
Abstract
The fucosyltransferase 8 gene (FUT8) encodes an enzyme that transfers fucose to the innermost N-acetylglucosamine unit of N-glycan chains. Recent study showed that fut8-deficient mice develop pathological and physiological phenotypes resembling pulmonary emphysema (PE). The role of FUT8 in human PE is not known. A non-synonymous single-nucleotide polymorphism at the amino-acid position of 267 in FUT8 (rs35949016; C/A, C allele=Thr, A allele=Lys) was genotyped in a total of 1149 consecutive autopsies of elderly Japanese. A following study included 182 outpatients with chronic obstructive pulmonary disease, whose emphysematous changes were assessed quantitatively as the percentage of low attenuation area (%LAA) by high-resolution computed tomography. PE was detected in 163 of 1149 autopsy subjects (14.2%). Comparison of patient with vs without PE indicated that the FUT8 A allele was associated with PE (AA+AC vs CC; odds ratio=1.74, 95% confidence intervals=1.19–2.56, P=0.005). In the clinical study, presence of the FUT8 A allele significantly correlated with %LAA after adjustment (AA+AC vs CC=37.5±14.7 vs 32.7±13.9, P=0.02). The FUT8 gene Thr267Lys polymorphism is associated with human PE, and the Lys allele is the risk. The core fucosylation might be involved in the molecular pathogenesis of human PE.
Similar content being viewed by others


Introduction
Pulmonary emphysema (PE) is characterized by the destruction of lung parenchyma. The combination of PE and small airway disease is the major pathological feature of chronic obstructive pulmonary disease (COPD). PE is usually defined by pathological identification of permanent abnormal airspace enlargement distal to the terminal bronchioles. It is accompanied by destruction of the alveolar walls without obvious fibrosis.1 Pathogenesis of PE involves chronic inflammation and oxidative stress due to cigarette smoking and imbalance between protease and anti-protease activity in the lung.2 Recent literature suggests that many genetic factors may have a role in disease susceptibility, but a detailed mechanism is yet to be clarified.3 Recent study showed that mice with fucosyltransferase 8 gene (fut8) disruption develop emphysematous destruction of the lung.4 The fut8 gene encodes α1,6-fucosyltransferase, which catalyzes the transfer of a fucose residue to N-linked oligosaccharides on glycoproteins in the mammals.5 The emphysematous change in the lung of fut8-null mice was accompanied by overexpression of matrix metalloproteinases and marked dysregulation of transforming growth factor (TGF)-β receptor signaling.4 As mice fut8 and human FUT8 genes are the only genes responsible for core fucosyl transfer, and both are 96.5% identical at the amino-acid level, we hypothesized that the human FUT8 gene would be a biologically plausible candidate for PE susceptibility.
In this study we searched for non-synonymous single-nucleotide polymorphisms (SNPs), of which minor allele frequency is >10%, and selected the FUT8 Thr287Lys polymorphism. We studied the association between this SNP and PE prevalence, which was categorized on the basis of macroscopic scores in consecutive autopsy subjects, and then quantitatively assessed the emphysematous changes by computed tomography in clinical subjects.
Materials and methods
Study population
Details of the first population are described elsewhere.6 Briefly, the initial study subjects were recruited from consecutive autopsies samples (n=1536) performed at the Department of Pathology, Tokyo Metropolitan Geriatric Medical Center, Tokyo, Japan, between 1995 and 2004. Among them, the pathological data for emphysema were available in 1335 samples. We genotyped all of the samples but as there were degraded and deficient DNA samples; genotypes of 1228 samples were determined. Of these, the overlapping 1149 samples with relevant information of pathological findings of the lung were used in this study. The characteristics of the study population are available at the Internet-based database, ‘The Japanese SNP database for geriatric research (JG-SNP)’.7 For the second study, 182 outpatients who attended the Respiratory Care Clinic, Nippon Medical School between April 2007 and April 2009 for ambulatory treatment of cough, expectoration, and/or dyspnea, and diagnosed with COPD and emphysema were invited to participate. All of these patients were current or ex-smokers.
Pathological assessment of PE
For the autopsy study, both the excised lungs were inflated with a 10% formalin solution at a constant pressure of 25 cm H2O. The lungs were dissected in the sagittal plane to obtained 2-cm-thick slices. A pulmonary pathologist (KT) macroscopically assessed the type and severity of emphysematous changes in all lung slices. Emphysema was classified as centrilobular emphysema, panlobular emphysema or focal emphysema, and its severity was scored on a scale of 0–4 for each slice using a previously described method,1, 8 where 0=no changes, 1=minimal changes, 2=mild changes, 3=moderate changes and 4=severe changes. In this study, emphysema was defined as centrilobular emphysema changes with a score of >2 and/or panlobular emphysema changes with a score of ⩾1, as centrilobular emphysema changes are known to be mainly caused by smoking, and panlobular emphysema with centrilobular emphysema is considered an extension of severe centrilobular emphysema.9 Patients whose emphysema scores for both the lungs differed by more than two points were excluded from the study, because in general, bilateral severity of emphysema is closely correlated in COPD,9 and such wide differences might be due to additional unusual effects. The mean scores for slices from both the lungs were calculated and employed for association analysis. Pathological assessment and genotyping were performed at different institutions in a double-blind manner.
Pulmonary function parameters
In the clinical sample, post-bronchodilator forced expiratory volume in 1 sec (FEV1), carbon monoxide-diffusing capacity (diffusing capacity divided by alveolar volume (DLCO/VA)), vital capacity and forced vital capacity were measured according to the American Thoracic Society guidelines10 using the equipment for lung function testing (CHESTAC; CHEST Co., Tokyo, Japan). We used the reference values of post-bronchodilator FEV1 and vital capacity specified by the Japanese Respiratory Society. A significant bronchodilator response was defined according to the Global Initiative for Obstructive Lung Disease criteria as a 12% and 200 ml improvement in FEV1 compared with baseline values.11
Quantitative assessment of emphysema by high-resolution computed tomography of the chest
In the clinical study, helical CT scans were acquired using 1.25-mm collimation, a scan time (rotation time) of 0.8 s, 120 kV and 100∼600 mA with a Light Speed Pro16 CT scanner (GE Co., Tokyo, Japan). Severity of emphysema was determined from high-resolution computed tomography (HRCT) scans obtained at full inspiration. The percentage of the low attenuation area (%LAA) was calculated to assess the extent of emphysema, using HRCT scans according to Nakano et al.,12 with a minor modification. Briefly, six slices were obtained from the bilateral lungs at the upper, middle and lower lobes. The upper slice was obtained at the aortic arch, a middle section was taken at the carina, and a lower section was taken ∼3 cm above the top of the diaphragm, and the mean value was used as a representative value of %LAA. Voxels within each field were categorized as emphysema based on their reconstructed image grey scale using Hounsfield units (HU). A density histogram obtained from the distribution of the number of voxels at each HU within the lung was plotted and values less than −940 HU were calculated. As it is reported that the threshold of the density mask is approximately −950 HU, −910 HU and −850 HU for severe, moderate and mild PE, respectively,13 our threshold corresponds to the detection of moderate-to-severe emphysema.
SNP selection and Genotyping
Human FUT8 is a large gene that comprises 333 kb in size, and there are 2937 SNPs in the FUT8 gene region according to dbSNP data.14 We sought for tag SNPs of FUT8 gene by Haploview and still there were 80 tag SNPs. Thus, we focused on non-synonymous SNPs. Among four non-synonymous SNPs in the FUT8 gene, we further selected rs35949016 as the minor allele frequency was >10%. Genomic DNA of the autopsy subjects was extracted from the renal cortex by the phenol–chloroform method and stored at −20 °C until use. For the clinical subjects, DNA was isolated from whole blood by using a QIAamp DNA Blood Mini Kit (Qiagen K.K., Tokyo, Japan). Genotype analysis was performed using 10 ng aliquots of DNA which were placed into 384-well PCR plates using an automated fluid dispenser (BioMek2000; Beckman Coulter Inc., Fullerton, CA, USA) and then dried in a dry chamber. Taqman Assay for rs35949016 was done in a total of 10 μl reaction mixture according to the vender's instruction (Applied Biosystems Inc., Foster City, CA, USA). Call for genotype was made by Lightcycler480 (Roche Diagnostics, Mannheim, Germany) and accompanying software.
Statistical analysis
A total 1149 autopsy subjects and 182 clinical subjects were included. Allelic and genotype test were used to detect genetic contributions. Multiple logistic regression analyses were performed to examine the emphysema-related differences in the patients’ clinical backgrounds and to calculate the odds ratio (OR) and 95% confidence intervals (CIs) for data on autopsy subjects. The effect of the SNP genotype on %LAA was calculated using a linear regression model with dominant, recessive or additive models on the minor allele to analyze the data on clinical subjects. The estimates were adjusted for gender, smoking status, pack-years and age at the time of death, when appropriate. Hardy–Weinberg equilibrium was tested using the χ2 goodness-of-fit test. A P-value of < 0.05 was considered statistically significant. This study had a statistical power of 0.8 to detect an OR of 1.5 in carriers of at least on polymorphic allele, compared with carriers of homozygous wild-type alleles. Statistical analyses were performed with SPSS II for Windows (SPSS Japan Inc., Tokyo, Japan).
Ethical consideration
The ethical committees of Nippon Medical School, Tokyo Medical and Dental University, and Tokyo Metropolitan Geriatric Hospital approved the current study. Written informed consent was obtained from bereaved family for autopsy subjects, and directly from the clinical patients.
Results
Association with emphysema in consecutive autopsy subjects
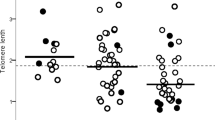
Table 1 shows characteristics of the autopsy subjects. Mean age at the time of death was 80.2 years. Of the 1149 subjects with relevant pathological records of the lung, 163 subjects (14.2%) met the pathological diagnosis of PE, and 986 subjects (85.8%) had no detectable PE. PE was more common among men than women (21.0% vs 6.7%, P<0.0001). This is likely to be due the significant gender difference for the history of smoking (men vs women: 84.9% vs 15.1%, P<0.0001). Centrilobular emphysema changes with the mean score of both lungs >2 were defined as PE as usual practice (Figure 1). A total of 157 subjects with genotyped data were defined as PE. In all, 14 subjects had panlobular emphysema changes and 8 of them were overlapped with centrilobular emphysema. Thus, a total of 163 subjects were categorized as PE.

Pathological score of centrilobular emphysema in autopsy subjects. The severity was scored on a scale of 0–4 for each slice as described in the text. 0=no change, 1=minimal changes, 2=mild changes, 3=moderate changes and 4=severe changes.
We examined the association between the rs35949016 SNP and emphysema employing these subjects (Table 2). The frequencies of the minor allele (A) and major allele (C) in the total population were 14.9% and 85.1%, respectively. The distribution of the genotype was not deviated from Hardy–Weinberg equilibrium (P=0.917). We performed allele count analysis under the contingency table of A/C alleles, and in subjects with PE (PE(+)) or without PE (PE(−)). Allele frequencies in PE(+) and PE(−) subjects were 18.7% and 14.2%, respectively; the difference was statistically significant (P=0.036). Genotype analysis under the additive model revealed that the prevalence of PE was significantly higher in AC heterozygotes compared with CC homozygotes (AC vs CC; OR=1.792, 95%CI=1.21–2.65; P=0.004), but not in AA homozygotes compared with CC homozygotes (AA vs CC; OR=1.172, 95%CI=0.32–4.36; P=0.812). As the minor A allele appeared to be the risk allele, we examined genotype association by adopting the A allele dominant model. The prevalence of PE in AA+AC genotypes was significantly higher than in the CC genotype (AA+AC vs CC; OR=1.50, 95%CI=1.06–2.13; P=0.024). This remained positive after adjustment for age, gender and smoking status (AA+AC vs CC; OR=1.74, 95%CI=1.19–2.56; P=0.005). There was no significant difference in adjusted recessive model (AA vs AC+CC; OR=1.01, 95%CI=0.27–3.717; P=0.992).
Association with emphysema in COPD subjects
To determine the effect of the FUT8 polymorphism in clinical subjects, we studied COPD patients with PE, which was evaluated by HRCT. The index of %LAA, which correlates with PE, was calculated.15 Subjects with bronchodilator response were excluded because the asthma or asthmatic component affects the %LAA value.16 Altogether, 182 subjects were investigated regarding the association between the genotype and %LAA as the extent of emphysema (Table 3). The minor A allele frequency was 14.8%, and the distribution of the genotype obeyed Hardy–Weinberg equilibrium (P⩾0.05). There was no difference in age, gender ratio, smoking status and pulmonary function between the AA+AC vs CC genotypes (Table 3). However, there was a significant difference in %LAA (AA+AC vs CC=37.5±14.7 vs 32.7±13.9, P=0.04) and it remained significant when adjusting for age, gender and pack-years (P=0.02).
DiscussionWe found that a non-synonymous SNP, Thr267Lys, of the FUT8 gene is associated with human PE in two different settings: one in consecutive autopsy subjects in which PE was pathologically confirmed, and another in clinical COPD patients, all of whom underwent assessment of PE by HRCT. In both studies, the A allele carriers were associated with more severe PE compared with the CC genotype. To our knowledge, this is the first report to demonstrate that polymorphism of FUT8, an enzyme involved in glycosyl transfer, may be linked to the pathogenesis of human PE.
In the pathological examination, we found considerable number of mild emphysematous changes with the score of 1 and 2 (Figure 1). These mild changes were excluded from the current analysis, as our usual practice. If these were included, the association would be negative (data not shown). Thus, the association of the polymorphism appears to be found only in moderate-to-severe PE. The observation using clinical subjects also are in the same line, as %LAA detected by −940 HU in our analysis corresponds to moderate-to-severe emphysema. According to a criteria by Hoffman et al.,13 the threshold of the density mask is approximately −950 HU, −910 HU and −850 HU for severe, moderate and mild PE, respectively.
FUT8 encodes α1,6-fucosyltransferase, which catalyzes the addition of a fucose residue to N-linked oligosaccharides on glycoproteins by means of an α1,6-linkage to form core fucosylation.5, 17 The FUT8 product is the sole enzyme that catalyzes this transfer in mammals. Core glycoprotein fucosylation is widely distributed in mammalian tissues and is seen in various proteins including TGFβ receptor,4 epidermal growth factor receptor,18 alpha-1-antitrypsin19 and integrins.20
Recently, mice with a disrupted fut8 gene were developed.4 They showed severe growth retardation and early death during postnatal development; surviving mice developed PE-like changes in the lung. Compared with wild type or hetero-knockout mice, these fut8-null mice displayed generalized air space enlargement and dilated alveolar ducts on histological assessment, and higher static lung compliance as measured by pressure–volume relationships. A marked overexpression of matrix metalloproteinase 12 and matrix metalloproteinase 13 and a downregulation of elastin, an extracellular matrix protein, were seen in the lungs of fut8-null mice. The correlation of phenotypes in fut8-null mice to human PE is not currently clear. Destruction of the lung in fut8-null mice might be due to impairment of developmental process of the lung, whereas that of human PE is clearly due to pathogenic processes in mature lung tissue that occurs later in life. Nevertheless, it is surprising that the same gene product underlies a similar phenotype and suggests that regulation of core fucosylation might be an important step in development of human PE.
An intriguing observation in fut8-null mice is that intraperitoneal administration of TGFβ rescued the PE phenotype. This implies that decreased TGFβ signaling, probably due to defect in the core fucosylation of the TGFβ receptor, contributed to the pathological phenotypes.4 TGFβ is known to have a role in the pathogenesis of COPD at various levels.21 Although increased TGFβ signaling triggers the development of small airway disease, several lines of evidences indicate that decreased TGFβ signaling leads to PE.22, 23 Taken together, it is tempting to speculate that the FUT8 gene product with 267Lys is less active than that with 267Thr, and thus impairs the TGFβ pathway and leads to PE rather than small airway disease. This is in agreement with our observation that the Thr267Lys polymorphism was associated with PE but not other parameters of small airway disease in COPD (Table 3). Although functional consequence of the amino-acid change of threonine to lysine at position 267 of FUT8 is not currently clear, Thr267Lys resides in the proline-rich region (261 amino acids to 340 amino acids), which is conserved among glycosyltransferases.24, 25 Thus, it is speculated that this substitution may have some effect on enzymatic function. Biochemical analysis of enzyme variants with 267Thr and 267Lys is warranted to clarify the activity and role of FUT8 in the pathogenesis of PE.
Limitations in the current study include that PE samples in the autopsy study are all those of old aged, and thus the study population may have been skewed towards individuals with mild disease because of survival bias. Also, we could not obtain clinical information on pulmonary function tests for these subjects.
In conclusion, we demonstrated that the Thr267Lys polymorphism in the FUT8 gene is associated with moderate-to-severe PE. The A allele, which encodes 267Lys, appears to be the risk allele for emphysema. Further clarification of the function of FUT8 may shed light on novel pathogenic pathways in human PE.
References
Report of a National Heart, Lung, and Blood Institute, Division of Lung Diseases workshop.. The definition of emphysema. Am. Rev. Respir. Dis. 132, 182–185 (1985).
Stockley, R. A. Neutrophils and protease/antiprotease imbalance. Am. J. Respir. Crit. Care Med. 160, S49–S52 (1999).
Wan, E. S. & Silverman, E. K. Genetics of COPD and emphysema. Chest 136, 859–866 (2009).
Wang, X., Inoue, S., Gu, J., Miyoshi, E., Noda, K., Li, W. et al. Dysregulation of TGF-beta1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc. Natl Acad. Sci. USA. 102, 15791–15796 (2005).
Miyoshi, E., Noda, K., Yamaguchi, Y., Inoue, S., Ikeda, Y., Wang, W. et al. The alpha1-6-fucosyltransferase gene and its biological significance. Biochim. Biophys. Acta. 1473, 9–20 (1999).
Fujimoto, K., Ikeda, S., Arai, T., Tanaka, N., Kumasaka, T., Ishii, T. et al. Polymorphism of SERPINE2 gene is associated with pulmonary emphysema in consecutive autopsy cases. BMC Med. Genet. 11, 159 (2010).
The Japanese SNP database for geriatric research (JG-SNP).. http://www.tmghig.jp/jg-snp/english/E_top.html.
Yamanaka, A. Morphopathology of chronic pulmonary emphysema. Acta. Pathol. Jpn. 15, 33–39 (1965).
Thurlbeck, M. William Chronic airflow obstruction. in Thurlbeck's Pathology of the Lung. 3rd edition, (eds. Churg, A.M. et al.) 675–742 (Thieme, New York, 2005).
Standardization of Spirometry, 1994 Update.. American Thoracic Society. Am. J. Respir. Crit. Care Med. 152, 1107–1136 (1995).
Rabe, K. F., Hurd, S., Anzueto, A., Barnes, P. J., Buist, S. A., Calverley, P. et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 176, 532–555 (2007).
Nakano, Y., Muro, S., Sakai, H., Hirai, T., Chin, K., Tsukino, M. et al. Computed tomographic measurements of airway dimensions and emphysema in smokers. Correlation with lung function. Am. J. Respir. Crit. Care Med. 162, 1102–1108 (2000).
Hoffman, E. A., Simon, B. A. & McLennan, G. State of the Art. A structural and functional assessment of the lung via multidetector-row computed tomography. Phenotyping chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 3, 519–534 (2006).
The National Center for Biotechnology Information dbSNP database. http://www.ncbi.nlm.nih.gov/projects/SNP/.
Sakai, N., Mishima, M., Nishimura, K., Itoh, H. & Kuno, K. An automated method to assess the distribution of low attenuation areas on chest CT scans in chronic pulmonary emphysema patients. Chest 106, 1319–1325 (1994).
Mitsunobu, F., Ashida, K., Hosaki, Y., Tsugeno, H., Okamoto, M., Nishida, K. et al. Complexity of terminal airspace geometry assessed by computed tomography in asthma. Am. J. Respir. Crit. Care Med. 167, 411–417 (2003).
Takahashi, M., Kuroki, Y., Ohtsubo, K. & Taniguchi, N. Core fucose and bisecting GlcNAc, the direct modifiers of the N-glycan core: their functions and target proteins. Carbohydr. Res. 344, 1387–1390 (2009).
Matsumoto, K., Yokote, H., Arao, T., Maegawa, M., Tanaka, K., Fujita, Y. et al. N-Glycan fucosylation of epidermal growth factor receptor modulates receptor activity and sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 99, 1611–1617 (2008).
Nakagawa, T., Uozumi, N., Nakano, M., Mizuno-Horikawa, Y., Okuyama, N., Taguchi, T. et al. Fucosylation of N-glycans regulates the secretion of hepatic glycoproteins into bile ducts. J. Biol. Chem. 281, 29797–29806 (2006).
Zhao, Y., Itoh, S., Wang, X., Isaji, T., Miyoshi, E., Kariya, Y. et al. Deletion of core fucosylation on alpha3beta1 integrin down-regulates its functions. J. Biol. Chem. 281, 38343–38350 (2006).
Königshoff, M., Kneidinger, N. & Eickelberg, O. TGF-beta signaling in COPD: deciphering genetic and cellular susceptibilities for future therapeutic regimen. Swiss Med. Wkly. 139, 554–563 (2009).
Morris, D. G., Huang, X., Kaminski, N., Wang, Y., Shapiro, S. D., Dolganov, G. et al. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 422, 169–173 (2003).
Bonniaud, P., Kolb, M., Galt, T., Robertson, J., Robbins, C., Stampfli, M. et al. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J. Immunol. 173, 2099–2108 (2004).
Uozumi, N., Yanagidani, S., Miyoshi, E., Ihara, Y., Sakuma, T., Gao, C. X. et al. Purification and cDNA cloning of porcine brain GDP-L-Fuc:N-acetyl-beta-D-glucosaminide alpha1 → 6fucosyltransferase. J. Biol. Chem. 271, 27810–27817 (1996).
Javaud, C., Dupuy, F., Maftah, A., Michalski, J. C., Oriol, R., Petit, J. M. et al. Ancestral exonic organization of FUT8, the gene encoding the alpha6-fucosyltransferase, reveals successive peptide domains which suggest a particular three-dimensional core structure for the alpha6-fucosyltransferase family. Mol. Biol. Evol. 17, 1661–1672 (2000).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yamada, M., Ishii, T., Ikeda, S. et al. Association of fucosyltransferase 8 (FUT8) polymorphism Thr267Lys with pulmonary emphysema. J Hum Genet 56, 857–860 (2011). https://doi.org/10.1038/jhg.2011.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.118
Keywords
This article is cited by
-
Loss of core-fucosylation of SPARC impairs collagen binding and contributes to COPD
Cellular and Molecular Life Sciences (2022)
