
Abstract
Copy number variations can be identified using newer genotyping arrays with higher single nucleotide polymorphisms (SNPs) density and copy number probes accompanied by newer algorithms. McCarroll et al. (2008) applied these to the HapMap II samples and identified 1316 copy number polymorphisms (CNPs). In our study, we applied the same approach to 859 samples from three Singapore populations and seven HapMap III populations. Approximately 50% of the 1291 autosomal CNPs were found to be polymorphic only in populations of non-African ancestry. Pairwise comparisons among the 10 populations showed substantial differences in the CNPs frequencies. Additionally, 698 CNPs showed significant differences with false discovery rate (FDR)<0.01 among the 10 populations and these loci overlap with known disease-associated or pharmacogenetic-related genes such as CFHR3 and CFHR1 (age related macular degeneration), GSTTI (metabolism of various carcinogenic compounds and cancers) and UGT2B17 (prostate cancer and graft-versus-host disease). The correlations between CNPs and genome-wide association studies–SNPs were investigated and several loci, which were previously unreported, that may potentially be implicated in complex diseases and traits were found; for example, childhood acute lymphoblastic leukaemia, age-related macular degeneration, breast cancer, response to antipsychotic treatment, rheumatoid arthritis and type-1 diabetes. Additionally, we also found 5014 novel copy number loci that have not been reported previously by McCarroll et al. (2008) in the 10 populations.
Similar content being viewed by others
Introduction
The term copy number variation (CNV) was first introduced in 2006 and it is generally defined as additions or deletions in the number of copies of a particular segment of DNA (larger than 1 kb in length) when compared with a reference genome sequence.1 The ubiquitous nature of CNVs in the human genome was underappreciated until 2004,2, 3 when these reports stimulated a series of efforts to detect and characterise CNVs in different populations.4, 5, 6, 7, 8 This development has also resulted in several new terminologies such as copy number polymorphisms (CNPs), which have been defined as common CNVs with a population frequency of at least 1%.4
CNVs can be detected using microarray-based methods, but these have relatively poor resolution when compared with sequencing-based approaches.9, 10 The low resolution of microarray-based methods also led to imprecise mapping of the breakpoints. This is important when constructing copy number loci to estimate population frequencies. Newer genotyping arrays, such as the Illumina Human 1M Beadchip (Illumina, San Diego, CA, USA) and the Affymetrix SNP Arrays 6.0 (Affymetrix, Santa Clara, CA, USA), have higher single nucleotide polymorphisms (SNPs) density and copy number probes, resulting in improved performance of microarray-based methods to detect CNVs. However, even with higher resolution arrays, the challenge of identifying common breakpoints still remains. This is largely due to the early CNV-calling algorithms that identified breakpoints sample-by-sample, resulting in significant variation of breakpoints. The Canary algorithm in the Birdsuite software overcomes this problem by calling CNPs simultaneously across multiple individuals at pre-defined genomic locations.11 McCarroll et al.4 used the Canary algorithm to identify 1316 CNPs in the HapMap Phase II populations. These CNPs were well validated and their sizes were in agreement with the results from the fosmid paired-end sequencing experiment.9
To provide a more global map of CNPs, our study aims to determine integer copy numbers of the 1316 CNPs set of three Southeast Asian populations in Singapore, namely Chinese (Sing–Chinese), Malay (Sing–Malay) and Asian Indian (Sing–Indian), and the seven populations from the HapMap Phase III.12 The HapMap III populations studied are people of African ancestry in the southwestern USA (ASW), the Chinese community in Metropolitan Denver, Colorado, USA (CHD), Gujarati Indians in Houston, Texas, USA (GIH), the Luhya in Webuye, Kenya (LWK), people of Mexican ancestry in Los Angeles, California, USA (MEX), the Maasai in Kinyawa, Kenya (MKK) and the Tuscans in Italy (TSI). The characteristics of CNPs in the 10 populations will be described and compared. In addition, the correlation between CNPs and SNPs in the 10 populations will also be characterised and compared. A special emphasis will be given to studying the correlation between SNPs in the genome-wide association studies (GWAS) catalog (GWAS–SNPs) and CNPs in the 10 populations. Additionally, novel copy number loci that have not been reported previously by McCarroll et al.4 will also be reported on from the 10 populations.
Materials and methods
DNA samples and genotyping
The detailed information on the sources of DNA samples, demographic data of the samples, sample selection and the origin and migration history of the three Singapore populations (Chinese, Malay and Asian Indian) have been described in our previous publication.8, 13 This study was approved by the National University of Singapore Institutional Review Board (Reference Code: 07-199E). In total, 292 DNA samples (99 Chinese, 98 Malay and 95 Indian) were genotyped using the Affymetrix Genome-Wide Human SNP Array 6.0. Of the 292 samples, 27 were excluded from subsequent analysis. The final set of 265 samples (93 Chinese, 88 Malays and 84 Indians) was available for analysis using Birdsuite. There were 135 females and 130 males in the final dataset. The detailed information on the quality control and sample filtering have also been described in our previous papers.8, 13
HapMap III samples
The CEL-files of the Affymetrix SNP Array 6.0 for the seven populations in HapMap III were downloaded from the ftp site (ftp://ftp.ncbi.nlm.nih.gov/hapmap/raw_data/hapmap3_affy6.0/). All the samples were analysed by Birdsuite, with only unrelated samples included in our study; that is, family-related samples were removed using the ‘relationships’ file provided by the International HapMap Project. After the sample exclusion step, a total of 594 unrelated samples from the seven HapMap III populations were analysed: ASW (n=52), CHD (n=89), GIH (n=89), LWK (n=90), MEX (n=53), MKK (n=132) and TSI (n=89).
CNP calling using Canary
The Birdsuite software was used to analyse the Affymetrix SNP Array 6.0 dataset, which consisted of two components for detecting copy number changes. The first component, Canary, was used to determine the integer copy number at each of the predefined 1316 CNPs identified by McCarroll et al.4 in the HapMap II samples. These CNPs were found in more than one HapMap II individual and the sizes of these CNPs were also determined. The 1316 CNPs were distributed in all the autosomes and sex chromosomes. However, 25 CNPs located in the sex chromosomes were removed, as CNP calling in sex chromosomes is more problematic and less accurate. Therefore, the results reported in this study comprised of only 1291 CNPs in the 22 autosomes. Confidence statistics was used to identify poor quality calls and only integer copy numbers detected with high confidence (confidence score <0.1) were reported and used for subsequent analyses. We performed the Hardy–Weinberg equilibrium analysis as a quality control measure for biallelic CNPs in all 10 populations. It is recommended that the samples should be analysed on the basis of the genotyping batches using Birdsuite; therefore, the samples for Singapore and HapMap III populations were analysed by batch without separating the samples into each specific population.
FDR correction for population comparisons of the integer copy numbers of the CNPs
Population differences in the integer copy numbers were examined using the Fisher's exact test as implemented by the ‘fisher test’ command in R. The false discovery rate (FDR) was used in place of the P-value to account for the multiple-testing problem. We calculated the FDR using the Benjamini and Hochberg method. We performed two different test procedures: (1) comparing the integer copy numbers among the 10 populations simultaneously and (2) pairwise comparisons of the integer copy numbers among the 10 populations. For each procedure, FDR was computed once to control for all the tests (that is, in the second procedure, we calculated the FDR once by combining the P-values from 45 × 1291 tests).
Correlation analysis
All the correlation analyses of CNPs and nearby SNPs were done separately for each of the 10 populations. For each autosomal CNP (restricted to biallelic CNPs with MAF⩾5%), SNPs in close proximity with the CNP; that is, within a 200-kb window from the start- and end-position of the CNP were considered. The square of the Pearson correlation coefficient (r2) for each of the SNPs (excluding the SNPs used for CNP-calling) found within the 200-kb windows of the respective CNP was then calculated.
The r2 is the square of the Pearson correlation coefficient between the copy number genotypes and the SNP genotypes. The copy number genotypes were obtained using Canary in the Birdsuite algorithm. The SNP genotypes were obtained using Larry Bird in the Birdsuite algorithms. Larry Bird outputs the number of allele A (0, 1, 2) and number of allele B (0, 1, 2) for each SNP. We used the number of allele A for the calculation. Larry Bird generates the number of allele A and number of allele B for each SNP. As each SNP has two alleles in total, knowing the number of allele A will inform the number of allele B; for example, if the number of allele A is 2, then number of allele B should be 0.
The same r2 calculations used for the autosomal CNPs and the SNPs identified by GWAS were used to explore the potential associations of CNPs with human diseases and traits. The list of GWAS–SNPs was downloaded from the National Human Genome Research Institute's website (http://www.genome.gov/gwastudies/) on 24 May 2010.
Copy number loci calling using Birdseye and validation
The Birdseye component in Birdsuite was used to detect additional copy number loci located outside the 1316 CNPs in the 10 populations. Similarly, only the copy number loci in autosomal chromosomes were detected because of the inaccuracy of Birdseye in detecting copy number loci in the sex chromosomes. Copy number calls with low confidence (confidence score <5) were removed. On the basis of the copy number calls generated by Birdseye, we constructed novel copy number loci using the methods that we developed previously.14 All the downstream analyses after Canary and Birdseye were performed using the software package R (http://www.r-project.org/). The novel copy number loci identified by Birdseye were compared with data from the Database of Genomic Variants (http://projects.tcag.ca/variation/) as a validation step. We defined a copy number locus overlapped with the Database of Genomic Variants, if the locus overlapped by >50% of its length with one or more entries in the Database of Genomic Variants.
Results
Characteristics of CNPs in the 10 populations
In each of the 10 populations, among the polymorphic CNPs (Table 1), most were biallelic, where the integer copy numbers were either exclusively deletions (copy number=0, 1) or exclusively duplications (copy number=3, 4). Among the biallelic CNPs, the majority did not show significant deviation from Hardy–Weinberg equilibrium with less than 2% failing a Hardy–Weinberg equilibrium test at P-value <0.01 in all except three populations—Sing–Chinese (2.2%), ASW (4.2%) and LWK (2.8%).
In terms of the proportion of non-polymorphic loci and loci with varying population frequencies, the Singapore populations were similar to the HapMap III populations of non-African descent (CHD, GIH, MEX and TSI) (Table 1 and Supplementary Figure 1). More than half of the CNPs were non-polymorphic in the Singapore and HapMap III populations of non-African descent. This was in contrast to the populations of African descent (ASW, LWK and MKK), where only 26.41–37.72% of the CNPs were not polymorphic. They also had higher proportions of CNPs with frequencies ranging from 1 to 10%, ASW (45.86%), LWK (35.48%) and MKK (39.27%), compared with the other populations (ranging from 24.63 to 27.50%). In addition, among all the populations, there were no substantial differences in the proportion of CNPs with a population frequency >10%. The discrepancy between populations of African descent and others is largely due to these populations having a larger number of rarer CNPs with a population frequency <10%. Hence, the differences between populations of African descent and the others were primarily in the proportion of non-polymorphic loci and those with population frequencies <10%. It is also worth noting that the Sing–Indian and Sing–Chinese populations have almost similar distributions of polymorphic loci, when compared with the HapMap III populations with whom they share a similar ancestry (that is, GIH and CHD, respectively) (Table 1 and Supplementary Figure 1).
The proportion of common (MAF⩾0.05) biallelic CNPs that were highly correlated with at least one SNP (r2>0.8) was approximately 50% for non-African populations, but a lower proportion for African populations; that is, ASW (35.34%), LWK (34.84%) and MKK (37.39%). The majority of the common biallelic CNPs were ‘deletions’. There was a substantial difference in the proportion that was highly correlated with at least one SNP for CNPs categorised as ‘deletions’ and ‘duplications’. However, this substantial difference could be biased because of the small number of ‘duplications’ (Table 2). The strength of correlation or the r2 value decreased with distance between the CNP and SNP (Supplementary Figure 2).
We further investigated whether CNPs that were not well tagged were located in the genomic regions where SNP markers are sparse. The correlation patterns did not seem to be affected by the number of nearby SNPs and the MAF of CNPs. There was no apparent difference in the number of nearby SNPs and the MAF of CNPs between (a) the CNPs that were in strong correlation (r2>0.8) and (b) CNPs that were not in strong correlation with SNPs (Supplementary Figures 3a and b). However, smaller sizes of CNPs were generally in strong correlation with more SNPs than the larger CNPs (Supplementary Figure 3c). These results were consistent across the 10 populations.
Population differences in the integer copy numbers of the CNPs
Out of the 698 CNPs (FDR<0.01) that differed between the 10 populations, several loci encompassed known disease- or traits-associated or pharmacogenetic-related genes (Supplementary Table 1). These included WWOX, ERBB4 and TP63 (cancers), ADAMTSL3 (height), CFHR3 and CFHR1 (age-related macular degeneration), GSTT1 (metabolism of various carcinogenic compounds and cancers), UGT2B17 (prostate cancer and graft-versus-host disease) and CYP2A6 (metabolism of various drugs). There was a large interpopulation difference in the frequencies of some of the CNPs overlapping these genes. For example, CNP2203, which overlaps with the tumour suppressor gene WWOX, was not polymorphic in CHD, whereas it had a deletion frequency of 2.38% in Sing–Chinese and 7.32% in Sing–Malay (Table 3 and Supplementary Table 2). In contrast, the deletion frequency was 51.81% in Sing–Indian and 48.86% in GIH. Similarly, CNP147, which overlaps with the CFHR3 and CFHR1 genes, had deletion frequencies in Sing–Chinese (10.75%), Sing–Malay (12.64%) and CHD (15.73%) that was substantially lower than the other populations.
Another CNP of interest was CNP2560, a 46-kb deletion that overlaps with GSTT1. GSTT1 is an important detoxification enzyme and has a key role in metabolism of carcinogenic compounds. The total deletion frequency of this CNP was high in all the 10 populations ranging from 56.63 to 96.77% (Table 3 and Supplementary Table 2). Sing–Indians had a considerably lower total deletion frequency (56.63%) than Sing–Malays (85.06%) and Sing–Chinese (96.77%). This difference is attributable to two-copy deletion, as the difference in two-copy deletion frequency ranged from 15.66% in Sing–Indian, 32.18% in Sing–Malay and 46.24% in Sing–Chinese. The two Chinese populations had the highest two-copy deletion frequency (CHD, 41.57%). Conversely, both the Indian populations had the lowest two-copy deletion frequency (GIH, 17.98%).
CNP603 is a 125-kb deletion that overlaps with TMPRSS11E and UGT2B17. The entire UGT2B17 gene is within the deletion locus, but only one exon from TMPRSS11E was deleted. The deletion frequency of CNP603 was very different in Asian and non-Asian populations (Table 3 and Supplementary Table 2). Asian populations (Sing–Chinese, Sing–Malay, Sing–Indian, CHD and GIH) had higher frequencies, which ranged from 82.14 to 100%, when compared with populations of European and African ancestry (48.08–67.18%). The differences were even more apparent for two-copy deletions with the highest frequencies in CHD (70.79%), Sing–Chinese (65.59%) and Sing–Malay (52.87%), followed by the two Indian populations, GIH (37.04%) and Sing–Indian (30.95%), whereas the European and African populations were in the lower end of the spectrum with frequencies <20%. Generally, this trend was reversed for the frequency of one-copy deletions especially in the Singapore populations (Sing–Chinese 33.33%, Sing–Malay 42.53% and Sing–Indian 51.19%).
The number of CNPs that showed significant differences (FDR<0.01) in pairwise comparisons of the 10 populations are shown in Table 4. Only 19 CNPs showed significant differences between Sing–Chinese and CHD, and 12 CNPs between Sing–Indian and GIH, suggesting that the CNPs profile in the two Chinese and two Indian populations were very similar (Supplementary Figure 4). Through these pairwise comparisons (Table 4 and Supplementary Figure 4), the 10 populations can be divided into three groups representing Asian, European and African ancestry: (a) Sing–Chinese, Sing–Malay and CHD, (b) Sing–Indian, GIH, MEX and TSI, (c) ASW, LWK and MKK. The CNPs profiles of Sing–Indian and GIH were closer to European populations (MEX and TSI).
Correlation analysis between CNPs and GWAS-SNPs
To investigate the potential role of CNPs in the aetiology of complex diseases or traits, we computed the r2 between CNPs and the SNPs in the National Human Genome Research Institute GWAS catalog. Out of the >2500 GWAS–SNPs that have been found to be associated with various complex diseases and traits, only 17 GWAS–SNPs were found to be in strong correlation with 12 CNPs (Table 5 and Supplementary Tables 3 and 4). In this analysis, we defined a strong correlation as r2>0.5, following Conrad et al.5 These 17 SNPs were reported to be associated with 14 diseases or traits and the notable phenotypes that were observed consistently across the populations were body mass index, Crohn's disease, multiple sclerosis, myocardial infarction and prostate cancer. Several SNPs were in strong correlation with a single CNP; for example, three SNPs (rs13361189, rs1000113, rs11747270) were found to be in strong correlation with CNP874. Of the 33 copy number loci identified by Conrad et al.,5 which were in strong correlation with GWAS–SNPs, seven were also identified in our study which had >50% overlap in length. The remaining five CNPs in our study were associated with childhood acute lymphoblastic leukaemia, age-related macular degeneration, breast cancer, response to antipsychotic treatment, rheumatoid arthritis and type-1 diabetes (Table 5 and Supplementary Tables 3 and 4).
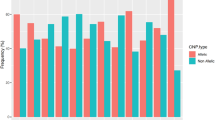
Several SNPs were consistently found to be in strong correlation with four CNPs (CNP60, CNP874, CNP877 and CNP333) in all populations. The most notable was rs2815752 near the NEGR1 gene (associated with body mass index), which is in perfect correlation (r2=1) with CNP60 in all the 10 populations (Table 5 and Supplementary Table 3). This locus is a 42-kb deletion located in chromosome 1, which did not overlap with any of the UCSC (University of California, Santa Cruz) genes and it is located only 1.3 kb away from the SNP. The total deletion frequency in the three Singapore populations was high (Figure 1a and Supplementary Table 5). There were, however, differences in the frequency of two-copy deletion. More than 80% of the Sing–Chinese and Sing–Malay samples were deleted in both copies, but only about 41% for the Sing–Indian samples. The pattern is similar between Sing–Chinese and CHD, as well as Sing–Indian and GIH. The frequency of two-copy deletion frequency varied substantially across the 10 populations, from the lowest in the LWK population (26.97%) to the highest in Sing–Chinese (87.10%). A significant difference in the two-copy deletion frequency of CNP60 was seen between Asian populations (>80% for Sing–Chinese, Sing–Malay and CHD) compared with African populations (<35% for ASW, LWK and MKK), whereas the frequency of the Sing–Indian and GIH resembles European populations (MEX and TSI) (Supplementary Table 5).

Total, two-copy and one-copy deletion frequencies of (a) CNP60, (b) CNP874 and (c) CNP877 in 10 populations.
CNP874 was found to be in strong correlation with three GWAS–SNPs located near the IRGM gene, which is associated with Crohn's disease. This strong correlation pattern was consistent across the 10 populations (Table 5). Most of the individuals carried either deletions or had a diploid copy. This locus spans 13 kb in chromosome 5 and did not overlap with any of the UCSC genes. The SNPs were located 4.8 kb (rs13361189), 21.4 kb (rs1000113) and 40.2 kb (rs11747270) away from the deletion. The differences in the frequency of two-copy deletion of CNP874 appeared to divide the 10 populations into two clusters. The populations of European ancestry (MEX and TSI) and Indian populations (Sing–Indian and GIH) had a frequency ⩽6.41%, but the other populations had higher frequencies, which ranged from 10% to 20.69% (Figure 1b and Supplementary Table 5). We also found a substantially lower frequency of two-copy deletion in the Sing–Indian (6.41%) compared with the Sing–Chinese (15.22%) and the Sing–Malay (11.49%) populations.
The CNP877 locus has been implicated in multiple sclerosis. It was however not polymorphic in the Sing–Chinese (Figure 1c and Supplementary Table 5). The total deletion frequencies for Sing–Malay and CHD were 2.30 and 1.14%, respectively. However, we found a much higher total deletion frequency for the other seven populations, which ranged from 17.05 to 42.53%.
Novel copy number loci in the 10 populations
The second component of the Birdsuite software, Birdseye, was used to identify novel copy number loci in the 10 populations. We subsequently found 5947 copy number loci, of which 933 loci were excluded because of overlap with the 1291 autosomal CNPs identified by McCarroll et al.4 As a result, only 5014 were novel copy number loci; that is, had not been previously found by McCarroll et al.4 Of these, 1448 loci were detected in two or more individuals in the 10 populations (Table 6). The list of these loci is available in Supplementary Table 6. Using a more stringent definition of ‘common’ novel copy number loci (population frequency ⩾1%), there were only 170 loci and of these, 42 loci had a population frequency ⩾5%.
Of the 1448 novel copy number loci, 763 (52.69%) were found to overlap with the data from the Database of Genomic Variants. Although for the 170 loci, the overlap was 78.82% (Table 6). Additionally, we also found that 86.54% of the 1448 loci were biallelic; that is, these loci contained either deletions (48.76%) or duplications (37.78%). The remaining loci were found to have both deletions and duplications. The majority of these loci did not overlap with the UCSC genes (62.43%). Of the 170 loci, 37.06% contained both deletions and duplications and the majority of these loci also did not overlap with the UCSC genes (52.35%).
Discussion
The finding that approximately 50% of the CNPs identified by the McCarroll et al.4 study were not polymorphic in all of the three Singapore populations and the HapMap III populations (CHD, GIH, MEX and TSI) suggests that the CNPs found in the ‘reference’ HapMap II populations are not necessarily polymorphic or common in other populations. This finding, together with the identification of novel copy number loci other than those found using the HapMap II populations, highlights the importance of characterising CNPs in different populations.
In addition, we also found several hundred CNPs that showed significant differences in integer copy numbers among the 10 populations. More interestingly, many of these loci encompass genes of medical relevance. For example, we found a markedly lower deletion frequency at CNP2203 (which is associated with the WWOX gene) in Sing–Chinese and Sing–Malay compared with other populations. WWOX is a tumour suppressor gene affected in multiple cancers.15 On the other hand, deletion of the UGT2B17 gene was also been found to be associated with an increased risk of prostate cancer.16, 17 The functional role of the UGT2B17 enzyme is clear in prostate cancer, as it is involved in steroid hormone (androgen) metabolism. The mismatch of UGT2B17 copy numbers in donors and recipients of stem cell transplantation were also associated with an increased risk of graft-versus-host disease.18 This gene is contained within CNP603, which show substantial differences between the Singapore and HapMap III populations. Although a direct association between the CNPs and phenotypic differences is not established in our study, collectively our results suggest that CNPs distributions are substantially different between populations and thus, may account for phenotypic differences between them.
We found 12 CNPs that may have potential implications in various diseases and traits; however, only five of them have not been reported by Conrad et al.,5 who found evidence of correlations for 33 copy number loci with GWAS–SNPs at r2>0.5. The difference in the number of loci found to be in correlation with GWAS–SNPs between our study and the Conrad et al.5 study is likely due to the limitation that we only focused on the 1291 CNPs, whereas Conrad et al.5 studied the whole genome. Furthermore, it could also be due to the difference in the marker density of the microarrays used in our study and the Conrad et al.5 study. We used the Affymetrix SNP Array 6.0, whereas they used a set of 20 oligonucleotide-CGH arrays, comprising 42 million probes. The differences in marker density will contribute to the differences in sensitivity of detection.5
Several previous studies have reported correlations between CNVs and GWAS–SNPs. For example, deletions near IRGM and NEGR1 genes, which were in perfect linkage disequilibrium (LD) with the GWAS–SNPs, were identified for Crohn's disease and body mass index, respectively.19, 20 Our study also showed strong correlations between CNPs and GWAS–SNPs near IRGM and NEGR1 in all 10 populations, but the deletion frequencies varied substantially among the populations. GWAS–SNPs are potentially indirect markers of disease variants, which include CNPs. This may have important clinical implications if these deletions are true disease variants.
A recent paper published by the International HapMap Consortium also studied CNPs in the HapMap III populations.12 However, they merged and analysed the probe-level intensity data from both the Affymetrix SNP Array 6.0 and the Illumina 1M Beadchip arrays. In contrast, we only analysed the Affymetrix SNP Array 6.0 data and focused primarily on the 1291 CNPs identified previously, as only the raw signal intensity files of this array were available from the HapMap website. A total of 1610 CNPs with an estimated frequency of at least 1% of the cohort were identified in the HapMap III populations by the International HapMap Consortium. They also found that most CNPs also occurred at a low frequency.12 This was consistent with our study where among the polymorphic CNPs, the majority also occurred at a low frequency (<10%). Similarly, the finding that the frequency spectrum of common CNPs (>10%) was similar across populations by the International HapMap Consortium was in good agreement with our results (Table 1).
References
Freeman, J. L., Perry, G. H., Feuk, L., Redon, R., McCarroll, S. A., Altshuler, D. M. et al. Copy number variation: new insights in genome diversity. Genome Res. 16, 949–961 (2006).
Iafrate, A. J., Feuk, L., Rivera, M. N., Listewnik, M. L., Donahoe, P. K., Qi, Y. et al. Detection of large-scale variation in the human genome. Nat. Genet. 36, 949–951 (2004).
Sebat, J., Lakshmi, B., Troge, J., Alexander, J., Young, J., Lundin, P. et al. Large-scale copy number polymorphism in the human genome. Science 305, 525–528 (2004).
McCarroll, S. A., Kuruvilla, F. G., Korn, J. M., Cawley, S., Nemesh, J., Wysoker, A. et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 40, 1166–1174 (2008).
Conrad, D. F., Pinto, D., Redon, R., Feuk, L., Gokcumen, O., Zhang, Y. et al. Origins and functional impact of copy number variation in the human genome. Nature 464, 704–712 (2010).
Yim, S. H., Kim, T. M., Hu, H. J., Kim, J. H., Kim, B. J., Lee, J. Y. et al. Copy number variations in East-Asian population and their evolutionary and functional implications. Hum. Mol. Genet. 19, 1001–1008 (2010).
Park, H., Kim, J. I., Ju, Y. S., Gokcumen, O., Mills, R. E., Kim, S. et al. Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat. Genet. 42, 400–405 (2010).
Ku, C. S., Pawitan, Y., Sim, X., Ong, R. T., Seielstad, M., Lee, E. J. et al. Genomic copy number variations in three Southeast Asian populations. Hum. Mutat. 31, 851–857 (2010).
Kidd, J. M., Cooper, G. M., Donahue, W. F., Hayden, H. S., Sampas, N., Graves, T. et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453, 56–64 (2008).
Korbel, J. O., Urban, A. E., Affourtit, J. P., Godwin, B., Grubert, F., Simons, J. F. et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 318, 420–426 (2007).
Korn, J. M., Kuruvilla, F. G., McCarroll, S. A., Wysoker, A., Nemesh, J., Cawley, S. et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 40, 1253–1260 (2008).
International HapMap 3 Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010).
Teo, Y. Y., Sim, X., Ong, R. T., Tan, A. K., Chen, J., Tantoso, E. et al. Singapore Genome Variation Project: a haplotype map of three Southeast Asian populations. Genome Res. 19, 2154–2162 (2009).
Mei, T. S., Salim, A., Calza, S., Seng, K. C., Seng, C. K. & Pawitan, Y. Identification of recurrent regions of Copy-Number Variants across multiple individuals. BMC Bioinformatics 11, 147 (2010).
Lewandowska, U., Zelazowski, M., Seta, K., Byczewska, M., Pluciennik, E. & Bednarek, A. K. WWOX, the tumour suppressor gene affected in multiple cancers. J. Physiol. Pharmacol. 60, 47–56 (2009).
Park, J., Chen, L., Ratnashinge, L., Sellers, T. A., Tanner, J. P., Lee, J. H. et al. Deletion polymorphism of UDP-glucuronosyltransferase 2B17 and risk of prostate cancer in African American and Caucasian men. Cancer Epidemiol. Biomarkers Prev. 15, 1473–1478 (2006).
Karypidis, A. H., Olsson, M., Andersson, S. O., Rane, A. & Ekström, L. Deletion polymorphism of the UGT2B17 gene is associated with increased risk for prostate cancer and correlated to gene expression in the prostate. Pharmacogenomics J. 8, 147–151 (2008).
McCarroll, S. A., Bradner, J. E., Turpeinen, H., Volin, L., Martin, P. J., Chilewski, S. D. et al. Donor-recipient mismatch for common gene deletion polymorphisms in graft-versus-host disease. Nat. Genet. 41, 1341–1344 (2009).
McCarroll, S. A., Huett, A., Kuballa, P., Chilewski, S. D., Landry, A., Goyette, P. et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat. Genet. 40, 1107–1112 (2008).
Willer, C. J., Speliotes, E. K., Loos, R. J., Li, S., Lindgren, C. M., Heid, I. M. et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 41, 25–34 (2009).
Acknowledgements
This study was supported by the Yong Loo Lin School of Medicine, the Life Science Institute and the Office of Deputy President (Research and Technology), National University of Singapore. We also acknowledge the technical and financial support of the Genome Institute of Singapore and Agency for Science, Technology and Research, Singapore.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Ku, CS., Teo, SM., Naidoo, N. et al. Copy number polymorphisms in new HapMap III and Singapore populations. J Hum Genet 56, 552–560 (2011). https://doi.org/10.1038/jhg.2011.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.54
