
Abstract
Retinitis pigmentosa (RP) is a heterogenous group of inherited retinal degenerations caused by mutations in at least 50 genes. To identify genetic mutations underlying autosomal recessive RP (arRP), we performed whole-exome sequencing study on two consanguineous marriage Indian families (RP-252 and RP-182) and 100 sporadic RP patients. Here we reported novel mutation in FAM161A in RP-252 and RP-182 with two patients affected with RP in each family. The FAM161A gene was identified as the causative gene for RP28, an autosomal recessive form of RP. By whole-exome sequencing we identified several homozygous genomic regions, one of which included the recently identified FAM161A gene mutated in RP28-linked arRP. Sequencing analysis revealed the presence of a novel homozygous frameshift mutation p.R592FsX2 in both patients of family RP-252 and family RP-182. In 100 sporadic Indian RP patients, this novel homozygous frameshift mutation p.R592FsX2 was identified in one sporadic patient ARRP-S-I-46 by whole-exome sequencing and validated by Sanger sequencing. Meanwhile, this homozygous frameshift mutation was absent in 1000 ethnicity-matched control samples screened by direct Sanger sequencing. In conclusion, we identified a novel homozygous frameshift mutations of RP28-linked RP gene FAM161A in Indian population.
Similar content being viewed by others

Introduction
Retinitis pigmentosa (RP) is a group of hereditary degenerative diseases of the retina displaying a significant phenotypic and genotypic variability and characterized by the progressive decrease of rod and cone photoreceptor function.1 The disease is highly heterogeneous with a prevalence of 1:4000 in the world2 and it can be inherited as autosomal recessive (50–60%), autosomal dominant (30–40%) or X-linked (5–15%).3 Initial symptoms include night blindness and a gradual constriction of peripheral vision caused by rod photoreceptor death. Secondary loss of cone photoreceptors may subsequently lead to impairment of central vision and ultimately legal blindness.4 Characteristic clinical features are elevated final dark adaptation thresholds and reduced and delayed full-field electroretinogram signals, which measures the electrical response of the retina to flashes of light.5 In recent years enormous efforts have been made in research and an early recognition of retinal degenerations has become possible.
At present, according to the RetNet database (https://sph.uth.edu/retnet/), over 60 genes have been associated with non-syndromic RP.6 The functions of these genes include components of the phototransduction cascade and the visual cycle, the regulation of retinal gene expression and general cellular processes like splicing, protein modification and degradation.7 In the recent years, numbers of RP genes encode proteins localized at the base of and within the connecting cilium as well as the outer segment axoneme of photoreceptor cells are growing.8 Nevertheless, these mutations account for only ~50–60% of the existing disease alleles.3
Enormous efforts have been made in identifying genetic mutations contributive to inherited retinal diseases since 1980s. However, most of these techniques are relatively inefficient, expensive and labor intensive. These days, the whole-exome sequencing has been proven to be the most efficient tool for identifying novel autosomal recessive RP (arRP) genes and mutations.9, 10 On account of its remarkable characteristic, whole-exome sequencing can contribute to novel disease gene identification and more efficient and accurate molecular diagnosis.
In Indian population, family-based disease gene analysis is highly effective, because of relatively high levels of consanguinity, large number of siblings in families and presence of isolated subpopulations. As a part of an international cooperation, we used exome sequencing to identify the disease causing genes in families from Indian population with RP. Here we report the identification of a novel homozygous frameshift mutation p.R592FsX2 in the FAM161A gene in two consanguineous Indian families and one sporadic Indian RP patient.
Materials and methods
Patients and controls
Two patients and three unaffected family members from two consanguineous marriage families in South India were recruited from Aravind Eye Hospital of India. 100 sporadic Indian RP patients and 1000 control individuals with no history of retinal degeneration were also from Aravind Eye Hospital of India. This research was carried out in accordance with the tenets of the Declaration of Helsinki and was approved by the Aravind Medical Research Foundation and Aravind Eye Hospital of India and Hospital of the University of Electronic Science and Technology of China and Sichuan Provincial People's Hospital. Written informed consent was obtained from the patients who participated in this study or from the legal guardians in case of minors.
DNA samples and exome sequencing
All genomic DNA samples were extracted from peripheral blood leukocytes of relative RP patients and control individuals using a blood DNA extraction kit according to the protocol provided by the manufacturer (TianGen, Beijing, China). Exome sequencing was performed on DNA samples of the index patients by Macrogen Inc, Seoul, Korea. DNA Illumina (San Diego, CA USA) TruSeq Exome Capture System (62 Mb) was used to collect the protein coding regions of human genome DNA. It covered 20 794 genes and 20 1121 exons in the Consensus Coding Sequence Region database (http://www.illumina.com/applications/sequencing/targetedresequencing.ilmn).
Data analysis
High-quality sequencing reads were aligned to the human reference genome (NCBI build 37.1/hg19) with SOAPaligner (soap2.21). Based on the SOAP alignment results, SOAPsnp v1.05 was used to assemble the consensus sequence and call genotypes in target regions. Data were provided as lists of sequence variants (single-nucleotide polymorphisms (SNPs) and short indels). We filtered SOAPsnp results following several steps as described previously.11 Briefly, for SNP quality control, we filtered SOAPsnp results as follows: (i) base quality is more than 20; (ii) depth is between 4 and 200; (iii) estimate copy number is ⩽2; and (iv) the distance between two SNPs must be longer than 4. Small indel detection was performed using the Unified-Genotyper tool from GATK (version v1.0.4705) after all high-quality reads were aligned to the human reference genome using BWA (version 0.5.9-r16). SNP and Indel detection were performed only on the targeted exome regions and flanking regions within 200 bp. The detected variants were annotated and filtered based on four databases, that is, NCBI CCDS (http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi), RefSeq (http://www.ncbi.nlm.nih.gov/RefSeq/), Ensembl (http://www.ensembl.org) and Encode (http://genome.ucsc.edu/ENCODE). Four major steps were taken to prioritize all the high-quality variants: (i) variants within intergenic, intronic and UTR regions and synonymous mutations were excluded from downstream analysis; (ii) variants in dbSNP137 (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genome project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp), YH Database (http://yh.genomics.org.cn/), HapMap Project (ftp://ftp.ncbi.nlm.nih.gov/hapmap) and our in-house database, which was generated by our laboratory using 1600 whole-exome sequencing data, were excluded; (iii) possible damaging impacts of each variant on protein structure/function were predicted by SIFT (http://sift.bii.a-star.edu.sg/) and Polyphen2 (http://genetics.bwh.harvard.edu/pph2/); and (iv) gene ontology (http://www.geneontology.org) and KEGG pathway annotations (http://www.ebi.ac.uk./clustalw) were used to predict the biological function of each putative gene.
Mutations validation
After reads calling, mapping and filtering against multiple databases, genomic variants were identified. Validation of the mutations in patients and their relatives was performed by Sanger sequencing on an ABI 3730XL Genetic Analyzer using the following primers: FAM161-p.R592FsX2-F, 5′-GAGGTAAACTGTGAACTGTGCT-3′; FAM161-p.R592FsX2-R, 5′-CACCTCAGCCTCCCAAGTAG-3′. Sequencing data were used to determine whether the remaining mutations co-segregated with the disease in these families and whether the frameshift mutations were confirmed in the 100 sporadic RP patients.
Results
Patients and clinical information
Two patients affected with RP from two consanguineous marriage Indian families RP-252 and RP-182 were recruited (Figure 1). These two RP patients presented with an early-onset increasing difficulty to adapt in the dark and gradually decreased vision acuity in both eyes and typical RP features were observed. The fundus examination of patient IV:4 in RP-252 family was shown in Figure 2. Other examinations, including main complaints, history, ocular examination and electroretinogram examination were shown in Supplementary Data 1. All sporadic Indian RP patients recruited in this study were under normal ophthalmological examinations.

Pedigrees of Indian families RP-252 and RP-182 with arRP. (a) Arrow indicates the proband IV:1 in family RP-252. (b) Arrow indicates the proband IV:1 in family RP-182. Solid symbol indicate affected individual and open symbols indicate unaffected individuals.

Fundus examination of patient IV:1 in family RP-252. (a and b) Fundus examination for right eye. (c and d) Fundus examination for the left eye. Both retina showed typical signs of RP: pigment formation. A full color version of this figure is available at the Journal of Human Genetics journal online.
Exome sequencing analysis
By exome sequencing of patient IV:1 in family RP-252 and patient IV:1 in family RP-182, we generated 5.2 billion and 5.3 billion bases of sequence with average throughput depth of target regions 103.2 × and 104.3 ×, respectively. 64 744 SNPs and 4755 indels that may affect amino acid sequence were identified in patient IV:1 of family RP-252 and 67 494 SNPs and 5609 indels that may affect amino acid sequence were identified in patient IV:1 of family RP-182 (Table 1).
In this study, we firstly compared these total variants with reported retina genes (https://sph.uth.edu/Retnet/). Screening though the RP gene database, we identified 47, 37 variants of several known RP genes in patients of family RP-252 and family RP-182, respectively. We then compared these variants with the dbSNP135, 1000 Genome Project, HapMap project, YH database and our in-house database, described previously.11 Therefore, we can exclude the variants with high frequency in normal population. By excluding the heterozygous variants and synonymous mutations, we found one variant in each patient. The variants screened by several database were show in Table 1 (candidate mutations filtered with 1000 genome database for RP-252 and RP-182 were listed in Supplementary Table 1).
After these steps, we identified a homozygous frameshift mutation p.R592FsX2 in the known RP gene FAM161A in both patients of family RP-252 and family RP-182. We checked the p.R592FsX2 mutation in the human gene mutation database (http://www.hgmd.org/) and found that the mutation is novel. We also checked the mutation in the newly available ExAC database of 63 000 control exomes (http://exac.broadinstitute.org/) and no variants were reported in this locus of FAM161A gene.
Mutation validation and analysis
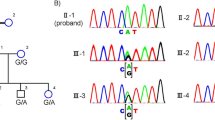
Sanger sequencing confirmed that the homozygous frameshift mutation p.R592FsX2 was in the affected wife and demonstrated that her son was an unaffected carrier, while her husband carried no mutations in family RP-252 (Figure 3a). In family RP-182, the frameshift mutation p.R592FsX2 was homozygous in the affected son and heterozygous in his unaffected father (Figure 3b). The genotypes of the family members were shown in Table 2.

Mutation identification of FAM161A gene in families RP-252 and RP-182. (a) Sanger sequencing analysis of FAM161A in family RP-252 showing the homozygous frameshift mutation p.R592FsX2 co-segregated with the phenotype. Patient IV:1 harbored homozygous frameshift mutation p.R592FsX2 of the FAM161A gene. The heterozygous mutation p.R592FsX2 was carried by her son V:2, while her husband IV:2 did not carry this mutation. (b) Sanger sequencing analysis of FAM161A in family RP-182 showed patient IV:1 harboring the homozygous p.R592FsX2 mutation while the father harboring the heterozygous mutation. (c) Sanger sequencing analysis of sporadic RP patient ARRP-S-I-46 showed the homozygous frameshift mutation p.R592FsX2. A full color version of this figure is available at the Journal of Human Genetics journal online.
Sanger sequencing result showed complete co-segregation of the mutations with the disease phenotype in the two consanguineous Indian families. We then screened these variants in 100 sporadic Indian RP patients by whole-exome sequencing. The homozygous frameshift mutation p.R592FsX2 was identified in one sporadic patient ARRP-S-I-46 (figure 3c). This mutation described above was absent in 1000 ethnicity-matched control samples screened by direct Sanger sequencing. In summary, we totally analyzed 14 India families with arRP and 100 sporadic Indian ARRP patients and we found two pedigrees with the FAM161A p.R592FsX2 homozygous mutation and one sporadic patient with this mutation. Thus, the frequency of FAM161A mutations in Indian RP patients form our result is similar to that of Israel and the Palestinian territories. These data, together with the clinical information, demonstrated that homozygous frameshift mutation p.R592FsX2 in the gene FAM161A was responsible for these three arRP patients from Indian.
The mutation p.R592FsX2 in the gene FAM161A introduced a frameshift, according to GenBank accession number NM_001201543.1. As shown in Figure 4, the amino acid changes affected highly conserved residues in five species. Therefore, the novel frameshift mutation was likely to affect the FAM161A protein function.

Protein alignment showed conservation of residues of FAM161A p.R592FsX2 across five species. This mutation abolished evolutionarily conserved amino acids. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
RP is an inherited disease of the retina, which leads to vision impairment due to progressive photoreceptor cell death. There are more than 50 known disease genes responsible for RP and there are still many new RP disease genes yet to be identified. For the mapping of new loci and subsequent identification of new genes for all inherited forms of RP, the whole-exome sequencing has been shown to be a powerful method. In this study, we found a novel homozygous frameshift mutation p.R592FsX2 in the known RP gene FAM161A in the Indian population with arRP using the whole-exome sequencing technology.
The novel mutation p.R592FsX2 in FAM161A identified in this study introduces a frameshift from exon 4, which truncates 124 amino acids from the FAM161A protein (716 amino acids). Therefore, the novel mutation is likely to affect the FAM161A protein function, thus resulting in the occurrence of arRP.
FAM161A gene mutation was initially identified in a study of RP patients in 2010.12 The previous study showed FAM161A mutations a major cause of arRP in Israel and the Palestinian territories, probably 12%, meanwhile they appear to be less frequent in other parts of the world, for example, Germany and the USA, about 1.5–3%.12, 13 In our result, the frequency of FAM161A mutations in Indian RP patients form our result is close to that of Israel and the Palestinian territories. Quite a few known mutations cluster in exon 3 of the FAM161A gene (Figure 5), which is by far the largest coding exon of the whole gene.12, 14 These mutations are predicted to result in a truncated polypeptide that lacks the carboxyl-terminus of the genuine protein, therefore only little of the truncated protein is expected to be synthesized.15 FAM161A-associated retinal disease displays typical features of arRP but shows a wide phenotypic variability in terms of age of onset and the severity of clinical symptoms.14

Localization of currently known RP-associated mutations in FAM161A gene. The novel p.R592FsX2 mutation found in this study is shown in red. Blue bars indicate coding exons or exon segments whereas white bars represent the 5′ and 3′ untranslated region (UTR), respectively. A full color version of this figure is available at the Journal of Human Genetics journal online.
The human FAM161A gene consists of seven exons and spans ~30 kb of genomic DNA on chromosome 2p15 with two proteins coding transcript variants, 660 amino acids (76 kDa) and 716 amino acids (83 kDa), respectively. Recent studies have shown that FAM161A localizes to the connecting cilium, the basal body and the adjacent centriole in mammalian photoreceptors. FAM161A was also present in synaptic layers and ganglion cells of the retina,12, 14 suggesting that this molecule participates in the maintenance of microtubule tracks. In addition, FAM161A directly binds to microtubules and increases the acetylation of α-tubulin and execution of microtubule-dependent functions in the non-photoreceptors.16 Moreover, FAM161A was found post-synaptic to the ribbon synapses in retinal second-order neurons similar to some IFT complex B proteins and the KIF17 motor.17, 18 Taken together, loss of FAM161A function results in mislocalization and accumulation of cargo vesicles in the inner segment as well as disturbed synaptic transmission, eventually causing retinal cell degeneration.
In this report, we identified a novel homozygous frameshift mutation p.R592FsX2 in the Indian population affected by RP disease. This study enriched the mutation spectrum of FAM161A gene for RP disease and confirmed that a loss of FAM161A function leads to RP disease in India population.
Accession codes
References
Koenekoop, R. K. Why do cone photoreceptors die in rod-specific forms of retinal degenerations? Ophthalmic Genet. 30, 152–154 (2009).
Bunker, C. H., Berson, E. L., Bromley, W. C., Hayes, R. P. & Roderick, T. H. Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 97, 357–365 (1984).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Bovolenta, P. & Cisneros, E. Retinitis pigmentosa: cone photoreceptors starving to death. Nat. Neurosci. 12, 5–6 (2009).
Berson, E. L. Retinitis pigmentosa. The Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 34, 1659–1676 (1993).
Daiger, S. P., Bowne, S. J. & Sullivan, L. S. Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 125, 151–158 (2007).
Ridley, A. J. & Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399 (1992).
Karlstetter, M., Sorusch, N., Caramoy, A., Dannhausen, K., Aslanidis, A., Fauser, S. et al. Disruption of the retinitis pigmentosa 28 gene Fam161a in mice affects photoreceptor ciliary structure and leads to progressive retinal degeneration. Hum. Mol. Genet. 23, 5197–5210 (2014).
Estrada-Cuzcano, A., Neveling, K., Kohl, S., Banin, E., Rotenstreich, Y., Sharon, D. et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am. J. Hum. Genet. 90, 102–109 (2012).
El Shamieh, S., Neuillé, M., Terray, A., Orhan, E., Condroyer, C., Démontant, V. et al. Whole-exome sequencing identifies KIZ as a ciliary gene associated with autosomal-recessive rod-cone dystrophy. Am. J. Hum. Genet. 94, 625–633 (2014).
Zhou, Y., Tao, S., Chen, H., Huang, L., Zhu, X., Li, Y. et al. Exome sequencing analysis identifies compound heterozygous mutation in ABCA4 in a Chinese family with Stargardt disease. PLoS One 9, e91962 (2014).
Langmann, T., Di Gioia, S. A., Rau, I., Stöhr, H., Maksimovic, N. S., Corbo, J. C. et al. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am. J. Hum. Genet. 87, 376–381 (2010).
Venturini, G., Di Gioia, S. A., Harper, S., Weigel-DiFranco, C., Rivolta, C. & Berson, E. L. Molecular genetics of FAM161A in North American patients with early-onset retinitis pigmentosa. PLoS ONE 9, e92479 (2014).
Bandah-Rozenfeld, D., Mizrahi-Meissonnier, L., Farhy, C., Obolensky, A., Chowers, I., Pe'er, J. et al. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 87, 382–391 (2010).
Zobor, D., Balousha, G., Baumann, B. & Wissinger, B. Homozygosity mapping reveals new nonsense mutation in the FAM161A gene causing autosomal recessive retinitis pigmentosa in a Palestinian family. Mol. Vis. 20, 178–182 (2014).
Zach, F., Grassmann, F., Langmann, T., Sorusch, N., Wolfrum, U. & Stöhr, H. The retinitis pigmentosa 28 protein FAM161A is a novel ciliary protein involved in intermolecular protein interaction and microtubule association. Hum. Mol. Genet. 21, 4573–4586 (2012).
Sedmak, T. & Wolfrum, U. Intraflagellar transport molecules in ciliary and nonciliary cells of the retina. J. Cell. Biol. 189, 171–186 (2010).
Kayadjanian, N., Lee, H. S., Pina-Crespo, J. & Heinemann, S. F. Localization of glutamate receptors to distal dendrites depends on subunit composition and the kinesin motor protein KIF17. Mol. Cell. Neurosci. 34, 219–230 (2007).
Acknowledgements
We thank all subjects who participated in this study. This study was supported by grants from the Natural Science Foundation of China (81271007, 81470668 (XZ); 81400437 (YZ); 81300802 (LH); and 81100693 (QC)), Department of Science and Technology of Sichuan Province (2014FZ0122, (XZ); and 2014SZ0169, 2015SZ0052, SZ20120209 (ZY)), Sichuan Provincial Outstanding Youth research grant (14QNJJ0089 (XJZ)), the 54th China Postdoctoral Science Foundation (2013M542300 (YZ)) and the Department of Sichuan Provincial Health (130176 (YZ)). We alone are responsible for the content and the writing of the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Zhou, Y., Saikia, B., Jiang, Z. et al. Whole-exome sequencing reveals a novel frameshift mutation in the FAM161A gene causing autosomal recessive retinitis pigmentosa in the Indian population. J Hum Genet 60, 625–630 (2015). https://doi.org/10.1038/jhg.2015.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.92
This article is cited by
-
A new mouse model for retinal degeneration due to Fam161a deficiency
Scientific Reports (2021)
-
Unique combination of clinical features in a large cohort of 100 patients with retinitis pigmentosa caused by FAM161A mutations
Scientific Reports (2020)
-
Genotypic spectrum and phenotype correlations of ABCA4-associated disease in patients of south Asian descent
European Journal of Human Genetics (2017)
