
Abstract
Hypospadias is a common congenital malformation in boys in which the urethral meatus opens on the underside of the penis. It is considered a complex disorder with several genes involved and the molecular etiology is just beginning to be revealed. As more than 85% of Opitz G/BBB syndrome (OS) patients with MID1 mutations are manifested with hypospadias, we have investigated the association between the MID1 gene and hypospadias. DNA from 114 hypospadias cases was analyzed with direct sequencing of the MID1 gene. Genotyping analysis was performed for the single-nucleotide polymorphism (SNP) c.1230G>A in 370 individuals with varying degrees of hypospadias and compared with 759 healthy controls. We identified one nonsense mutation c.712G>T (p.E238X), one missense mutation c.1679A>G (p.K560R) and two synonymous variants c.1230G>A (p.S410S) and c.1284T>G (p.V428V). We also detected a significant difference in the rare allele frequency of SNP c.1230G>A in hypospadias patients as compared with controls (P=0.016). Our finding suggests that hypospadias associated with hypertelorism is the mildest phenotype in OS caused by MID1 mutations.
Similar content being viewed by others

Introduction
Hypospadias is one of the most common birth defects and is characterized by an incomplete fusion of the urethral folds. In Sweden, since the beginning of the 1970s, the prevalence of hypospadias is 3 per 1000 male births according to the annual Swedish Malformation Registry. Other international studies have suggested an increasing incidence of hypospadias.1, 2, 3, 4 The etiology is now beginning to be elucidated as mutations in both the MAMLD1 and the ATF3 genes have been identified to cause hypospadias.5, 6 However, most cases of hypospadias remain unexplained.
Normal urethral development begins in the sixth week of gestation with the formation of the urogenital sinus. Two edges of urethral groove fuse in a proximal-to-distal direction, forming the urethra. Failed seam formation during fusion of the urethral folds between fetal weeks 8 and 16 result in hypospadias.7 Any gene having a role during the development of urogenital organs in the critical period is therefore a candidate gene for hypospadias. Mutations in the MID1 gene cause the X-linked form of Opitz G/BBB syndrome (OS, characterized by midline abnormalities such as hypertelorism, cleft lip/palate, tracheo–esophageal abnormalities, cardiac defects and hypospadias).8, 9, 10 The function of MID1 is highly conserved in vertebrates, and experiments conducted in mice and chicken showed that MID1 expression correlated well with the tissues affected in OS.11, 12 In situ hybridization studies on human embryos showed that the expression of MID1 was localized in undifferentiated cells of midline structures including the urogenital system.13 Hypospadias is one common finding in OS cases (>85%) with MID1 mutations, thus MID1 is a strong candidate gene for hypospadias.10
The MID1 protein consists of 667 amino acids and belongs to the tripartite motif family.14, 15 The N-terminus of the protein is composed of a RING finger motif (R), two B-box zinc-fingers (B1 and B2) and a coiled coil region.14 The C-terminus consists of a COS box, fibronectin type 3 domain and a B30.2 domain (PRY+SPRY).16 As an E3 ubiquitin ligase, MID1 has been shown to regulate phosphatase 2A catalytic subunit degradation.15, 17 In addition, MID1 is involved in the formation of multiprotein complex serving as anchor points and associated with microtubuli.18, 19 Given the molecular knowledge about the MID1 protein, a disruption in either the N- or the C-terminus of the protein would have some influences on its interaction with microtubules in the cytoplasm, which is hypothesized to be the underlying mechanism for the midline defects including hypospadias.20
The aim of this study was to perform direct sequencing of the MID1 gene in hypospadias cases to elucidate the role of this gene in the pathogenesis of isolated hypospadias.
Materials and methods
Patients
Altogether, 378 male patients with different degrees of severity of hypospadias were recruited for sequencing and genotyping analysis. A total of 98 patients had additional family members with hypospadias and 280 were sporadic cases. Among them, a subgroup consisting of 20 patients, also suffered from other accompanying midline malformations, including cardiac anomalies, esophageal atresia and/or anal atresia. For these patients, the clinical findings were however not enough for making clinical diagnosis of OS. As a control group for direct sequencing, placenta samples from 95 healthy babies were included. A total of 759 healthy voluntary anonymous blood donors (410 males and 349 females) at the Karolinska University Hospital were recruited as controls for genotyping. Informed consent was obtained from all individuals included in this study. DNA was extracted from peripheral blood, placenta or penile skin obtained during surgery and using standard phenol–chloroform extraction protocols. The research ethics committee at Karolinska Institutet approved the study.
Mutational analysis
We amplified all exons and flanking intronic sequences (100 bp on each side) of the MID1 gene in 114 hypospadias cases using a standard PCR protocol. Primers and amplification conditions are available upon request. Direct sequencing was performed using the Big Dye terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA), and was run on an ABI3130 Sequencer (Applied Biosystems). Primers used in sequencing were the same as used in the PCR amplification. The sequencing reactions were carried out according to the manufacturer's recommendations. Sequence chromatograms were analyzed using SeqScape v3.7 (Applied Biosystems) and compared with reference sequence ENSG00000101871.
Genotyping
The single-nucleotide polymorphism (SNP), rs16986145 (c.1230G>A), located in exon 8 of the MID1 gene, was selected for an association study after the sequencing result. Genotyping of 370 cases and 759 blood donor controls was performed using 5′-nuclease allele discrimination TaqMan assay with fluorescent probes and the standard protocol. The samples were analyzed on an ABI7900HT (Applied Biosystems). Only successful samples are reported. All samples with the minor allele were confirmed by direct sequencing including four relatives of familial cases carrying the minor allele c.1230A.
Splice site and protein structure prediction
HomoloGene detected the putative homologs of the human MID1 gene and alignments were made using ClustalW2 (Hinxton, UK). The impacts of mutations on 5′ and 3′ splice sites were predicted using Alamut v1.5 (Rouen, France). In Alamut, splicing predictions are based on four different algorithms (SpliceSiteFinder, MaxEntScan, NNSPLICE, GeneSplicer); therefore our conclusion was based upon consistency of results from at least two of the algorithms. We only consider predictions in which the score from the mutant variant differ with at least 20% from the wild-type variant. Potential exonic splicing enhancer was also investigated using Alamut.
Statistical analysis
Odds ratio was calculated using Mantel–Haenszel method. χ2-test and Yates’ correction were used for association study. P<0.05 was considered significant.
Results
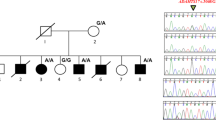
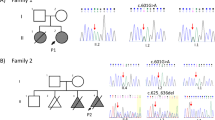
All coding exons and their flanking introns of the MID1 gene were screened for mutations in 114 cases of hypospadias, and exons in which DNA variants were found were also sequenced in 95 placenta controls. Mutation analysis demonstrated one nonsense mutation (c.712G>T; p.E238X) causing premature protein truncation in two brothers with hypospadias (Table 1). The two brothers were both operated because of penoscrotal hypospadias in combination with chordee and they did not suffer from any accompanying anomalies. However, reexamination of the family revealed hypertelorism in both brothers but not in the carrier mother.
In one case, we detected a novel missense mutation c.1679A>G (p.K560R) involving a lysine residue within the B30.2 (SPRY) domain (Table 1). This mutation disrupted one potential splicing site, whereas it created one new splicing site and one exonic splicing enhancer site for the SFRS6 (SRp55) protein. This boy was reexamined and is healthy, but has a clear hypertelorism and no other midline defect.
A unique T–G substitution at position c.1284 (p.V428V) was identified in one case, but neither protein sequence nor splicing pattern of MID1 was affected (Table 1). None of the controls exhibited this nucleotide substitution.
Homology analysis of the MID1 protein sequence showed that all the above changes were located at conserved positions (Figure 1). None of these alterations were detected in any of the 95 controls.

Alignments of homologous MID1 protein sequences. Partial alignments are presented and mutations detected in this study are indicated.
The polymorphism c.1230G>A (p.S410S) was found in five patients (Table 1). In the 95 placenta control samples, we found 3 females, who carried the polymorphism in heterozygous form, but we did not find any male A-allele carrier. Splice site prediction showed that the polymorphism c.1230G>A disrupted exonic splicing enhancer site for the SFRS1 (SF2, ASF). Unfortunately, because of lack of tissue from the patients, we cannot study the mRNA expression and splicing pattern in these patients. This polymorphism was also selected for genotyping in a larger cohort as the rare A allele was more common in cases than in controls after sequencing. Overall, 9 patients among 366 hypospadias successfully genotyped carried the A allele, whereas there were 8 allele A carriers (one male and seven heterozygous females) among 754 blood donor controls (males and females), which is a significant difference (P=0.016). Our data further showed that the A allele is significantly more frequent among hypospadias cases than in normal male controls. The risk of hypospadias in c.1230A carriers was increased significantly, with an odds ratio of 10.21 (P=0.017) (Table 2). Among the nine patients carrying the A allele, four cases were familial, with more than one affected man within the family. The affected brothers of two index familial cases were found to carry allele A, whereas in the third familial case, the affected brother was a G carrier. In the fourth familial case, the affected maternal cousin was not available for DNA analysis, however the mother of the proband carried the mutation in the heterozygous form. The nine patients with the rare A allele were also screened for mutations in the MID1 gene, and no mutation was detected.
Discussion
Hypospadias is a relatively common congenital anomaly, but the molecular etiology of hypospadias remains to be completely solved. As hypospadias forms part of OS,8 we hypothesized that the gene responsible for the X-linked form of OS, the MID1 gene, might be involved in the development of hypospadias as a mild form of OS.9, 10 We identified one nonsense mutation, one missense mutation and two synonymous variants. We also found there was significant difference between the rare allele frequency of SNP c.1230G>A in cases as compared with controls.
The mutation c.712G>T (p.E238X) was found in two brothers in our study. Both of them had severe hypospadias and hypertelorism. This mutation c.712G>T resulted in the premature introduction of a stop codon, giving a truncated protein at amino acid 238 in the CC domain. This mutation has been reported before in one OS patient.21 The mutated protein lacks the whole C-terminus portion and a previous study has indicated that the C-terminus portion maybe have an important role in the association between MID1 and the microtubular apparatus.22 A microtubule co-assembly assay revealed that, the MID1 protein from OS fetal fibroblasts with the mutation in C-terminal exon lacks microtubule-binding ability.23 In another study conducted on avian embryos, the mutated MID1 construct without C-terminus portion failed to affect the left–right axis pattern, suggesting that the mutated construct is a loss-of-function construct.24, 25 Therefore, it is likely that the nonsense mutation found in our two patients leads to the loss of function of MID1, which may be the underlying mechanism of hypospadias. Furthermore, MID1 and MID2 have been reported to have overlapping functions and therefore MID1-MID2 redundancy could be the reason for the mild phenotype in these boys compared with OS cases.25
One boy with isolated penoscrotal hypospadias and hypertelorism carries the mutation c.1679A>G (p.K560R) that results in a lysine residue change into arginine within the B30.2 domain. Arginine and lysine have the similar properties, including being polar and positive charged. The prediction programs showed that c.1679A>G may exert some influence on splicing patterns. This variant was not found in any controls and in view of the clinical presentation is probably a true mutation.
The third substitution c.1284 T>G (p.V428V) found in a boy with isolated penoscrotal hypospadias, is not predicted to affect either amino acid or splicing sites. This is also novel but is presently regarded as a rare polymorphism.
The genotyping experiments revealed nine cases carrying the rare A allele of SNP c.1230 G>A in exon 8. To evaluate the contribution of this SNP for development of hypospadias, we compared allele frequencies between hypospadias patients and male controls, 0.025 and 0.002, respectively. These findings suggest that carriers of the A-allele of the c.1230 G>A SNP have an increased risk for hypospadias. To predict the consequence of this nucleotide change, Alamut software was used. The analysis results showed that this substitution might disrupt one serine/arginine-rich (SR) protein-binding site. This could indicate that the c.1230 G>A SNP may alter the expression of MID1. It is also possible that this SNP may be in linkage disequilibrium with a functionally important genetic variant elsewhere in the MID1 gene and therefore could have the role of a marker for a functionally important sequence change. However, we noted that five of the nine cases had at least one parent born in North Africa, where the A allele is more prevalent, which speaks against the significance of the result.
In addition, we cannot exclude that there are functional mutations in other parts of the MID1 gene, such as regulatory elements. Genes coding proteins that regulate or interact with MID1 could be interesting as candidate genes. Our study indicates that the mildest spectrum of OS can indeed be hypospadias associated with hypertelorism. The polymorphism c.1230 G>A can be yet another risk factor for hypospadias and needs further replication in other materials.
References
Källén, B. & Winberg, J. An epidemiological study of hypospadias in Sweden. Acta Paediatr Scand Suppl. 293, 1–21 (1982).
Paulozzi, L. J. International trends in rates of hypospadias and cryptorchidism. Environ Health Perspect. 107, 297–302 (1999).
Pierik, F. H., Burdorf, A., Nijman, J. M., de Muinck Keizer-Schrama, S. M., Juttmann, R. E. & Weber, R. F. A high hypospadias rate in The Netherlands. Hum Reprod. 17, 1112–1115 (2002).
Nelson, C. P., Park, J. M., Wan, J., Bloom, D. A., Dunn, R. L. & Wei, J. T. The increasing incidence of congenital penile anomalies in the United States. J Urol. 174, 1573–1576 (2005).
Fukami, M., Wada, Y., Miyabayashi, K., Nishino, I., Hasegawa, T., Nordenskjöld, A. et al. CXorf6 is a causative gene for hypospadias. Nat Genet. 38, 1369–1371 (2006).
Beleza-Meireles, A., Töhönen, V., Söderhäll, C., Schwentner, C., Radmayr, C., Kockum, I. & Nordenskjöld, A. Activating transcription factor 3: a hormone responsive gene in the etiology of hypospadias. Eur J Endocrinol. 158, 729–739 (2008).
Baskin, L. S., Erol, A., Jegatheesan, P., Li, Y., Liu, W. & Cunha, G. R. Urethral seam formation and hypospadias. Cell Tissue Res. 305, 379–387 (2001).
Opitz, J. M. G syndrome (hypertelorism with esophageal abnormality and hypospadias, or hypospadias-dysphagia, or ‘Opitz-Frias’ or Opitz-G’ syndrome): perspective in 1987 and bibliography. Am. J. Med. Genet. 28, 275–285 (1987).
Quaderi, N. A., Schweiger, S., Gaudenz, K., Franco, B., Rugarli, E. I., Berger, W. et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet. 17, 285–291 (1997).
Fontanella, B., Russolillo, G. & Meroni, G. MID1 mutations in patients with X-linked Opitz G/BBB syndrome. Hum Mutat. 29, 584–594 (2008).
Dal Zotto, L., Quaderi, N. A., Elliott, R., Lingerfelter, P. A., Carrel, L., Valsecchi, V. et al. The mouse Mid1 gene: implications for the pathogenesis of Opitz syndrome and the evolution of the mammalian pseudoautosomal region. Hum Mol Genet. 7, 489–499 (1998).
Richman, J. M., Fu, K. K., Cox, L. L., Sibbons, J. P. & Cox, T. C. Isolation and characterisation of the chick orthologue of the Opitz syndrome gene, Mid1, support a conserved role in vertebrate development. Int J Dev Biol. 46, 441–448 (2002).
Pinson, L., Auge, J., Audollent, S., Mattei, G., Etchevers, H., Gigarel, N. et al. Embryonic expression of the human MID1 gene and its mutations in Opitz syndrome. J Med Genet. 41, 381–386 (2004).
Reymond, A., Meroni, G., Fantozzi, A., Merla, G., Cairo, S., Luzi, L. et al. The tripartite motif family identifies cell compartments. EMBO J. 20, 2140–2151 (2001).
Trockenbacher, A., Suckow, V., Foerster, J., Winter, J., Krauss, S., Ropers, H. H. et al. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat Genet. 29, 287–294 (2001).
Short, K. M. & Cox, T. C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J Biol Chem. 281, 8970–8980 (2006).
Liu, J., Prickett, T. D., Elliott, E., Meroni, G. & Brautigan, D. L. Phosphorylation and microtubule association of the Opitz syndrome protein mid-1 is regulated by protein phosphatase 2A via binding to the regulatory subunit alpha 4. Proc Natl Acad Sci USA. 98, 6650–6655 (2001).
Schweiger, S. & Schneider, R. The MID1/PP2A complex: a key to the pathogenesis of Opitz BBB/G syndrome. Bioessays. 25, 356–366 (2003).
Aranda-Orgillés, B., Trockenbacher, A., Winter, J., Aigner, J., Köhler, A., Jastrzebska, E. et al. The Opitz syndrome gene product MID1 assembles a microtubule-associated ribonucleoprotein complex. Hum Genet. 123, 163–176 (2008).
Hsieh, E. W., Vargervik, K. & Slavotinek, A. M. Clinical and molecular studies of patients with characteristics of Opitz G/BBB syndrome shows a novel MID1 mutation. Am J Med Genet A. 146, 2337–2345 (2008).
Ferrentino, R., Bassi, M. T., Chitayat, D., Tabolacci, E. & Meroni, G. MID1 mutation screening in a large cohort of Opitz G/BBB syndrome patients: twenty-nine novel mutations identified. Hum Mutat. 28, 206–207 (2007).
Cainarca, S., Messali, S., Ballabio, A. & Meroni, G. Functional characterization of the Opitz syndrome gene product (midin): evidence for homodimerization and association with microtubules throughout the cell cycle. Hum Mol Genet. 8, 1387–1396 (1999).
Schweiger, S., Foerster, J., Lehmann, T., Suckow, V., Muller, Y. A., Walter, G. et al. The Opitz syndrome gene product, MID1, associates with microtubules. Proc Natl Acad Sci USA 96, 2794–2799 (1999).
Granata, A. & Quaderi, N. A. The Opitz syndrome gene MID1 is essential for establishing asymmetric gene expression in Hensen's node. Dev. Biol. 258, 397–405 (2003).
Granata, A., Savery, D., Hazan, J., Cheung, B. M., Lumsden, A. & Quaderi, N. A. Evidence of functional redundancy between MID proteins: implications for the presentation of Opitz syndrome. Dev Biol. 277, 417–424 (2005).
Acknowledgements
The Swedish Research Council, the Foundation Frimurare Barnhuset, HRH Crownprincess Lovisas Foundation, the Stockholm City Council and the Karolinska Institutet supported the work. We thank all the participating patients and their families.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, X., Chen, Y., Zhao, S. et al. Hypospadias associated with hypertelorism, the mildest phenotype of Opitz syndrome. J Hum Genet 56, 348–351 (2011). https://doi.org/10.1038/jhg.2011.17
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.17
