrome (ERGS; OMIM #204690), an autosomal recessive disorder caused by mutations in FAM20A (Golgi-associated secretory pathway pseudokinase), is characterized by nephrocalcinosis, nephrolithiasis, amelogenesis imperfecta, hypoplastic type, gingival fibromatosis, and other dental abnormalities, including hypodontia, and unerupted teeth with large dental follicles. There can be heterotopic calcifications in gingiva, dental follicles, dental pulp chambers, periodontal ligament and lungs.1, 2, 3, 4, 5, 6, 7, 8
FAM20A is a member of evolutionarily conserved Family with sequence similarity 20 (FAM20) that in mammals there are three members: Fam20a, Fam20b and Fam20c.9, 10 These Fam20 genes encode proteins that contain a conserved C-terminal putative kinase domain and a glycine-rich loop, a signature of protein kinases. These FAM20 kinases phosphorylate secreted proteins and proteoglycans. FAM20C is a Golgi lumen protein that serves as a protein kinase that phosphorylate secreted proteins on Ser-Xaa-Glu/phospho-Ser motifs,11, 12 while FAM20B phosphorylates xylosyl residues in proteoglycans.13 In contrast, FAM20A appears to be an inactive kinase, which instead appears to bind to and activate FAM20C in tissues expressing large amounts of secreted phosphoproteins.14 FAM20A has Glycine at amino acid position 258 (Gln258) which lacks negatively charged side chain and may not functionally substitute its ortholog Glu306 in FAM20C, which is required for kinase activity. FAM20A is therefore a pseudokinase.14, 15 Mutations in the FAM20A and FAM20C genes have been reported to be associated with disorders of biomineralization in humans and mice, indicating the physiological importance of secreted protein phosphorylation.4, 10, 16
We report three patients with homozygous or compound heterozygous mutations in FAM20A and findings that extend the phenotypic spectrum of this disorder, showing that protein truncation is associated with greater clinical severity.
In addition, we suggest a possible role for calcium ion flux abnormalities based on a known role for calcium channel blockers in causing gingival hyperplasia, in addition to dysregulation of inhibitory effects on biomineralization. We also suggest that periodontitis is more than just a coincidental finding in ERGS, and that calcium ion flux may have a pathogenic role here.
Materials and methods
Ethic statement
The study was conducted with informed consent from all patients and their parents. The study was approved by the Human Experimentation Committees of the Faculty of Medicine and Faculty of Dentistry, Chiang Mai University.
Subjects
Patient 1
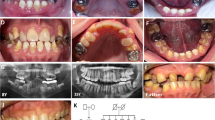
A 12-year-old Thai boy presented to the Dental Department, Lampang Hospital, Lampang because of mobility of the mandibular central incisors (Figure 1a). He was the first child of healthy consanguineous parents. His younger brother was healthy. Medical history and general physical findings were unremarkable. Oral examination showed amelogenesis imperfecta, hypoplastic type, prolonged retention of deciduous canines and molars, and generalized enlargement of the gingival tissue (Figures 1b–d). Panoramic radiograph showed generalized thin or absent enamel and severe alveolar bone loss around the mandibular incisors, mandibular right second premolar and all first permanent molars. Unerupted maxillary and mandibular permanent teeth were displaced apically and surrounded by very large dental follicles with sclerotic margins. Pulpal calcifications were evident in both erupted and unerupted teeth (Figure 1e). Renal ultrasonography showed multiple small hyperechoic lesions scattered in the parenchyma bilaterally, indicating nephrocalcinosis (Figure 1f). A diagnosis of ERGS with periodontitis was made. All other family members were investigated with renal ultrasonography and results were unremarkable.

Patient 1 at age 12 years (a). Intraoral photographs show amelogenesis imperfecta, hypoplastic type, gingival fibromatosis, retained deciduous teeth and delayed eruption of permanent teeth (b–d). Panoramic radiograph shows severe alveolar bone loss around the mandibular central incisors and all first permanent molars (arrows), generalized thin and absent enamel, multiple unerupted teeth with large dental follicles and pulp stones (e). Renal ultrasonograph of the right kidney shows multiple small hyperechoic lesions in renal parenchyma (f).
Patient 2
An 11-year-old Malay girl presented to the School of Dental Service, Health Promotion Board, Singapore, with the chief complaint of swollen and bleeding gingiva. Her parents and elder brother were healthy. Consanguineous marriage was denied. Medical history and general physical findings were unremarkable. Oral examination showed generalized enamel hypoplasia and generalized enlargement of gingiva. Teeth appeared yellowish and lacked proximal contacts because of a generalized absence of enamel (Figures 2a, b and d). Generalized periodontal disease with probing depth of 5–8 mm was detected mainly around the maxillary and mandibular incisors along with periodontal abscesses seen around the over-retained primary canines. There was also marked delayed eruption of maxillary canines and first permanent molars as a result of overlying hyperplastic gingiva. Increased overbite and marked reduction in vertical dimension was present secondary to the delayed eruption of the upper left and lower right first permanent molars. Radiographic examination showed generalized absence of enamel, absence of proximal contacts, severe alveolar bone loss around the maxillary and mandibular incisors, odd shaped pulp canals with early obliteration at the apical third of the root canals, and taurodontism of the permanent molars (Figures 3a–d). Histopathological studies of the gingiva revealed hyperplastic parakeratinized stratified squamous epithelium covering underlying connective tissue, which exhibits fibrous collagenous connective tissue hyperplasia. A few areas of the lesional fibrous connective tissue show a focally distributed chronic inflammatory cell infiltrate of mainly plasma cells. Small foci of psammomatoid calcifications are seen in the lesion (Figure 2c). Findings were consistent with amelogenesis imperfecta, hypoplastic type and generalized gingival fibromatosis, and a diagnosis of ERGS with periodontitis was made. Suggested renal ultrasonography was refused.

Patient 2 at age 11 years Amelogenesis imperfecta, hypoplastic type, generalized gingival fibromatosis and delayed eruption of permanent teeth (a, b and d). Histopathological study of gingiva shows psammomatoid calcifications (original magnification, × 40) (c).

Patient 2. Panoramic radiograph shows severe alveolar bone loss around the maxillary and mandibular incisors, generalized thin and absent enamel, multiple unerupted teeth with large dental follicles and pulp stones (a). Periapical radiographs show severe alveolar bone loss around incisors (b–d). Unusual shape of root canals (c).
Patient 3
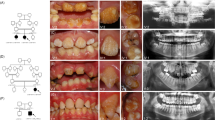
An 11-year-old Turkish boy presented to the Department of Pedodontics, Istanbul University, with problems with deciduous and permanent teeth. He was the first child of non-consanguineous parents (Figures 4a and e). No family history was reported of similar dental conditions.

Patient 3 at age 11 years (a). Intraoral photographs show amelogenesis imperfecta, hypoplastic type, mild gingival fibromatosis, retained deciduous teeth, infraclusion of deciduous first molars and delayed eruption of permanent teeth (b–d). Pedigree of patient 3 (e). Panoramic radiograph shows generalized loss of enamel thickness, multiple unerupted teeth with very large dental follicles, intrapulpal calcifications (arrowheads) and underdeveloped roots of the unerupted mandibular permanent second molars with very large dental follicles. Note thin lower border of the mandible (arrows) (f).
Oral examination revealed amelogenesis imperfecta, hypoplastic type in his early mixed dentition. Mild generalized gingival enlargement was observed. All of the deciduous teeth had erupted, with infra-occlusion of the maxillary right first deciduous molar and both mandibular first deciduous molars, and some deciduous teeth retained beyond the normal ages for exfoliation. Marked delayed eruption of the first permanent molars and mandibular left central permanent incisor was noted (Figures 4b–d).
A panoramic radiograph showed generalized loss of enamel thickness of erupted and unerupted teeth, delayed eruption of permanent teeth, and multiple unerupted permanent incisors, premolars and molars with hyperplastic dental follicles. Poorly developed roots of the unerupted mandibular permanent second molars appeared at the lower border of the mandible, which was eroded at those areas. The mandibular second permanent molar dental follicles were very large. Intrapulpal calcifications were observed in erupted and unerupted teeth (Figure 4f). No calcification was observed on the patient’s renal ultrasound and he was otherwise healthy. His parents declined kidney ultrasounds.
Mutation analysis
Informed consent and 1 ml of saliva sample of the patients and their parents were obtained and approved by the Human Experimentation Committee of the Faculty of Dentistry, Chiang Mai University and in full accordance with ethical principles, including the World Medical Association Declaration of Helsinki. Genomic DNA was extracted from saliva using Oragene Saliva collecting kits (DNA Genotek, Ottawa, Canada). Mutation analysis of FAM20A was performed using sequencing primers (Supplementary Tables S1–S4). Sequencing data were analyzed using the Sequencher software (Gene Codes Corporation, Ann Arbor, MI, USA) for the identification of the point mutations.
Results
Mutation analysis of FAM20A (Genbank accession no. NM_017565.3; NP_060035.2) of patient 1 revealed the homozygous mutation c.349_367delCTGGCCAGCCAGGAGGCGC in exon one (Supplementary Figure S1). Both parents were heterozygous for the mutation. The wild-type FAM20A protein comprises 541 amino acids (O'Sullivan et al.1). The c.349_367delCTGGCCAGCCAGGAGGCGC mutation is predicted to cause a frameshift at position 117, leading to a premature termination codon at position 138 (p.Leu117Cysfs*22; Supplementary Figure S1). Mutation analysis of FAM20A of patient 2 revealed compound heterozygous mutations of FAM20A, c.915_918delCTTT and c.976_977insG in exons 6 and 7, respectively. The c.915_918delCTTT mutation is predicted to cause a frameshift at position 305, leading to a premature termination codon at position 380 (p.Phe305Leufs*76; Supplementary Figure S2). The pathogenicity of the c.915_918delCTTT mutation was confirmed by the presence of this mutation in the previously reported patient (Kantaputra et al.).7 The c.976_977insG mutation is predicted to cause a frameshift at position 326, leading to a premature termination codon at position 379 p.Glu326Glyfs*54 (Supplementary Figure S3). This mutation has been reported in Genbank (rs761066773) but has not been reported to be associated with any genetic disorder.
Mutation analysis of FAM20A of patient 3 identified a novel homozygous missense mutation in exon 10. A base substitution from G to A (c.1307G>A) caused an amino acid change to glutamic acid instead of glycine at position 436 in the protein (p.Gly436Glu; Supplementary Figure S4). In accordance with the sequencing, this nucleotide change was identified not only in the proband, but also in heterozygous form in his parents, suggesting the mutation had been vertically transmitted from the parents to their offspring. Alignments of FAM20A amino acid sequences demonstrated that the glycine at position 436 was highly conserved during evolution (Supplementary Figure S5).
Pathogenicity of all mutations was supported by their absence in 100 unrelated healthy controls (200 chromosomes). The mutations were not found in the gene variant public databases, including NCBI, 1000 genome, and EXOME variant databases. The molecular alterations were predicted to be disease-causing variants by mutation prediction software, including SIFT, Polyphen2, MutationTaster and HANSA.
Mutant protein model
The replacement of the conserved Gly436 by the Glu residue is likely to disrupt the active site structure, due to the insertion of a larger side chain in a compact space and the introduction of an additional negative charge to imbalance the charge. Comparison to the structure of CeFAM20C and a derived homology model of FAM20A isoform a indicates that Gly436 is in a turn toward the end of the long active site loop that includes the kinase active site DFG motif (‘DHG’ in CeFAM20 and ‘DNA’ in FAM20A) and Arg433, which is expected to form a salt-bridge hydrogen bond with Asp410 (which would be the catalytic aspartate in an active kinase), as shown in Figure 5, and is likely to interact with the gamma-phosphate of ATP. Therefore, this mutation is likely to displace several residues around the ATP-binding site to destabilize the protein and impede FAM20A binding of ATP, which appears to be required for optimal activation of FAM20C by FAM20A.14

Model of the FAM20A protein active site with the p.G435E mutation. The homology model of the FAM20A (yellow) is superimposed with the X-ray crystal structure of the CeFAM20C with Mn-ADP. The mutation of Gly436 to Glu is shown in green and the clashes this mutation makes with the otherwise salt-bridging pair of Asp256 and Arg259 (Asp211 and Arg214 in CeFAM20C) are shown as pink dashes. The bridge between these two residues positions the active site residue Gln258 (Glu214 in CeFAM20C). Although the side chain of Glu436 could move elsewhere to avoid these clashes, the space is too tight and it will clash with other residues. Also, it is closely linked to Arg433 (Arg390 in CeFAM20C), which salt bridges to the catalytic Asp (Asp410 in FAM20A and Asp366 in CeFAM20C) to position it for catalysis, and other residues in the catalytic loop, such as Asp430 (Asp387). Putative hydrogen bonds between Arg433 and Asp410 of FAM20A and the beta-phosphate of ADP in the active site from the structure of CeFAM20C are shown as dark gray dashes. A likely position for the gamma-phosphate of ATP is indicated in red. The human FAM20A isoform a model generated with M4T4245 and CeFAM20C (PDB: 4KQB)15 are shown in cartoon representation with relevant amino acid side chains shown in stick representation (drawn with PyMol, Schrodinger LLC). Models made with other programs, including RastorX, SwissModel, Phyre2 and IntFold2 gave essentially the same structure in this part of the model, although they diverged in less conserved areas.
Discussion
FAM20A-associated syndrome delineation has long been an issue, and these newly discovered mutations extend the phenotype and illuminate the diverse functions of the protein. Amelogenesis imperfecta with gingival fibromatosis syndrome (OMIM #614253) and enamel-renal syndrome or amelogenesis imperfecta with nephrocalcinosis (ERS; OMIM #204690) were originally described as separate disorders, but both are caused by FAM20A mutations, and the name ‘enamel-renal-gingival syndrome’ was coined to recognize this situation.6 The characteristic features of this autosomal recessive disorder are nephrocalcinosis, nephrolithiasis, amelogenesis imperfecta, hypoplastic type, gingival fibromatosis and other dental abnormalities, including hypodontia and unerupted teeth with large dental follicles.1, 2, 3, 4, 5, 6, 7, 8 Heterotopic calcifications in gingiva, dental follicles, dental pulp chambers, periodontal ligament and lungs have also been reported.6
Phenotypically, de la Dure-Molla5 suggested that ‘fibrous gingival hyperplasia is a pathognomonic feature of the syndrome with variable severity.’ However, patient 3, with a novel homozygous c.1307G>A mutation, presented with mild gingival fibromatosis despite displaying manifestations of the typical amelogenesis imperfecta, hypoplastic type, tooth eruption issues, large dental follicles and dental pulp calcifications which strongly suggested the diagnosis.5 This seems to reflect a generally milder phenotype: gingival fibromatosis was mild, and no renal calcifications were seen on ultrasound, which is unusual. Previously, the youngest patient without this finding was in family 1 of Wang et al.8, at age 11 years. Jaureguiberry et al. (2013) ‘speculated that all individuals with biallelic FAM20A mutations will eventually show nephrocalcinosis,’ and our patient obviously needs to be followed, but there may be exceptions–certainly we would have felt the same about the gingival findings.3
This probably reflects a genotype–phenotype correlation with the missense mutation having a milder phenotype than a protein truncation. Still, significant effects on function are not unexpected. The c.1307G>A base substitution replaces a conserved glycine with glutamic acid at position 436 (p.Gly436Glu; Supplementary Figure S4), which is likely to destabilize the protein and disrupt the ATP-binding site structure by insertion of a larger side chain in a compact space, and the introduction of an additional negative charge. As shown in Figure 5, Gly436 is in the active site loop that includes several ATP-binding site residues, including Arg433, which is positioned to interact with the gamma-phosphate of ATP. Although FAM20A appears inactive as a kinase, the binding of ATP by FAM20A appears to be necessary for its strong activation of FAM20C.14 Because a FAM20A variant mutated to regain kinase and ATPase activity was a weaker activator of FAM20C, Cui et al. 15 posited that the clearance of ATP from the FAM20A active site cause a weaker activation of FAM20C than that of wild-type FAM20A, which binds ATP without hydrolyzing it.14 The mild symptoms suggest that the Gly436Glu variant may still activate FAM20C, but less effectively than wild-type FAM20, perhaps due to decreased binding of ATP. However, other mutations might have even milder manifestations, and the full spectrum remains uncertain.
Most of previously reported patients with ERGS had mutations that caused protein truncations. Only three missense mutations of FAM20A have been reported. These include Leu173Arg,3 Gly331Asp4 and Asp403Asn.8 However homozygous missense mutation (Leu173Arg) has only been reported only once (Family 9).3 As for the p.Gly436Glu mutation found in Patient 3, because FAM20A is most likely a loss-of-function mutation like all other FAM20A mutations identified so far and not catalytically active. It is likely that this mutation undermined structural integrity of FAM20A and made it unstable and unable to allosterically form complex with FAM20C, or FAM20A may be unstable or misfolded, so as to be cleared by the ER-associated protein degradation pathway.14, 15
Periodontitis found in our patients is worth emphasizing as a possible part of the disorder, and may be associated with certain mutations more than others. According to the most recent classification of periodontal diseases, the periodontal disease of our patients would be diagnosed as periodontitis as a manifestation of systemic and genetic disorder.17 The c.349_367delCTGGCCAGCCAGGAGGCGC mutation in patient 1 was reported with ERGS with periodontitis and compound heterozygosity along with a 915_918delCTTT mutation.7 The same c.915_918delCTTT mutation was also found in patient 2 with compound heterozygosity, who had periodontitis as well.
The observation of periodontitis which had early onset in patients 1 and 2, and in previously reported patients, including two Thai and one Israeli, the latter without gene analysis,7, 18 presents another interesting issue in phenotype definition. Periodontitis is very rare in the young population, but gingival fibromatosis may be a contributing factor in patients 1 and 2. However, there are several suggestions that more is involved. First, it can be seen in specific families, and specific mutations seem associated, suggesting a genotype–phenotype correlation. Second, it is a particularly virulent form, with a young age of onset and a rapid clinical course. Third, Fam20a knockout mice also show periodontal inflammation, even though gingival fibromatosis was not noted.19, 20
In humans, mutations in FAM20C (Golgi-associated secretory pathway kinase) have been reported to cause autosomal recessive lethal osteosclerotic bone dysplasia or Raine syndrome (OMIM #259775), characterized by craniofacial dysmorphism, osteosclerosis, gingival hyperplasia, amelogenesis imperfecta, periodontal disease and pulmonary hypoplasia.21, 22 The overlap of the phenotypes of Fam20a and Fam20c knockout mice and patients with FAM20A and FAM20C mutations including amelogenesis imperfecta, gingival fibromatosis, ectopic calcifications and periodontal disease supports that FAM20A and FAM20C are closely related structurally, functionally and developmentally. Fam20a controls extracellular localization of and allosterically interact with Fam20c,23 a Golgi casein kinase in the secretory pathway responsible for serine phosphorylation in the Ser-x-Glu/pSer motifs in several enamel matrix proteins, including ameloblastin and enamelin. In other words, the function of Fam20a is structural, rather than catalytic. It is therefore essential for Fam20a to adopt a well-folded and stable structure.14 This explains the finding of amelogenesis imperfecta in patients with FAM20A and FAM20C mutations and in mice lacking either Fam20a or Fam20c.19, 20, 24 The milder phenotype in patients with FAM20A mutations than that of FAM20C mutations can be explained by the lack of complete loss of Fam20c-dependent substrate phosphorylation in the absence of Fam20a in vitro.14
Fam20c is intensely expressed in the mineralized tissues, including enamel, dentin, cementum, bone and cartilage. Its expression has also been shown in soft tissues, including periodontal ligament, cerebral cortex, cranial nerve ganglia, lung, liver, heart and kidney.9, 20, 25 Fam20c secretion is tissue-specific23 and is crucial for the formation and mineralization of all mineralized tissues, consisting of enamel, dentin, cementum and bones. Fam20c knockout mice showed severe defective ameloblasts and enamel matrix with subsequent amelogenesis imperfecta similar to those in the ameloblastin or enamelin knockout mice. In addition to amelogenesis imperfecta, severe defects in dentin and bones were also observed, along with hypophosphatemia.4, 19, 26 Fam20c-knockout mice had severe loss of alveolar bone and cementum, periodontal inflammation and periodontal pockets. Histologically, dental pulp and dentin appeared abnormal19 and the alveolar bone showed dramatic abnormalities of osteocytes and lacuno-canalicular networks. The severity of periodontal disease progresses with age.20 The Fam20c-knockout mice had markedly thickened maxillae and mandibles,19 most likely caused by abnormal bone remodeling. Fam20c is important for bone remodeling because, in addition to enamel matrix proteins, Fam20c also phosphorylates a number of members of the secretory calcium-binding phosphoprotein (SCPP) family, including small integrin-binding ligand N-linked glycoproteins.12, 16 These small integrin-binding ligand N-linked glycoprotein proteins consist of bone sialoprotein, osteopontin, dentin matrix protein 1 and dentin sialophosphoprotein.27 The periodontal disease in the Fam20c-knockout mice may reflect periodontal deterioration due to the low levels of bone sialoprotein, osteopontin, dentin matrix proteins, dentin sialoprotein, periostin and fibrillin-1. These proteins are secreted into the extracellular matrix of certain mineralized and non-mineralized tissues28 and have crucial roles in periodontal formation and maintenance of a healthy periodontium.29, 30, 31 Periostin32, 33, 34 and fibrillin-1,35, 36, 37, 38 extracellular matrix proteins essential for periodontal tissues contain several Ser-x-Glu/pSer motifs, and thus are potential substrates of FAM20C. All lines of evidence have shown that FAM20C has important roles in osteoblast differentiation and is crucial for periodontal formation and maintenance. FAM20A interacts directly with FAM20C and function as an allosteric activator to activate FAM20C kinase activity in a dose-dependent manner.14 Therefore, lack of FAM20A activation of FAM20C in our patients with FAM20A mutations might result in inefficient phosphorylation of enamel and bone matrix proteins and led to amelogenesis imperfecta, abnormal bone remodeling and periodontitis.
Our previously reported patients with ERGS with periodontal disease had periodontal ligament psammomatoid calcifications.7 As periodontal disease is a disease of defective immune system.39 We hypothesize that periodontitis in the patients with ERGS is caused by defective immune system as a result of FAM20A mutations and the disease condition is aggravated by the presence of psammomatoid calcifications. There may be similar to what observed around calcium oxalate crystals in a patient with hyperoxaluria and oxalosis.40 Ectopic calcifications patients with ERGS is hypothesized to be caused by the loss of FAM20A kinase and phosphorylation activities which subsequently leads to abnormal regulation of inhibitory system on biomineralization.7, 19, 23 The better understanding on ectopic calcification-related FAM20A mutations might lead us to understand more on ectopic calcifications in other tissues such as heart valves and blood vessels, which are life-threatening in general population.
It is likely that other pathogenetic factors may also be involved. Although not previously remarked upon, it is noteworthy that the typical gingival findings also occur with calcium channel blockers–classically nifedipine, but others as well.41, 42 Also, periodontal disease damage involves T-cell subset balance,43 which in turn can be affected by calcium ion flux,44 and experience with gingival hyperplasia secondary to calcium channel blockers supports this particular mechanism. In addition, Fam20a has been reported to have important roles in hematopoietic differentiation,9 and comparison of the expression of FAM20A in human tissues showed intermediate levels in the thymus, which would be consistent with T-cell involvement.9
All previously reported patients with ERGS with periodontitis had protein truncations. FAM20 family proteins show a highly conserved sequence of ~350 amino acids in the C-terminal region,9 which is likely to be affected. Even though periodontal disease is influenced by multiple factors, genotype–phenotype correlation is suggested here.
References
O'Sullivan, J., Bitu, C. C., Daly, S. B., Urquhart, J. E., Barron, M. J., Bhaskar, S. S. et al. Whole-Exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am. J. Hum. Genet. 88, 616–620 (2011).
Cho, S. H., Seymen, F., Lee, K. E., Lee, S. K., Kweon, Y. S., Kim, K. J. et al. Novel FAM20A mutations in hypoplastic amelogenesis imperfecta. Hum. Mutat. 33, 91–94 (2012).
Jaureguiberry, G., De la Dure-Molla, M., Parry, D., Quentric, M., Himmerkus, N., Koike, T. et al. Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A mutations. Nephron. Physiol. 122, 1–6 (2012).
Wang, S. K., Aref, P., Hu, Y., Milkovich, R. N., Simmer, J. P., El-Khateeb, M. et al. FAM20A mutations can cause enamel-renal syndrome (ERS). PLoS Genet. 9, e1003302 (2013).
de la Dure-Molla, M., Quentric, M., Yamaguti, P. M., Acevedo, A. C., Mighell, A. J., Vikkula, M. et al. Pathognomonic oral profile of Enamel Renal Syndrome (ERS) caused by recessive FAM20A mutations. Orphanet J. Rare. Dis. 9, 84 (2014).
Kantaputra, P. N., Bongkochwilawan, C., Kaewgahya, M., Ohazama, A., Kayserili, H., Erdem, A. P. et al. Enamel-Renal-Gingival syndrome, hypodontia, and a novel FAM20A mutation. Am. J. Med. Genet. A 164A, 2124–2128 (2014a).
Kantaputra, P. N., Kaewgahya, M., Khemaleelakul, U., Dejkhamron, P., Sutthimethakorn, S., Thongboonkerd, V. et al. Enamel-renal-gingival syndrome and FAM20A mutations. Am. J. Med. Genet. A 164A, 1–9 (2014b).
Wang, S. K., Reid, B. M., Dugan, S. L., Roggenbuck, J. A., Read, L., Aref, P. et al. FAM20A mutations associated with enamel renal syndrome. J. Dent. Res. 93, 42–48 (2014).
Nalbant, D., Youn, H., Nalbant, S. I., Sharma, S., Cobos, E., Beale, E. G. et al. FAM20: an evolutionarily conserved family of secreted proteins expressed in hematopoietic cells. BMC Genomics 6, 11 (2005).
Tagliabracci, V. S., Pinna, L. A. & Dixon, J. E. Secreted protein kinases. Trends Biochem. Sci. 38, 121–130 (2013).
Kawasaki, K. & Weiss, K. M. SCPP gene evolution and the dental mineralization continuum. J. Dent. Res. 87, 520–531 (2008).
Tagliabracci, V. S., Engel, J. L., Wen, J., Wiley, S. E., Worby, C. A., Kinch, L. N. et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 336, 1150–1153 (2012).
Koike, T., Izumikawa, T., Tamura, J. & Kitagawa, H. FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem. J. 421, 157–162 (2009).
Cui, J., Xiao, J., Tagliabracci, V. S., Wen, J., Rahdar, M. & Dixon, J. E. A secretory kinase complex regulates extracellular protein phosphorylation. Elife 4, e06120 (2015).
Xiao, J., Tagliabracci, V. S., Wen, J., Kim, S. A. & Dixon, J. E. Crystal structure of the Golgi casein kinase. Proc. Natl Acad. Sci. USA 110, 10574–10579 (2013).
Ishikawa, H. O., Xu, A., Ogura, E., Manning, G. & Irvine, K. D. The Raine syndrome protein FAM20C is a Golgi kinase that phosphorylates bio-mineralization proteins. PLoS ONE 7, e42988 (2012).
López, R. & Baelum, V. Periodontal disease classifications revisited. Eur. J. Oral Sci. 123, 385–389 (2015).
Ashkenazi, M., Rafe, Z., Sarnat, H. & Levin, L. Nephrocalcinosis associated with continuous enamel hypoplasia and severe alveolar bone loss: a case report and literature review. Pediatr. Dent. 36, 250–253 (2014).
Vogel, P., Hansen, G. M., Read, R. W., Vance, R. B., Thiel, M., Liu, J. et al. Amelogenesis imperfecta and other biomineralization defects in Fam20a and Fam20c null mice. Vet. Pathol. 49, 998–1017 (2012).
Liu, P., Zhang, H., Liu, C., Wang, X., Chen, L. & Qin, C. Inactivation of Fam20C in cells expressing type I collagen causes periodontal disease in mice. PLoS ONE 9, e114396 (2014).
Simpson, M. A., Hsu, R., Keir, L. S., Hao, J., Sivapalan, G., Ernst, L. M. et al. Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am. J. Hum. Genet. 81, 906–912 (2007).
Acevedo, A. C., Poulter, J. A., Alves, P. G., de Lima, C. L., Castro, L. C., Yamaguti, P. M. et al. Variability of systemic and oro-dental phenotype in two families with non-lethal Raine syndrome with FAM20C mutations. BMC Med. Genet. 16, 8 (2015).
Ohyama, Y., Lin, J. H., Govitvattan, N., Lin, I. P., Venkitapathi, S., Alamoudi, A. et al. FAM20A binds to and regulates FAM20C localization. Sci. Rep. 6, 27784 (2016).
Rafaelsen, S. H., Raeder, H., Fagerheim, A. K., Knappskog, P., Carpenter, T. O., Johansson, S. et al. Exome sequencing reveals FAM20C mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J. Bone Miner. Res. 28, 1378–1385 (2013).
Wang, X., Hao, J., Xie, Y., Sun, Y., Hernandez, B., Yamoah, A. K. et al. Expression of FAM20C in the osteogenesis and odontogenesis of mouse. J. Histochem. Cytochem. 58, 957–967 (2010).
Wang, X., Wang, S., Lu, Y., Gibson, M. P., Liu, Y., Yuan, B. et al (2012) FAM20C plays an essential role in the formation of murine teeth. J. Biol. Chem. 287, 35934–35942.
Fisher, L. W., Torchia, D. A., Fohr, B., Young, M. F. & Fedarko, N. S. Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochem. Biophys. Res. Commun. 280, 460–465 (2001).
Qin, C., Baba, O. & Butler, W. T. Post-translational modifications of sibling proteins and their roles in osteogenesis and dentinogenesis. Crit. Rev. Oral Biol. Med. 15, 126–136 (2004).
Ye, L., Zhang, S., Ke, H., Bonewald, L. F. & Feng, J. Q. Periodontal breakdown in the Dmp1 null mouse model of hypophosphatemic rickets. J. Dent. Res. 87, 624–629 (2008).
Foster, B. L., Soenjaya, Y., Nociti, F. H. Jr., Holm, E., Zerfas, P. M., Wimer, H. F. et al. Deficiency in acellular cementum and periodontal attachment in bsp null mice. J. Dent. Res. 92, 166–172 (2013).
Gibson, M. P., Zhu, Q., Liu, Q., D'Souza, R. N., Feng, J. Q. & Qin, C. Loss of dentin sialophosphoprotein leads to periodontal diseases in mice. J. Periodontal. Res. 48, 221–227 (2013).
Coutu, D. L., Wu, J. H., Monette, A., Rivard, G. E., Blostein, M. D. & Galipeau, J. Periostin, a member of a novel family of vitamin K-dependent proteins, is expressed by mesenchymal stromal cells. J. Biol. Chem. 283, 17991–18001 (2008).
Romanos, G. E., Asnani, K. P., Hingorani, D. & Deshmukh, V. L. PERIOSTIN: role in formation and maintenance of dental tissues. J. Cell. Physiol. 229, 1–5 (2014).
Rios, H. F., Ma, D., Xie, Y., Giannobile, W. V., Bonewald, L. F., Conway, S. J. et al. Periostin is essential for the integrity and function of the periodontal ligament during occlusal loading in mice. J. Periodontol. 79, 1480–1490 (2008).
Pereira, L., D'Alessio, M., Ramirez, F., Lynch, J. R., Sykes, B., Pangilinan, T. et al. Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum. Mol. Genet. 2, 961–968 (1993).
Shiga, M., Saito, M., Hattori, M., Torii, C., Kosaki, K., Kiyono, T. et al. Characteristic phenotype of immortalized periodontal cells isolated from a Marfan syndrome type I patient. Cell Tissue Res. 331, 461–472 (2008).
Suda, N., Shiga, M., Ganburged, G. & Moriyama, K. Marfan syndrome and its disorder in periodontal tissues. J. Exp. Zool. B Mol. Dev. Evol. 312b, 503–509 (2009).
Ganburged, G., Suda, N., Saito, M., Yamazaki, Y., Isokawa, K. & Moriyama, K. Dilated capillaries, disorganized collagen fibers and differential gene expression in periodontal ligaments of hypomorphic fibrillin-1 mice. Cell Tissue Res. 341, 381–395 (2010).
Hajishengallis, G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 15, 30–44 (2015).
Panis, V., Tosios, K. I., Gagari, E., Griffin, T. J. & Damoulis., P. D. Severe periodontitis in a patient with hyperoxaluria and oxalosis: a case report and review of the literature. J. Periodontol. 81, 1497–1504 (2010).
Fardal, O. & Lygre, H. Management of periodontal disease in patients using calcium channel blockers—gingival overgrowth, prescribed medications, treatment responses and added treatment costs. J. Clin. Periodontol. 42, 640–646 (2015).
Trackman, P. C. & Kantarci, A. Molecular and clinical aspects of drug-induced gingival overgrowth. J. Dent. Res. 94, 540–546 (2015).
Ohlrich, E. J., Cullinan, M. P. & Seymour, G. J. The immunopathogenesis of periodontal disease. Aust. Dent. J. 54 (Suppl 1), 2–10 (2009).
Toldi, G., Kaposi, A., Zsembery, A., Treszl, A., Tulassay, T. & Vasarhelyi, B. Human Th1 and Th2 lymphocytes are distinguished by calcium flux regulation during the first 10 min of lymphocyte activation. Immunobiology 217, 37–43 (2012).
Rykunov, D., Steinberger, E., Madrid-Aliste, C. J. & Fiser, A. Improved scoring function for comparative modeling using the M4T method. J. Struct. Funct. Genomics 10, 95–99 (2009).
Acknowledgements
We thank our patients and their families for their kind cooperation and for allowing us to use their medical and dental information for the benefit of others. We would also like to thank Dr Yeo Jin Fei for carrying out the histopathology analysis for Patient 2 in this report. Drs Jixin Cui and Jack Dixon are thanked for helpful comments on the manuscript. This research project was supported by The Center of Excellence in Medical Genetics Research, Chiang Mai University; the Thailand Research Fund (TRF); Dental Association of Thailand, and The Faculty of Dentistry, Chiang Mai University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Kantaputra, P., Bongkochwilawan, C., Lubinsky, M. et al. Periodontal disease and FAM20A mutations. J Hum Genet 62, 679–686 (2017). https://doi.org/10.1038/jhg.2017.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.26
This article is cited by
-
Abnormal teeth and renal calcifications: Answers
Pediatric Nephrology (2023)
-
FAM20C directly binds to and phosphorylates Periostin
Scientific Reports (2020)
-
Aggressive periodontitis and NOD2 variants
Journal of Human Genetics (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}